
Abstract
Objectives: In the context of human immunodeficiency virus (HIV) treatment, the emergence of therapeutic failures with existing antiretroviral drugs presents a significant challenge. This study aims to employ advanced molecular modeling techniques to identify potential alternatives to current antiretroviral agents. Methods: The study focuses on three essential classes of antiretroviral drugs: nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), and protease inhibitors (PIs). Computational analyses were performed on a database of 3,343,652 chemical molecules to evaluate their binding affinities, pharmacokinetic properties, and interactions with viral reverse transcriptase and protease enzymes. Molecular docking, virtual screening, and 3D pharmacophore modeling were utilized to identify promising candidates. Results: Molecular docking revealed compounds with high binding energies and strong interactions at the active sites of target enzymes. Virtual screening narrowed down potential candidates with favorable pharmacological profiles. 3D pharmacophore modeling identified crucial structural features for effective binding. Overall, two molecules for class 1, 7 molecules for class 2, and 2 molecules for class 3 were selected. These compounds exhibited robust binding affinities, interactions with target enzymes, and improved pharmacokinetic properties, showing promise for more effective HIV treatments in cases of therapeutic failures. Conclusion: The combination of molecular docking, virtual screening, and 3D pharmacophore modeling yielded lead compounds that hold potential for addressing HIV therapeutic failures. Further experimental investigations are essential to validate the efficacy and safety of these compounds, with the ultimate goal of advancing toward clinical applications in HIV management.
Keywords
Introduction
Human immunodeficiency virus (HIV) infections remain a global health concern, and its effective management is of utmost importance in countries like Morocco. To fulfill its lifecycle and facilitate its replication, the HIV virus relies on two pivotal viral enzymes: reverse transcriptase and protease. Reverse transcriptase is responsible for converting the virus’s RNA genetic material into DNA, allowing it to integrate into the host cell's genetic makeup and instigate the production of viral components.1,2 Concurrently, protease assumes a critical role in the later stages of the HIV lifecycle by cleaving newly formed viral proteins into their functional forms. 3 This cleavage process is indispensable for the assembly of mature and infectious viral particles. The targeted inhibition of these enzymes has proven central to the development of antiretroviral therapies, which have significantly contributed to managing HIV infection and improving patient outcomes. When these enzymes are inhibited, the consequences for HIV are profound. Inhibition of reverse transcriptase prevents the formation of the essential viral DNA, rendering the virus unable to integrate into the host cell's genome and produce new viral particles. Similarly, inhibiting protease halts the formation of infectious viral particles, reducing the virus's ability to spread to new cells. Consequently, antiretroviral therapies target these enzymes precisely to hinder viral replication. The combination of drugs that inhibit reverse transcriptase and protease, among other targets, forms the backbone of HIV treatment regimens. These therapies have revolutionized the management of HIV infections, dramatically slowing the progression of the disease, improving the quality of life for those infected, and decreasing the transmission rates.
Over the years, significant progress has been made in the treatment of HIV with the introduction of antiretroviral therapy (ART), leading to improved outcomes and prolonged survival for people living with HIV. 4 Nevertheless, the occurrence of treatment failure has become a major obstacle in the fight against this devastating virus. 5
Treatment failure in HIV patients can be attributed to various common reasons. One significant factor is the development of drug resistance, where HIV mutates and becomes less susceptible to the effects of antiretroviral drugs, leading to diminished viral suppression.6,7 Additionally, poor adherence to medication is a crucial concern, as consistent adherence to antiretroviral therapy (ART) is essential for its efficacy. Failure to adhere to the prescribed treatment regimen can result in inadequate drug levels, allowing the virus to replicate and potentially develop resistance. 8 Moreover, drug toxicity is another challenge, as some antiretroviral drugs may cause side effects or toxicities that prompt patients to discontinue treatment or switch to alternative medications. 9 Intolerable side effects can negatively impact adherence and compromise treatment effectiveness. Furthermore, drug interactions pose a risk, with certain medications (including prescription drugs, over-the-counter medications, and herbal supplements) interacting with antiretroviral drugs and potentially affecting their efficacy. 8 Such interactions can lead to suboptimal drug levels and, consequently, treatment failure in HIV management.
