
Abstract
Autosomal dominant hyper-IgE syndrome (AD-HIES) is linked to dominant negative mutations of the STAT3 protein whose molecular basis for dysfunction is unclear and presenting with a variety of clinical manifestations with only supportive treatment. To establish the relationship between the impact of STAT3 mutations in different domains and the severity of the clinical manifestations, 105 STAT3 mutations were analyzed for their impact on protein stability, flexibility, function, and binding affinity using in Silico approaches. Our results showed that 73% of the studied mutations have an impact on the physicochemical properties of the protein, altering the stability, flexibility and function to varying degrees. In particular, mutations affecting the DNA binding domain (DBD) and the Src Homology 2 (SH2) have a significant impact on the protein structure and disrupt its interaction either with DNA or other STAT3 to form a heterodomain complex, leading to severe clinical phenotypes. Collectively, this study suggests that there is a close relationship between the domain involving the mutation, the degree of variation in the properties of the protein and the degree of loss of function ranging from partial loss to complete loss, explaining the variability of clinical manifestations between mild and severe.
Keywords
Introduction
Autosomal dominant hyper-IgE syndrome (AD-HIES), or Job’s syndrome, is a life-shortening primary immunodeficiency and multisystem disorder characterized by developmental abnormalities, poor wound healing, and recurrent infections. 1 It is caused by dominant-negative mutations in signal transducer and transcription activator 3 (STAT3), a transcription factor that plays a central role in the signal transduction pathway of several cytokines, growth factors and other peptide hormones. 2
STAT (signal transducers and activators of transcription) proteins are a group of latent cytoplasmic transcription factors involved in cytokine signaling. 3 In resting cells, STAT3 is primarily dimerized at its N-terminal to the cytoplasm terminal (head-to-head). 4 In cells stimulated by peptide hormones, STAT3 is recruited to phosphotyrosyl (pY) peptide motifs in receptor complexes activated via its Src homology domain 2 (SH2), becomes phosphorylated at Y705, and then dimerizes C-terminal- to C-terminal (tail to tail). Tail-to-tail STAT3 homodimers accumulate in the nucleus, where they bind to DNA and activate promoters of genes involved in tissue damage repair, inflammation and immunity. 5
AD-HIES syndrome is caused by mutations in the STAT3 gene and is characterized by great diversification at the level of the clinical tables generated by the disease ranging from moderate to severe. These various clinical manifestations can be related to the location of the mutation at the gene level according to the field.3,4
The stability and flexibility of folded proteins are critical to their biological function, so predicting the energetic effects of protein mutations can improve the fundamental understanding of structural biology, the molecular basis of diseases, and possible pathways to treat these diseases with drugs. 6 Studying the effects of a mutation on protein stability, flexibility, and function is important to elucidate the mechanism of a disease. To this extent many computational tools have been developed in recent years for in silico predictions of protein stability, flexibility and function, ranging from empirical sequence-based approaches to rigorous free-energy based methods. 7
To establish the relationship between different STAT3 mutations and variations in clinical phenotypes of AD-HIES pathology, we first collected all known STAT3 mutations from the literature. Then, we performed an in silico study to predict the impact of these mutations on the stability, flexibility, function and energy binding of this protein. Then, we explored the deleterious mutations and to examine how they induce severe clinical phenotypes.
Materials and Methods
Mutations collection
The selection of literature articles dating from 1999 to 2022 describing STAT3 mutations involved in AD-HIES was based on the following inclusion criteria: case reports, original articles and reviews; A search equation: (hyper IgE syndrome OR autosomal dominant hyper IgE syndrome) and (STAT3 mutations). These criteria resulted in collection of 105 point mutations (SNPs) of the STAT3 protein responsible for the development of AD-HIES. They were classified according to the STAT3 protein domains as follows: 12/105 mutations in the coiled coil domain (CC), 10/105 mutations in the linker domain (L), 34/105 mutations in the DNA binding domain (DBD), 36/105 mutations in the SH2 domain and 12/105 mutations in the transactivation (TA) domain.
