
Abstract
Keywords
Introduction
Autosomal Dominant Hyper-IgE Syndrome (AD-HIES) is a rare primary immunodeficiency disorder that affects multiple body systems, with a particularly significant impact on the immune system. 1 AD-HIES is characterized by a range of symptoms, most notably recurrent infections. Among these infections, pneumonia is particularly prevalent, often caused by specific bacteria that target the lungs and induce inflammation. 2
Hyper-IgE Syndrome (HIES), as a broader category, currently lacks a definitive and comprehensive treatment strategy. The therapeutic approaches available for HIES predominantly focus on managing symptoms and preventing infections. This involves a combination of antibiotics to treat bacterial infections, antifungal medications for fungal infections, and prophylactic measures to reduce the risk of future infections. Additionally, patients may receive immunoglobulin replacement therapy to boost their immune defenses.3,4 In diseases like HIES that present with allergic symptoms, mast cells play a pivotal role as key immune effector cells. These cells are critical in the body's allergic response and defense mechanisms. The activation of mast cells occurs through a well-defined process. It begins when antigen-bound IgE (Immunoglobulin E) binds to its high-affinity receptor (FcɛRI) located on the surface of mast cells. This interaction triggers a signaling cascade that involves the recruitment and activation of various kinases.5,6 One of the primary kinases involved in this process is Syk (spleen tyrosine kinase). Upon activation, Syk initiates a series of downstream signaling events that lead to the degranulation of mast cells. Degranulation is a process whereby mast cells release granules containing pro-inflammatory mediators, such as histamine, cytokines, and other chemokines, into the surrounding tissue. These mediators play a crucial role in orchestrating the inflammatory response, contributing to the symptoms observed in allergic reactions and in the pathophysiology of HIES.7,8
Syk (Spleen Tyrosine Kinase) is of paramount importance in the immune system, particularly in innate immune responses.9-12 While it was initially identified for its high expression in hematopoietic cells and its vital role in adaptive immune responses, recent research has revealed additional involvement of Syk in various biological functions, notably in innate immune responses.
13
Syk plays essential roles in macrophage-mediated inflammatory responses and is closely linked to innate immune activation.
13
Macrophages act as the frontline defense against invading pathogens, crucially initiating innate immune responses. In macrophage-mediated inflammatory responses, Syk enhances phagocytosis by stimulating the generation of reactive oxygen species and inhibiting SOCS1.
13
Additionally, Syk is essential for establishing an immediate immune response and plays a critical role in innate antiviral immunity.
14
Gain-of-function variants in SYK have been identified as causing immune dysregulation and systemic inflammation in humans and mice.
15
Overall, Syk is a critical immune signaling molecule and therapeutic target that plays a crucial role in innate immune responses.
16
SYK consists of two consecutive SH2 domains followed by a C-terminal tyrosine kinase domain. Connecting these domains are two linker regions: interdomain A, which lies between the two SH2 domains, and interdomain B, positioned between the C-terminal SH2 domain and the kinase domain shown in Figure 1; Located at the C-terminal end of the protein, the kinase domain of SYK (spleen tyrosine kinase) is responsible for catalyzing the tyrosine phosphorylation of multiple proteins.
17
Activation of SYK occurs when its tandem SH2 domains bind to immunoreceptor tyrosine-based activating motifs (ITAMs). This interaction triggers the activation of the kinase domain, initiating its catalytic function.
18
The protein tyrosine kinase Syk is of vital importance in facilitating FcɛRI signaling specifically in mast cells.19,20 The interaction between Syk and phosphorylated immunoreceptor tyrosine-based activation motifs (ITAMs) on the FcɛRI receptor causes structural alterations that interrupt its self-restrained configuration, consequently initiating the activation of Syk.
21
The activation of Syk is essential for the cellular processes that lead to alterations in the morphology of mast cells.