Among all these factors previously cited, the development of drug resistance remains the most important factor contributing to treatment failure. HIV can mutate and develop resistance to antiretroviral drugs, making them less effective at suppressing viral replication.7,10 HIV drug resistance emerges as a result of genetic alterations within the HIV virus, leading to a diminished ability of antiretroviral drugs to effectively inhibit viral replication.11,12 This resistance is essentially highlighted by the emergence of drug-resistant strains: The widespread implementation of antiretroviral therapy (ART) worldwide has been accompanied by the emergence of HIV strains that are resistant to drug treatments. Over the past years, the prevalence of drug resistance has been consistently rising, 11 and escalating levels of drug resistance, posing a significant challenge to effective treatment strategies. This upward trend in drug resistance levels underscores the need for vigilant monitoring and proactive measures to combat this issue. Transmitted drug resistance refers to the acquisition of drug-resistant strains of HIV by certain individuals prior to initiating antiretroviral therapy. In other words, some individuals may already possess HIV strains that are resistant to certain drugs, even before starting antiretroviral treatment.9,13 Things that can have detrimental consequences, including treatment failure where the efficacy of antiretroviral drugs in suppressing viral replication diminishes. Consequently, this can lead to a rise in the number of HIV infections, increased HIV-associated morbidity, and mortality rates.11,14
There are several molecular modeling approaches that can be used to design new antiretroviral drugs and identify potential drug candidates for the treatment of HIV. Pharmacophore modeling is a computational technique used to identify the essential features of a molecule that are responsible for its biological activity.15,16 Pharmacophore models can be generated based on the known structure of a target protein or ligand, and used for virtual screening to identify potential drug candidates17,18;then, virtual screening is a computational technique used to identify potential drug candidates from large databases of compounds. Virtual screening can be performed using various methods, including pharmacophore-based screening, shape-based screening, and docking-based screening,16–18 the docking is a computational technique used to predict the binding mode of a ligand to a target protein. Docking can be used to identify potential drug candidates and optimize their binding affinity to the target protein18,19
By using these techniques, we can optimize the drug properties of potential drug candidates and predict their interactions with the target protein, which can help accelerate drug discovery.
The main objective of this study is to address the limitations of current antiretroviral HIV/AIDS therapies by proposing new, highly effective, and potent therapeutic molecules to replace the failing antiretrovirals from the three therapeutic classes (NRTI, NNRTI, and PI) currently in use. By leveraging the cutting-edge approaches of molecular docking, virtual screening, and 3D pharmacophore building, we seek to resolve the persistent issue of therapeutic failure in the treatment of HIV/AIDS, by employing these advanced computational techniques.
Materials and methods
First part: 3D-Pharmacophore model building
Structures collection
Sixteen antiretrovirals have been confirmed in the literature as the standard treatment actually in use for HIV infections, and they were collected from the HIV French Resistance database. 20 Those antiretrovirals were subsequently classified into three therapeutic classes: seven for nucleotide reverse transcriptase inhibitors (NRTI), five for non-nucleotide reverse transcriptase inhibitors (NNRTI), and four for protease inhibitors (PI).
Subsequently, the Protein Data Bank 21 (PDB) database was used to gather the 3D crystal structures of antiretrovirals in complex with viral enzymes (reverse transcriptase and protease).