Structures preparation and mutagenesis
The STAT3 protein crystal and DNA were collected in (.pdb) format with an ID (=6QHD, =1A73) characterized by a resolution of (2.85 Å, 1.80 Å) from the PDB (Protein Data Bank).
Out of 105 mutations altering the 5 domains, only 27 and 28 deleterious mutations affecting DBD and SH2, respectively, were selected for a docking study and to confirm the results, neutral mutations were used as controls. For this purpose, 5 neutral mutations for the DBD domain and 8 for the SH2 domain were prepared for molecular docking. Chimera (1.15) was used as a tool to generate 68 mutated models of the STAT3 protein. 8
Stability, flexibility, and function predictions
To predict, the effect of the collected mutations on STAT3 protein stability, flexibility and function, the SDM tool was used to predict the effect of each mutation on the stability of the STAT3 protein by specifying the impact that translates into stabilizing mutations and other destabilizing mutations. 9 This tool is a computational method that analyses the variation in amino acid replacements occurring in specific structural environments that are tolerated within the family of homologous proteins of known 3-D structures and converts them into substitution probability tables.
DynaMut 10 was used to predict the impact of mutations on the flexibility of the STAT3 protein, including mutations that increase flexibility and others that decrease it. This tool is a web server implementing 2 distinct, well established normal mode approaches, which can be used to analyze and visualize protein dynamics by sampling conformations and assessing the impact of mutations on protein dynamics and stability resulting from vibrational entropy changes. 11
The functional effect of those variations was further predicted using PROVEAN (P = protein/V = variation/E = effect/An = analyzer), including single amino acid substitutions and small insertions/ deletions. PROVEAN is a web server that provides a rapid analysis of protein variants based on a score to predict whether the mutation is deleterious or neutral. 7
Docking and scoring
HDOCK was used for docking of wild-type and mutated models with the STAT3 protein. Twenty-seven deleterious and 5 neutral mutations in the DBD domain were docked with DNA and then 28 deleterious and 8 neutral mutations were docked with the SH2 domain to assess the effect of mutations on STAT3-DBD-DNA and STAT3-STAT3 (SH2-SH2) affinity, respectively. 12
Results
Mutational effect prediction
A total of 105 mutations affecting the STAT3 protein were collected from the literature, and their distribution according to their domains is represented in Figure 1, which shows that the majority of the mutations are located in the DBD and SH2 domains, representing the most important domains of the protein.

STAT3 Mutations distribution. The plot depicts a lollipop graph showing the location of each mutation over the protein domains (Uniprot id P04637).
After predicting the effect of each mutation on the stability, flexibility and function of the STAT3 protein structure, the results were divided according to the domain to which each mutation belongs to relate the effect of the mutation to the function of each domain (Supplemental Table 1S).
According to the scatter plot shown in Figure 2, representing the distribution of the stability and flexibility scores of the deleterious and neutral mutations in the DBD and SH2 domains, the 2 most important domains of the STAT3 protein, we observed that the neutral mutations were mainly located near 0 (point representing the wild type) for the 2 parameters studied and the deleterious mutations were distributed in different zones.

Scatterplot representing the distribution of stability and flexibility per domain among different mutations. Neutral mutations are represented in triangle, deleterious one in circle and the reference in square.
The coiled coil (CC) domain, contains 12 mutations that generate stability values between −3.2 and 0.35; according to SDM. Only 2 mutations (R152W/M206R) increase the stability of the CC domain, while 10 decrease it. The flexibility values generated by these mutations ranges between 0.10 and −0.52, which are all values indicating increased flexibility. According to the Provean tool 8 mutations are deleterious and 4 are neutral (Figure 3A).

Mutations contained in the different domains of the STAT3 protein. (A) CC domain, (B) DBD domain, (C) SH2 domain, (D) TA domain, (E) L domain.
The DNA binding domain (DBD) contains 34 mutations, 18 of which increase the stability of the protein structure and 17 of which decrease it, with stability values ranging from −3.77 to 3.78. Among the 34 mutations 24 increase the protein flexibility compared to 9 that decrease this it, with flexibility values ranging between −0.579 and 1.11. according to Provean 29/34 mutations are deleterious and 5/34 are neutral (Figure 3B).