20
The prevailing approach to comprehending the role of Syk has been through Syk mutation. It was shown that Syk plays a pivotal role not only in mast cells but also in other hematopoietic cells.22,23 Inhibition of Syk has therapeutic benefits for autoimmune and allergic diseases since Syk plays a critical role in triggering mast cell activation, a process central to autoimmune and allergic responses. Therefore, by inhibiting Syk, we can reduce mast cell activation, degranulation and the release of inflammatory mediators. The generation and release of inflammatory agents by mast cells may be reduced, leading to an alleviation of symptoms associated with these disorders.
24
Flowchart explaining the mechanism of Syk protein signaling.
This study seeks to comprehensively assess the impact of all identified SYK mutations to date on the stability and flexibility of molecular structures. Additionally, it specifically examines the influence of these mutations on the interaction between three known inhibitors and potent inhibitors extracted from the Binding Database, with the overarching objective of using in silico approaches to explore specific inhibitors targeting the kinase domain of SYK protein as potential therapeutic candidates for treating AD-HIES disease. In pursuit of this goal, an in silico approach was employed to distinguish between neutral and pathogenic mutations. Subsequently, virtual screening was conducted for the most potent inhibitors previously identified in the Binding Database, considering favorable ADMET properties. The selected molecules were then docked against the mutated models to investigate if these mutations influence the affinity.
Materials and methods
Mutations collection
Three databases, namely Clinvar, 25 Cosmic, 26 and TCGA Cancer, 27 were utilized to gather all the mutations SNPs that impact the Syk protein. Initially, 536 mutations were compiled, and subsequently, duplicates were eliminated, resulting in a final count of 329 unique substitution mutations. All mutations included in our analysis belong to the same type, which are missense variants. Missense mutations result in the substitution of one amino acid for another in the protein sequence, thereby changing the structure and potentially function of the protein. Exclusion criteria were applied to filter out mutations of other types, such as nonsense mutations (resulting in premature termination codons) and frameshift mutations, as these types would result in incomplete and consequently non-functional SYK proteins. Our objective was to identify mutations that are involved in resistance, thus we focused solely on missense mutations. 28
Target selection and preparation of wild-type and mutated models of Syk protein
The X-ray crystallographic structure of Syk protein (PDB ID:
Prediction of mutational effects
To ensure a dependable outcome, the functional impact of the mutations on the protein structure of Syk was predicted using two tools: PolyPhen-2 31 and Mutation-Assessor. 32
Ligands dataset preparation and filtration
To search ligands that have potential inhibitory activity against the Syk protein, A library of 997 compounds was selected using IC50s filter from Binding database, 33 the conversion to 3D conformation, the elimination of duplicate and then the conversion into a PDB file format was performed by using OpenBabel (https://openbabel.org/.) The reference inhibitors have been collected from the literature, and then downloaded with their 3D structures in (.sdf) format from the PubChem database. 17
The previously downloaded potential inhibitors underwent screening through multiple filters. The process began with the Drulito 18 software, which helped eliminate inhibitors that did not comply with Lipinski and Veber rules. Subsequently, the StopTox 34 tool was employed to retain only non-toxic molecules (https://stoptox.mml.unc.edu/).
To prepare ligands for molecular docking, the structures were prepared using Autodock Tool 1.5.71 30 by adding Gasteiger charges, and then finally, saved in the (.pdbqt) format.
Docking, scoring and visualization
Initially, we identified the active site residues of the target protein, which included Leu377, Ala451, Pro455, Arg498, Glu449, Ser511, Asp512, Asn499, Phe513, and Asn457. These residues were chosen based on their known involvement in the binding pocket of the protein and their importance in ligand recognition and binding. To prepare for docking simulations, we utilized AutoDock Tools 1.5.7 to set up the grid box. 30 The grid parameters were carefully selected, with a spacing of 1 Å for the Syk protein to ensure accurate representation of the binding site. The center coordinates of the grid box were fixed at (X = −26.609, Y = 6.211, Z = −2.729), and the dimensions were set to (X = 22, Y = 22, Z = 18). The grid settings file was obtained from the grid menu option, ensuring consistency and reproducibility in our docking experiments. To evaluate the binding affinity and bound conformation of the selected ligands, we employed Autodock Vina, a widely used molecular docking software known for its accuracy and efficiency. 35 Autodock Vina employs an empirical scoring function to predict the binding energy between ligands and the target protein, allowing us to prioritize ligands with lower affinity scores for further analysis. We used the Lamarckian Genetic Algorithm (LGA) as implemented in AutoDock.