For the purpose of conducting an extensive virtual screening on a vast molecular scale, a total of 3,344,652 molecules have been acquired in (.sdf) format from the Zinc database. 22
3D-Pharmacophore model generation
The actives compounds were collected from the literature and were aligned based on a set of common pharmacophoric features. The alignment process was performed in the Molecular Operating Environment (MOE) software, 23 utilizing the “Align Database” tool with appropriate parameters to maximize the structural overlap and match the pharmacophoric features. The aligned compounds were used to generate a pharmacophore model using the MOE software's “Pharmacophore Generation” module. The software analyzed the aligned structures to identify common chemical features, such as hydrogen bond acceptors, hydrogen bond donors, aromatic rings, hydrophobic regions, or any other relevant features.
Second part: Proposal of potential inhibitors
Virtual screening and docking
The molecules obtained after the initial screening using the Moe tool 23 exhibit the majority of the features contained in the pharmacophore model of each therapeutic class. After removing duplicates using the Open Babel software, 24 these molecules will undergo further screening using the Drulito software 25 to select the ones that comply with the Lipinski, Veber, and Ghose rules, known as drug-like molecules.
The docking process by Maestro from Schrödinger 26 was carried out on the previously screened molecules. The goal was to identify candidate structures with comparable or superior affinity to the molecules currently commercialized. A grid with a spacing of 1 Å was employed for each macromolecular protein, with fixed central coordinates of (X = 12, Y = 13, Z = 17) for NRTI and (X = 12, Y = 14, Z = 22) for IP. The grid size was defined as (X = 20, Y = 20, Z = 20). The grid settings file was obtained from the software’s grid menu option.
The 3D interactions between the protein and ligands, hydrogen bonds, and poses were visualized using Pymol, 27 a software tool for molecular visualization.