The SH2 domain contains 36 mutations, and according the SDM tool, 13 are stabilizing mutations and 23 are destabilizing mutations, with stability values ranging between −2.26 and 1.01. According to DynaMut, 25 mutations increase the flexibility of the domain and 11 mutations decrease it, with flexibility values ranging between −1.52 and 4.716. According to the Provean tool, of the 36 mutations, 27 are deleterious and 9 are neutral (Figure 3C).
The transactivation domain (TA) contains 12 mutations that are divided according to the SDM tool into 8 destabilizing, 2 stabilizing and 2 neutral mutations, with stability values ranging between −1.31 and 0.45 kcal/mol. According to DynaMut, an equal number of mutations (six) increase and decrease the flexibility of the domain, with values ranging between −4.387 and 4.16 kcal/mol. According to Provean, 2 mutations are deleterious and 10 are neutral (Figure 3D).
The Linker domain (L) contains 10 mutations, an equal number of which increase and decrease peotein stability according to SDM, with stability values ranging between −1.15 and 2.54 kcal/mol. According to DynaMut, these mutations are also divided equally between −1.048 and 0.71 kcal/mol. According to Provean, 9 mutations are deleterious and 1 is neutral (Figure 3E).
Of the 105 mutations studied, almost half (51 mutations) were determined to be deleterious and destabilizing, with stability scores ranging between −0.03 and −3.77 (Figure 4).
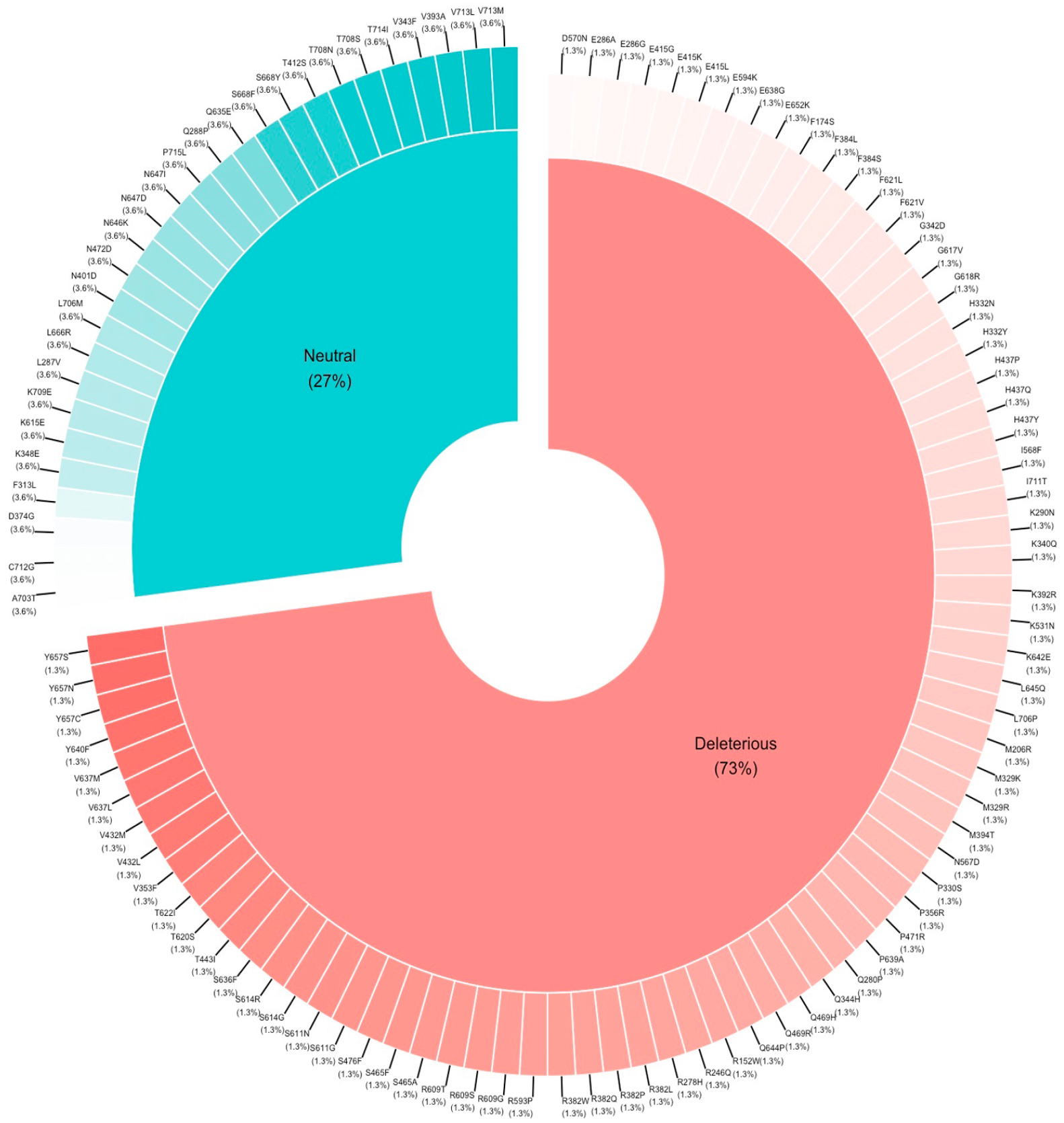
Distribution of mutations (neutral and deleterious).