Subsequently, we conducted a comprehensive analysis of all types of interactions between the protein and ligands using BIOVIA Discovery Studio 36 (https://discover.3ds.com/discovery-studio-visualizer-download). This software facilitated the visualization and interpretation of protein-ligand interactions, allowing us to identify key binding residues, hydrogen bonds, and other non-covalent interactions critical for ligand binding and recognition. To further englucidate the 3D protein-ligand interactions, hydrogen bonds, and ligand poses, we utilized PyMOL, 37 a powerful molecular visualization tool. 37 PyMOL (https://www.pymol.org/) enabled us to generate high-quality 3D images of the protein-ligand complexes, providing valuable insights into the binding mode and orientation of the ligands within the active site of the target protein.
Molecular dynamics simulation
In order to study the protein-ligand interaction energy, MD simulations using GROMACS v.2020.4 38 were performed. Syk protein with the reference ligand R343 and Syk protein with the four main potential ligands (ID: 10344820, 118558000, 118558008 and 118557092) at intervals of 100 ns for these complexes. Protein-solvent interactions were characterized by the CHARMM27 force field, while the TIP3P model was applied to water. Each system was placed in a dodecahedral simulation box with an edge length of 1.0 nm and neutralized by adding an equal number of positive and negative ions to achieve the target ionic concentration. The steepest descent algorithm was used to minimize system energy, and V scaling was used for 100 ps in the NVT ensemble to balance it at 300 K. Production cycles were performed using the NPT ensemble at a time step of 100 ps. The Parrinello-Rahman algorithm was then used to equilibrate at a pressure of 1 atm. Using the Verlet scheme, the LINCS algorithm was used to maintain distance limits during the simulation.
Molecular dynamics analysis
Using the gmx_rms tool in GROMACS.32, the RMSD of the atomic coordinates, radius of gyration (Rg), solvent accessible surface area (SASA), and H-bond parameters were determined in order to have a better understanding of the structural changes that take place during the simulation.
3D-pharmacophore model building
To investigate the three-dimensional (3D) distances between the shared features of the most 3 promising molecules, (1S,4R)-4-[5-[6-[(5-chloro-4-methylpyridin-2-yl)amino]-4-methylpyridin-2-yl]-1,3-thiazol-2-yl]-4-hydroxy-2,2-dimethylcyclohexane-1-carboxylic acid
The Molecular Operating Environment (MOE) software tool 39 (https://www.chemcomp.com/) (was selected for its robust capabilities in molecular modeling and pharmacophore analysis. To initiate the process, the builder option within MOE was utilized to construct a dedicated MOE database comprising the molecular structures of the most promising molecules. Subsequently, energy optimization procedures were applied to the molecules within the database to refine their geometric conformations and minimize steric clashes. This step ensures that the molecules are in energetically favorable conformations for subsequent analysis. Following energy optimization, the molecules were aligned to generate a comprehensive pharmacophoric map highlighting the common attributes shared among the molecules. During the alignment process, emphasis was placed on preserving the spatial arrangement of key chemical features, allowing for accurate comparison and analysis. Within the pharmacophoric map, specific queries were executed to identify and quantify the 3D distances between shared features, providing insights into the spatial relationships critical for ligand-receptor interactions. The distances between hydrogen bond donors/acceptors, aromatic rings, and other functional groups were systematically characterized, enabling a thorough assessment of the molecular interactions driving ligand binding and activity.
Results
Mutational effect prediction
A total of 539 mutations were gathered from the databases. After eliminating duplicates, 329 mutations remained, as shown in Supplemental data. The distribution of these mutations across the domains of the Syk protein shows a notable concentration within the Kinase domain, highlighting its significance (Supplemental data).