Subsequently, the Qikprop of Schrodinger 26 was utilized to calculate the ADME (Absorption, Distribution, Metabolism, and Excretion) descriptors. This step was crucial to conduct the pre-final screening, wherein only the molecules meeting the criteria of a validated therapeutic compound, based on their percentage of human oral absorption and predicted apparent Caco-2 cell permeability in nm/sec (QPPCaco) were retained.
Finally, these molecules will also undergo the last screening to eliminate toxic compounds using the pkCSM tool 28 : Predicting Small-Molecule Pharmacokinetic and Toxicity Properties.
Results
3D-Pharmacophore model generation
List of antiretroviral drugs used as standard treatment, classified into three therapeutic classes; NRTI, NNRTI, &PI.

The 3D pharmacophore model, created by the MOE software, describes the common features of the seven antiretrovirals (ARV) drugs in complex with reverse transcriptase. Figure 1 illustrates the shared common features of these compounds using the MOE software. The map presents two views: one with the alignment of candidate ligands (Figure 1(a)) and another without the ligands (Figure 1(b)). The 3D pharmacophore model comprises four main common features, namely F1 = Ani/Acc, F2 = Acc, F3 = Don, F4 = Ani/Acc, and F5 = Acc. The XYZ coordinates of all the three pharmacophore models generated are shown in Table 2. 3D pharmacophoric map of NRTI class ligands. (a), a simplified view of the model with aligned ligands. (b), a simplified view without aligned ligands. The XYZ coordinates of the generated pharmacophore models for NRTI, NNRTI, and PI classes.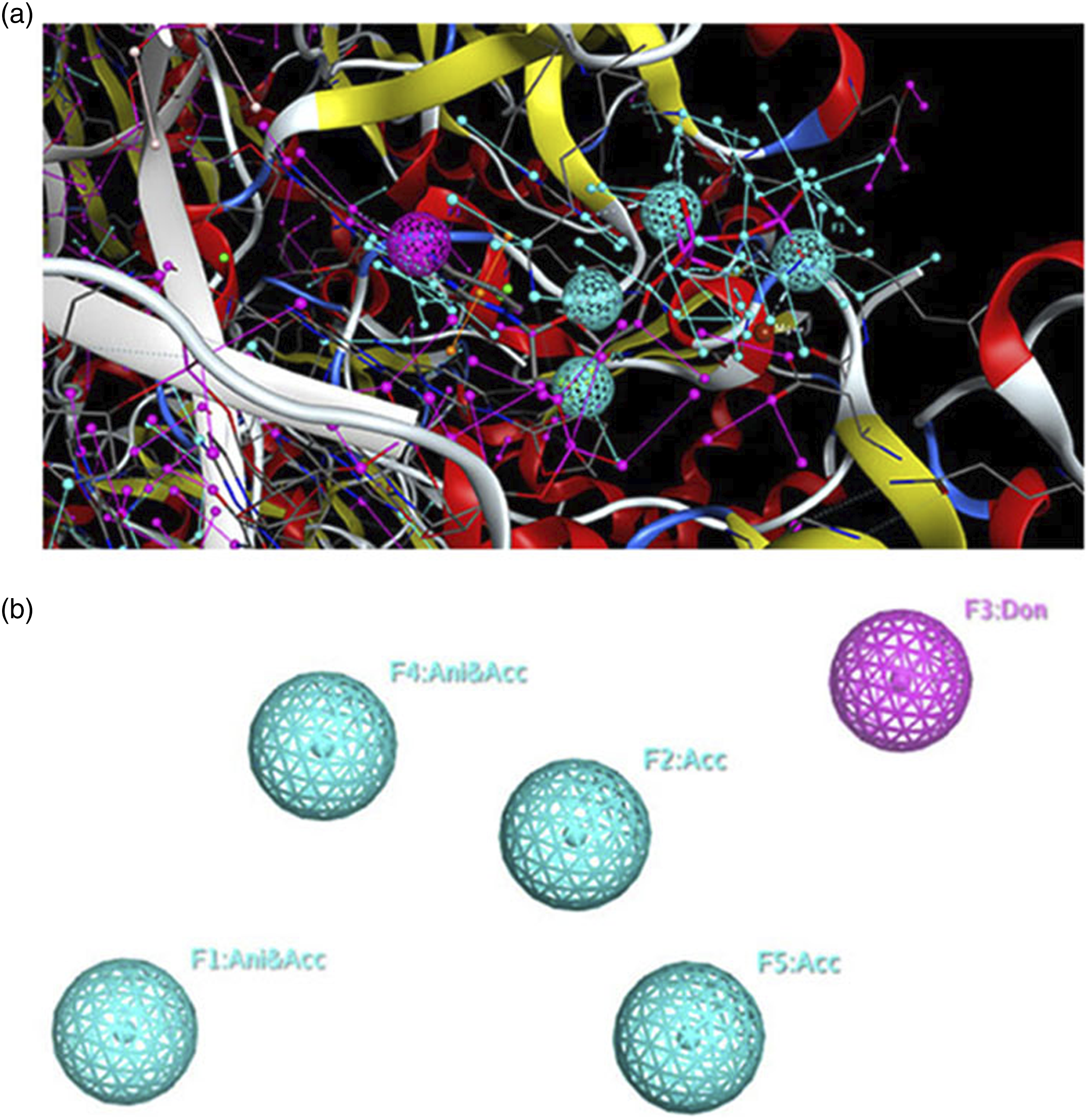

A pharmacophore model was created for five antiretroviral (ARV) drugs in complex with reverse transcriptase that fall under the category of non-nucleotide reverse transcriptase inhibitors (NNRTIs). The shared features of these compounds are depicted in Figure 2. The figure presents a simplified representation, both with the alignment of the five ligands (Figure 2(a)) and without the ligands (Figure 2(b)). The 3D pharmacophore model encompasses five common features: F1 = Don, F2 = Acc, F3 = Aro, F4 = Aro, and F5 = Aro, which are a combination of the features mentioned earlier. 3D pharmacophoric map of NNRTI class ligands. (a), a simplified view of the model with aligned ligands. (b), a simplified view without aligned ligands.