Docking and scoring
To better understand the effect of mutations on the interaction of the SH2 and DBD domains and subsequently on their cytoplasmic dimerization, which is necessary for nuclear translocation and DNA binding of the STAT3 protein (SH2-SH2 and DBD-DNA), docking studies of the deleterious mutations of the SH2 and DBD domains were first performed using the HDOCK tool first. Then docking studies of the neutral mutations were performed to verify the reliability of the results.
For the DBD domain, the binding affinity score of the wild type was −221.85, while the molecular docking scores of the deleterious mutated models ranged from −218.74 to −185.85, and those of the neutral models ranged between −494.77 and −510.58 (Figure 5A).
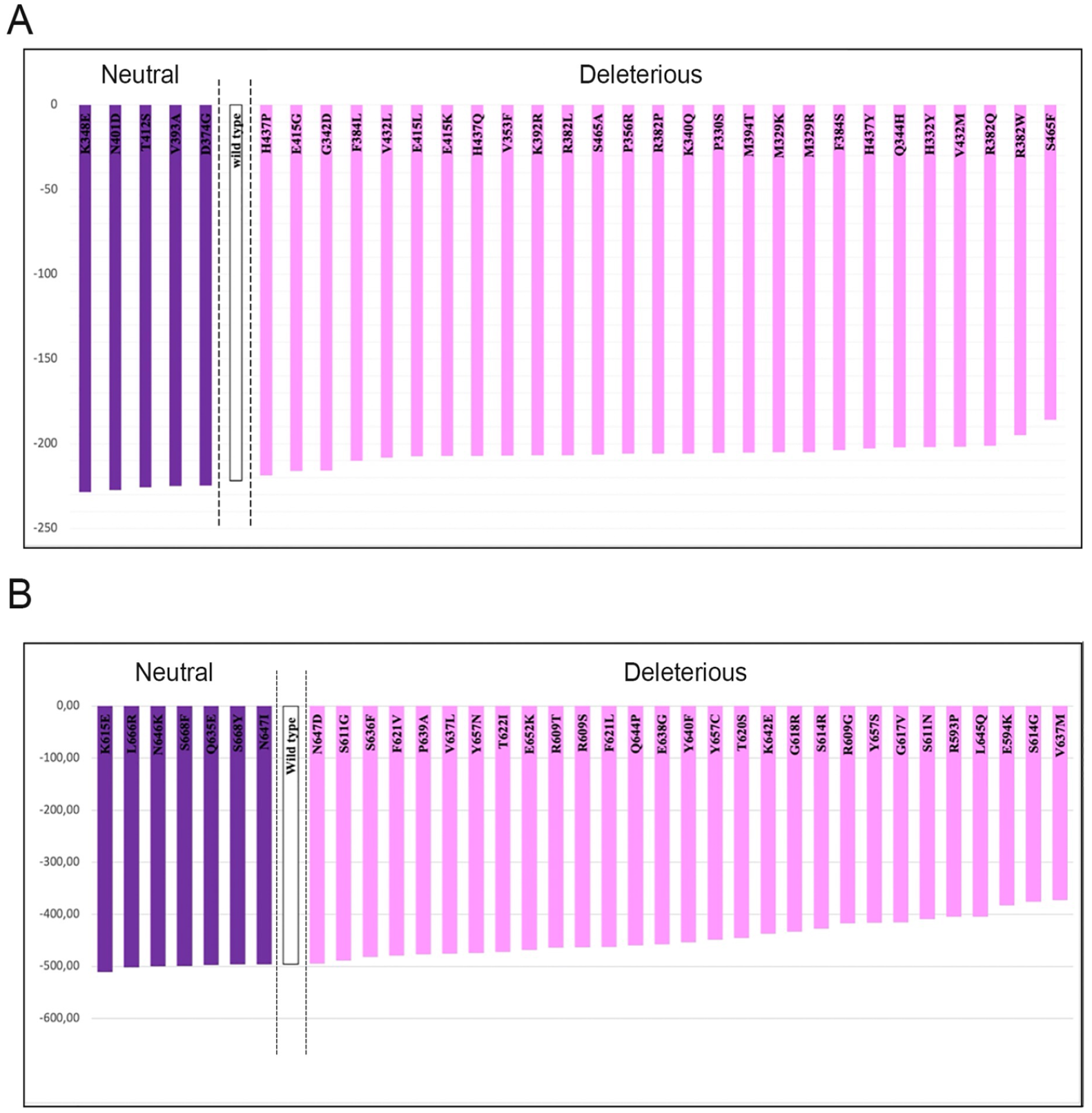
Binding energy of DNA binding domain (A) and Src Homology 2 (B) domain’s mutations.
For the SH2 domain, as shown in Figure 5B, the binding affinity score of the wild type was −495.44. The structures generated from the deleterious mutations showed decreased affinity energy, with values ranging between −489.21 and −372.80, while those generated from the neutral mutations increased the affinity energy, although not significantly, with scores ranging between −510.58 and −494.77.
Mutations and clinical signs
After studying the effects of the mutations on the protein structure, we investigated their clinical impact on patients by collecting clinical signs reported in the literature to have manifested in the presence of each mutation. Figure 6 shows the clinical signs that characterize each mutation, according to the protein domain. The deleterious mutations that encompass all or most of the clinical signs are located in the DBD and SH2 domains, while mutations in the other domains show only a few signs that are not severe. Neutral mutations present moderate clinical manifestations.

Bubble plot showing the clinical signs of some mutations that cause AD-HIES syndrome.