Only concordant results between 3 tools were taken into consideration when predicting the impact of mutations on Syk protein function. The analysis revealed a distribution of 130 neutral mutations, which do not alter function, and 199 deleterious mutations, representing 39.5% and 60.5% of the total, respectively, as shown in Figure 2(A). In the kinase domain, a total of 139 mutations were identified, classified according to their functional impact. Of these mutations, 45 were found to be neutral, while the remaining 94 were found to be deleterious and affect the function of the protein. (Figure 2(B)). Distribution of mutations. (Neutral and deleterious); (A) Syk protein, (B) kinase domain; the analysis revealed a distribution of 130 neutral mutations, which do not alter function, and 199 deleterious mutations, representing 39.5% and 60.5% of the total, respectively, as shown in (A). In the kinase domain, a total of 139 mutations were identified, classified according to their functional impact. Of these mutations, 45 were found to be neutral, while the remaining 94 were found to be deleterious and affect the function of the protein (B).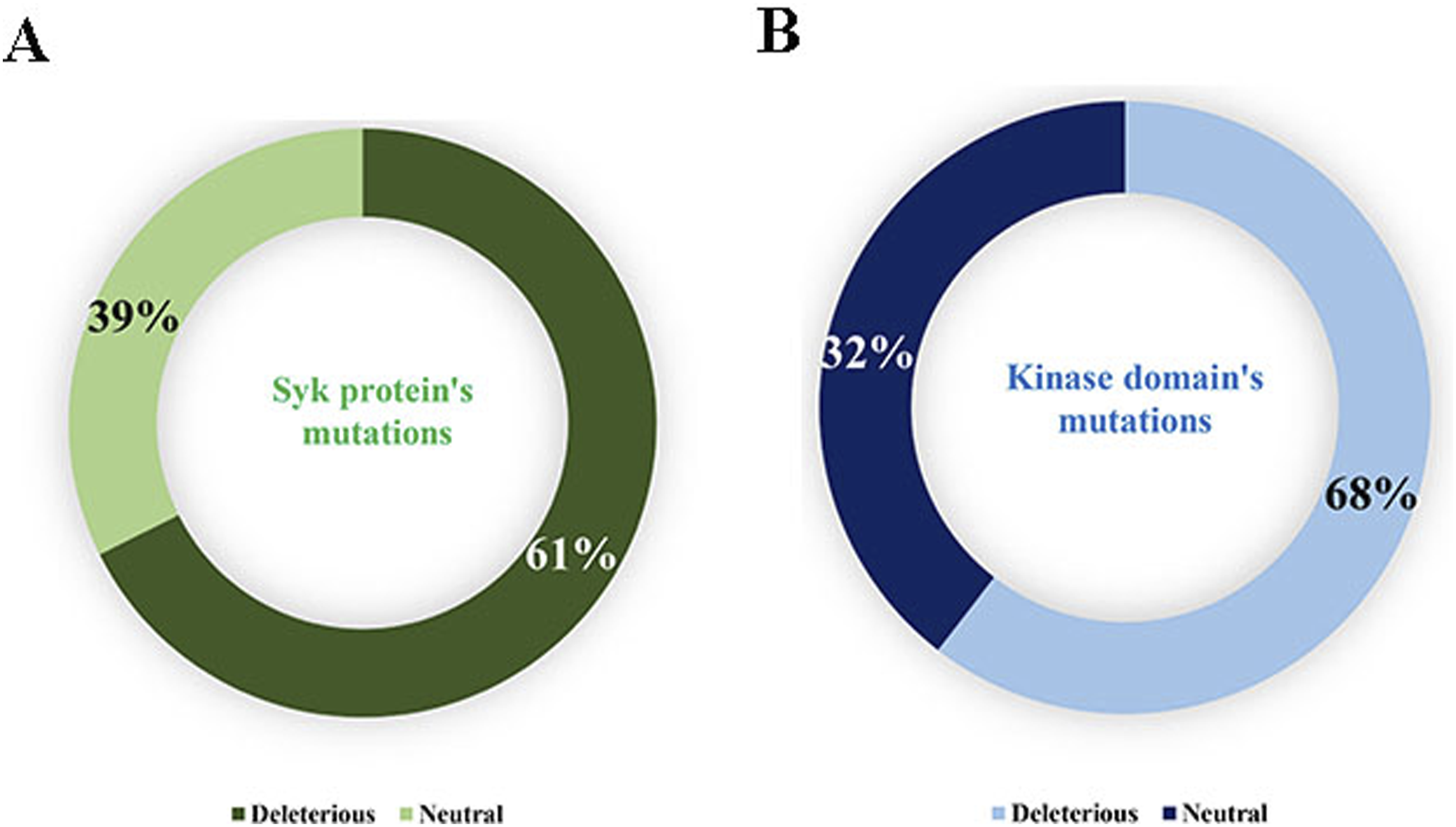
Compounds screening
A total of 997 inhibitors were initially selected based on their IC50 and went through different steps of screening as illustrated in Figure 3. These compounds underwent screening through multiple filters as shown in the Figure 2. The first filter applied was the rule of five of Lipinski and Veber, which is widely used in drug discovery to assess pharmacological effectiveness. This step yielded 280 orally bioavailable molecules. Subsequently, a second filter was employed to identify non-toxic molecules, resulting in 78 candidates. These 78 molecules were further evaluated based on their IC50 values using a reference value of 0.45 nM, which was the smallest value among the IC50 values of the three reference inhibitors. Following this step, 14 potential inhibitors were identified (Figure 3). For the reference inhibitors, it was found that the R112 inhibitor meets the criteria of the Lipinski, Veber rule and it is non-toxic orally. While the inhibitor R343 respects the Lipinski rule but does not respect the Veber rule and it is also non-toxic. For the PRT062607 inhibitor, it respects Lipinski's rule but does not respect Veber's rule because its polar surface area exceeds 140 Å, thus it is orally toxic. The information related to these inhibitors is listed in the Table 1. In silico study workflow. Overview of chemical identifiers, compound names, molecular formulas, 2D structures, and FDA approval status of reference Syk protein inhibitors.