The pharmacophore model for the four antiretroviral drugs (ARVs) in complex with the protease viral enzyme, belonging to the protease inhibitor (PI) class has been generated. Figure 3 illustrates the shared characteristics of these compounds. The figure provides a simplified view, both with the alignment of the four ligands (Figure 3(a)) and without the ligands (Figure 3(b)). The 3D pharmacophore model consists of six common features: (1) Acc; (2) Acc; (3) Don; (4) Acc; (5) Don and (6) Aro. 3D pharmacophoric map of PI class ligands. (a), a simplified view of the model with aligned ligands. (b), a simplified view without aligned ligands.
Virtual screening
For NRTI class 1, we conducted a large-scale virtual screening of 3,344,652 molecules, starting with a pharmacophore-based screening approach. From this initial screening, we obtained 3849 molecules that contained all five specified features. Subsequently, these molecules were further filtered using the Drulito tool, resulting in 2593 drug-like molecules that complied with the Veber, Lipinski, and Ghose rules. The selected molecular database was then docked with the viral enzyme, reverse transcriptase. From the docking results, we focused on the top 10% of compounds, which consisted of 260 molecules with affinity scores ranging from −11.71 to −8.779 kcal/mol. Importantly, these affinities were greater than the reference docking score performed between the reference drug zidovudine and the reverse transcriptase which was −7.512 kcal/mol.
To further refine the selection, a subsequent screening step was performed based on the human oral absorption parameter for zidovudine, which was 65%.29–31 This value was in close agreement with literature estimates of approximately 64%. Nine molecules were excluded from consideration as their absorption values did not surpass 56%. Consequently, from the remaining 251 molecules, we identified 89 that displayed a Predicted Apparent Caco-2 Cell Permeability (QPPCaco) exceeding 500 nm/sec. This value is conventionally deemed suitable for assessing a drug’s capability to permeate the Caco-2 cell monolayer, an in vitro model of intestinal epithelium. Leveraging Schrödinger’s QikProp module, we harnessed a comprehensive array of molecular descriptors encompassing properties such as LogP, polar surface area (PSA), hydrogen bond donors and acceptors, molecular weight, and more. These descriptors are pivotal in predicting characteristics associated with absorption, distribution, metabolism, and excretion (ADME). Consequently, a final screening step focused on in silico toxicity evaluation yielded 78 molecules as candidates. Ultimately, after the culmination of rigorous evaluations, a subset of five molecules emerged as the most promising contenders that met all the established parameters. Refer to Figure 4(c) for a graphical representation of this selection process, while Table 3 provides an intricate display of the five potential candidate structures. This table encompasses pertinent details including nomenclature, 2D and 3D structures, chemical formulas, and other relevant data. General process of the virtual screening of the three therapeutic classes; NRTI (a), NNRTI (b) and PI (c). Potential candidate structures and relevant information for novel compounds for the NRTI class.

Seven candidate molecules-information and representations.

For a comprehensive exploration of protease inhibitors (PIs), a virtual screening process was meticulously conducted. This screening was predicated on a meticulously crafted pharmacophore model comprising six distinct features. As highlighted in Figure 4(c), the initial phase of this screening yielded 19 molecules that impeccably exhibited the stipulated characteristics as per the pharmacophore model.