Discussion
In this study, all mutations of the STAT3 gene previously described in the literature that can cause hyper-immunoglobulin E syndrome were collected.2,13-16 These mutations were categorized according to the domain of the STAT3 protein to which they belong. Then using computer modeling, each mutation reported in AD-HIES patients that results in a single SNP substitution or deletion of an amino acid (104 known mutations) was analyzed to predict its effect on the stability, flexibility and function of the STAT3 protein structure, as well as to explore deleterious mutations that could alter the function of the STAT3 protein while determining the affected domain and the modified role. Docking studies were also carried out with the aim of highlighting the effect of the mutations on the interaction of the essential domains of the STAT3 protein, in particular SH2 and DBD.
A global in silico analysis of the mutations affecting the different domains showed the presence of 76 deleterious and 29 neutral mutations, 72% and 28% of the total mutations respectively. The deleterious mutations are distributed on the different domains of the STAT3 protein as follows: 9 for CCD, 28 for DBD, 28 for SH2, 2 for TA, and 9 for L. Thus, the majority of deleterious mutations affect the 2 most important domains of the protein. First, the DBD domain, which has a role, as its name indicates, in directing DNA binding and is also involved in nuclear translocation 17 and second the SH2 domain which is essential for the recruitment, dimerization and nuclear export of receptors. 18 According to our study, these deleterious mutations have an impact on the structure and flexibility of the protein; Thus, their deleterious effects could be explained by the modification of the three-dimensional structure of the protein, which is closely linked to the alteration of its function and thus the inhibition of the signaling pathway in which the protein is involved.