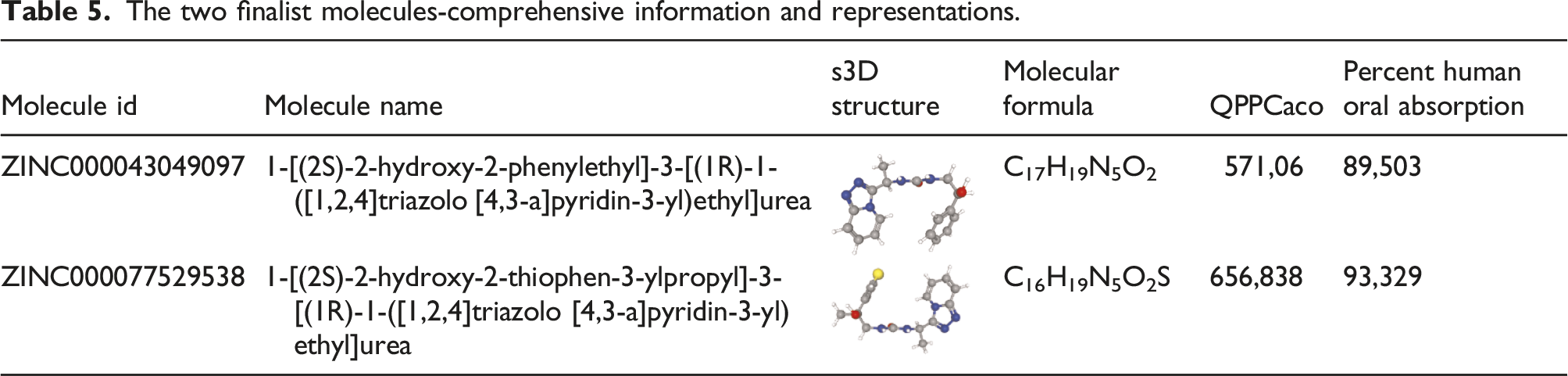
The two finalist molecules-comprehensive information and representations.
Overall, this multi-step virtual screening process has identified a promising subset of molecules that demonstrate favorable drug-like properties, enhanced binding affinity to the viral enzyme, and potential for improved oral absorption characteristics.
Discussion
In this study, a 3D pharmacophore model was developed using the MOE software to elucidate the shared features of antiretroviral (ARV) drugs while interacting with the reverse transcriptase and the protease enzymes. The pharmacophore model provides valuable insights into the key chemical features necessary for these drugs to effectively bind to their target.
For the first class of NRTI, the 3D pharmacophore model identifies four primary common features that we can explain as: F1 = Ani/Acc: This feature represents a dual nature, acting both as an aromatic ring (Ani) and as an acceptor (Acc) of hydrogen bonds during the drug-target interaction,16,32 then F2 = Acc: This feature denotes an acceptor group that can form hydrogen bonds with the target protein,33,34 F3 = Don: This feature signifies a donor group able to donate hydrogen bonds during the drug-target binding process, 35 and F4 = Ani/Acc: Similar to F1, this feature also possesses a dual characteristic, functioning as both an aromatic ring (Ani) and an acceptor (Acc) of hydrogen bonds.32,36 Additionally, the 3D pharmacophore model includes F5 = Acc, indicating another acceptor group in the ARV drugs that contributes to their interactions with the reverse transcriptase enzyme.15,35
Concerning the second class NNRTI, the obtained result highlights the development of a pharmacophore model specific to a group of five antiretroviral (ARV) drugs. This model aims to identify the common chemical features essential for their interaction with the reverse transcriptase enzyme. The 3D pharmacophore model includes five primary common features, each of which represents: F1 = Don: Represents a donor group capable of donating hydrogen bonds during the drug-target interaction, 34 F2 = Acc: Denotes an acceptor group that can form hydrogen bonds with the target protein, 37 F3 = Aro: Signifies an aromatic ring, which often plays a crucial role in hydrophobic interactions and π-π stacking with the target protein, F4 = Aro: Another aromatic ring feature, emphasizing the significance of additional hydrophobic interactions in the drug-target binding, and F5 = Aro: The third aromatic ring feature, further highlighting the importance of multiple aromatic interactions in the pharmacophore model. 38 These five common features combine the essential aspects identified earlier during the pharmacophore model creation.