Of the 76 deleterious mutations, 38 decreased stability and increased flexibility, 12 increased stability and decreased flexibility, 10 decreased stability and flexibility, and 14 increased stability and flexibility. These different impacts of the mutations on the flexibility and stability of the STAT3 protein can be explained by the type of substituted amino acids generated by the mutation, either in terms of the nature of its side chain or in terms of the polarity and charge of its side chain, which modify the function and structure of the protein differently.
The scatter plot presents the values of stability and flexibility of the deleterious and neutral mutations in the DBD and SH2 domains. The majority of the neutral mutations have flexibility values close to 0 with variation not exceeding 0.5, so they do not have a significant impact on the protein structure, and most of the deleterious mutations are located in different regions, mainly those far from the reference value of 0, which shows that they have an effect on the stability and flexibility of the protein structure and may alter protein function and subsequent clinical signs. Therefore, any mutation that decreases flexibility is deleterious.
For the first graph representing the distribution of mutations in the DBD domain, the majority of mutations are deleterious (circle) and are located in different areas altering stability and flexibility more significantly than the neutral mutations (triangle), which cluster mainly near 0 (square) and do not exceed a variation of 0.5 at the level of the 2 variables, excluding D374G, which increases stability, and V393A, which decreases it. It can be deduced that deleterious mutations have a direct impact on stability and flexibility but to different degrees and that these structural parameters are strongly linked to the function of the altered protein to different degrees and therefore have a different impact on the clinical phenotypes. We also note that any mutation decreasing flexibility is deleterious, which can be explained by a gain in rigidity.
The second part of Figure 2 represents the distribution of mutations at the level of the SH2 domain. Apart from mutation L666R, which increases flexibility, neutral SNPs (in blue) are located close to the origin, which explains their neutrality in terms of impaired protein function; however, most of the deleterious mutations (in red) alter stability and flexibility parameters, consistent with their deleterious effect on function, and the degree of change in structural parameters may be directly related to the severity of clinical manifestations.
According to our results, these deleterious mutations affect stability and flexibility to different degrees, so we suggest that they also affect other properties of the STAT3 protein to different degrees, since a deleterious genetic variation can affect the electrostatic nature of protein surfaces and introduce deleterious effects, such as changes in stability or folding, altering the specificity and affinity of the binding partner and modifying protein function. 19 This modification of function would depend closely on the type of mutation related to the type of amino acid substitution and the degree of variation in the properties of the protein. Thus, the loss of function is either total or partial.
Higher stability increases the rigidity of the protein, and lower stability increases the flexibility of the protein, 11 and 52.9% of the deleterious mutations studied followed this general rule, so a change in the flexibility pattern of functional residues could altered the functional region and activity of the mutant proteins. A decrease in overall flexibility and an increase in rigidity due to mutations may affect the binding properties of proteins.
We suggest that the 13 mutations that increase stability and decrease flexibility increase the rigidity of the protein and decrease its possible conformations, which can make the protein incapable of interacting with the target because it may lose the conformation suitable for this interaction and therefore lose its function completely. This is the example of the H332Y mutation, which considerably increases the stability of the protein and therefore its rigidity. This mutation is characterized by a severe clinical manifestation, which confirms our prediction 20 (Figure 6).
However, in the case of the 36 mutations that caused decreased stability, the flexibility was increased, and the possible conformations of the protein increased, which may decrease the chances of the protein interacting with its target and thus cause a partial loss of function.
Given that hyper IgEs syndrome is strictly related to loss-of-function (LOF) mutations in the STAT3 gene, 18 we suggest that the 12 LOF mutations mentioned above would cause a severe clinical manifestation, whereas the 50 mutations that generate partial loss of function would cause a less severe clinical manifestation. We also suggest that this variation in clinical outcome is strongly related to the domain of the protein affected by the mutation and that mutations affecting the DBD or SH2 domain will have a more deleterious effect than those affecting the other STAT3 domains. As an examples, the H332Y 20 and S636F 21 mutations which affect the DBD and SH2 domains respectively, and present a more severe clinical outcome than the S476F13,22 and I711T mutations which affect the linker and TA domains, respectively (Figure 6).