For the protease inhibitor (PI) class, the 3D pharmacophore model is composed of six key common features: acceptor (Acc), additional acceptor (Acc), donor (Don), another acceptor (Acc), another donor (Don), and aromatic ring (Aro). These features represent essential chemical aspects that play significant roles in the drug–protease interactions. By elucidating the structural requirements for effective binding to the protease enzyme, the pharmacophore model provides valuable insights and serves as a fundamental tool for rational drug design and optimization within the PI-class antiretroviral medications, offering potential advancements in the treatment of HIV infections. However, further experimental validation will be essential to confirm the practical applicability and effectiveness of the identified pharmacophore features in the drug development process.39,40
The results obtained from the virtual screening and pharmacophore-based approach can be linked to the broader problem of treatment failure in HIV patients. The research aimed to identify potential drug candidates for nucleotide reverse transcriptase inhibitors (NRTIs), for non-nucleotide reverse transcriptase inhibitors (NNRTIs) and protease inhibitors (PIs), which are crucial classes of antiretroviral drugs used in HIV treatment.
One of the common reasons for treatment failure is the development of drug resistance. HIV has the ability to mutate and become less susceptible to the effects of antiretroviral drugs, leading to diminished viral suppression. The virtual screening and pharmacophore-based approach focused on identifying drug candidates with strong binding affinities to the target viral enzymes (reverse transcriptase and protease) to mitigate the risk of resistance development.
Another important factor in treatment failure is poor adherence to medication. Consistent adherence to antiretroviral therapy (ART) is critical for its efficacy. The identified drug candidates possess favorable drug-like properties, and their high potential for oral absorption can potentially enhance patient adherence by providing more convenient dosing regimens.
Drug toxicity is also a challenge in HIV treatment, as some antiretroviral drugs may cause side effects or toxicities that prompt patients to discontinue treatment or switch to alternative medications. The screening process aimed to identify drug-like compounds that adhere to drug development criteria, such as the Veber, Lipinski, and Ghose rules, to minimize potential toxicities.
Moreover, drug interactions with other medications can also impact treatment effectiveness. The virtual screening and pharmacophore-based approach aimed to identify molecules with desirable pharmacological properties to reduce the risk of drug interactions and suboptimal drug levels, which can contribute to treatment failure.
Overall, the results of the virtual screening and pharmacophore-based approach provide valuable insights into potential drug candidates that could address the common reasons for treatment failure in HIV patients. These promising drug candidates with favorable drug-like properties and strong binding affinities to the target enzymes may have the potential to enhance HIV treatment outcomes by mitigating drug resistance, improving adherence, reducing toxicities, and minimizing drug interactions. However, further experimental validation and clinical studies will be essential to confirm their efficacy and safety for future therapeutic development in HIV management.
While our study presents promising alternative options for managing HIV treatment failures, it's important to note several inherent limitations. The reliance on computational modeling necessitates experimental validation to confirm the actual binding affinities and activities of identified lead compounds. Variations in scoring functions and parameters used in molecular docking and virtual screening could influence compound rankings and introduce uncertainties. 3D pharmacophore modeling's simplified representation may not fully capture the complexities of binding mechanisms. The transition of lead compounds to clinical applications requires comprehensive preclinical and clinical evaluations to ascertain safety, potential off-target effects, and interactions. In summary, our study offers valuable insights, but these limitations underline the need for cautious interpretation and further investigation to establish the practical viability of the identified compounds.
Conclusion and perspective
This study successfully utilized molecular modeling techniques, such as molecular docking, virtual screening, and 3D pharmacophore modeling, to propose novel therapeutic molecules as potential replacements for existing antiretroviral drugs in the treatment of HIV. The results indicated that the proposed molecules showed enhanced binding affinities and interactions with their target enzymes, suggesting improved therapeutic efficacy compared to current antiretrovirals. While these computational findings are encouraging, validation through in vitro testing is essential to confirm the functional activity of these molecules. The next steps will involve conducting rigorous experimental investigations to assess their inhibitory potential and safety profiles, which will eventually lead to preclinical and clinical evaluations. If successful, these new therapeutic agents could pave the way for more effective and personalized treatments for patients experiencing therapeutic failures with existing antiretroviral therapy.
Footnotes
Acknowledgments
The authors would like to thank the virology department personnel especially the HIV diagnosis labs.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.