To better understand the interaction and the affinity of the complexes (STAT3-STAT3 and STAT3-DNA), the mutated models of the STAT3 protein were generated to perform a docking study of the mutated protein and compare the results to those of the control docking study. For the DBD domain, the docking scores varied between −193.79 and −204.28, which are scores with minimal values compared to the reference value (−215.01), while the SH2 domain presents a very significant decrease (fall) in docking scores of the mutated types varying between −176.32 and −234.96 compared to the value of the reference score (−1085.03). A drop in docking scores signifies a weak affinity resulting in partial or total dysfunction of the protein, which may explain how these deleterious mutations can induce AD-HIES disease.
To determine the clinical impact of the mutations on the syndrome, the clinical signs of each mutation were collected from the literature. On the basis of this data, the majority of the mutations are deleterious and located in the DBD and SH2 domains. also, the mutations R382W and V637M located in the DBD and SH2 domains, respectively, the most important domains of the STAT3 protein are associated with all of the clinical signs of AD-HIES disease and are the most commonly reported mutations in the literature, This explains their severity, and their impact on the structural properties of the protein. 23
Neutral mutations present moderate clinical manifestations that do not encompass all clinical signs, especially severe ones. They are located mainly in the TA domain, which can explain why these mutations do not affect or do not significantly affect the symptomatology of the patients and the properties of the STAT3 protein.
Conclusion
In summary, HIES is a group of different diseases with primary immunodeficiency and multisystem disorders characterized by developmental abnormalities, poor wound healing, and recurrent infections. AD-HIES is caused by heterozygous mutations with a dominant negative effect on the STAT3 gene. STAT3 mutations are located mainly in the SH2 and DNA binding domains, with fewer occurring in the transactivation domain. The mutations are missense mutations or short in-frame deletions, and therefore, the STAT3 protein is present but abnormal, leading to diminished STAT3 signaling. The clinical characteristics of HIES are similar to those of patients with SH2 domain and DNA binding mutations. However, the exact mechanism by which these changes lead to the traits of HIES is not known. This study investigated the impact of AD-HES mutations on the structural stability, flexibility, and function of the STAT3 protein and their impact on patient clinical outcomes. The results obtained suggest the existence of a close relationship between the degree of variation in flexibility and stability and the clinical manifestation of AD-HIES disease related to a total or partial loss of STAT3 function.
Supplemental Material
sj-docx-1-evb-10.1177_11769343231169374 – Supplemental material for In Silico Analyses of All STAT3 Missense Variants Leading to Explore Divergent AD-HIES Clinical Phenotypes
Supplemental material, sj-docx-1-evb-10.1177_11769343231169374 for In Silico Analyses of All STAT3 Missense Variants Leading to Explore Divergent AD-HIES Clinical Phenotypes by Mariam Mansouri, Ghyzlane El Haddoumi, Houda Bendani, Nasma Boumajdi, Mohammed Hakmi, Hanane Abbou, El Mehdi Bouricha, Boutaina Elgharbaoui, Souad Kartti, Rachid El Jaoudi, Lahcen Belyamani, Ilham Kandoussi, Azeddine Ibrahimi and Naima El Hafidi in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out under national funding from the Moroccan Ministry of Higher Education and Scientific Research (COVID-19 program) to A.I. This work was also supported by a grant from the Moroccan Institute of Cancer Research and the PPR-1 program to A.I. and Biocodex grant to AI.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MM contributed to the conceptualization, data collection and analysis, and writing the initial draft and revisions. GEH, HB, NB, and MH contributed to the data curation, methodology, and visualization. HA, EMB, BE, and SK contributed to the conceptualization and draft revision. REJ, LB, and IK supervised, revised, and validated the last draft. AI and NEH conceived and supervised the study, contributed to writing revisions and validation. All authors have read and agreed to the published version of the manuscript.
Abbreviations
AD-HIES: Autosomal dominant hyper IgE syndrome; SDM: Site Directed Mutator; Provean: Protein Variation Effect Analyzer; STAT3: Signal transducer and activator of transcription3; molecular docking.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.