Docking and scoring
A total of 1615 (17 × 95) dockings were conducted in this study, involving the wild-type and the 94 mutated types of the Syk protein. These dockings included interactions with 14 potential inhibitors and 3 reference inhibitors.
The initial phase of the study focused on the molecular docking of the three reference inhibitors with both the wild-type and the mutated structures of the Syk protein. The results indicated that R343 exhibited the highest affinity with the wild-type Syk protein, scoring −9.8 kcal/mol in terms of energy. The mutated types showed scores ranging from −9.7 to −10.1 kcal/mol, compared to the other two reference inhibitors, R112, and PRT062607, which exhibited affinities of −8.7 and −8.6 kcal/mol, respectively (Supplemental data).
The affinity energy in (kcal/mol) between the Syk protein (wild and mutated) and the 3 reference ligands.

The results indicated that R343 exhibited the highest affinity with the wild-type Syk protein, scoring −9.8 in terms of energy. The mutated types showed scores ranging from −9.7 to −10.1, compared to the other two reference inhibitors, R112, and PRT06260, which exhibited affinities of −8.7 and 8.6, respectively.
Potential inhibitors for SYK and their relevant information.

Among these inhibitors, namely 118558008, 10344820, 118558000, and 118558092 demonstrated superior affinities compared to both the other potential inhibitors and the reference inhibitors for all mutations and the wild-type crystal structure, with affinities ranging between −9.9 Kcal/mol and −10.1 Kcal/mol.
Interaction visualization
The intricate diagram clarifies the hydrogen bonding network established between the Spleen Tyrosine Kinase protein, a pivotal player in cellular signaling, and its quartet of promising ligands. The hydrogen bonds, depicted as dashed lines, delineate the electron-rich donor-acceptor interactions essential for stabilizing the protein-ligand complexes. Such interactions, often characterized by their directionality and strength, orchestrate specific binding events, thereby influencing the protein's conformation and activity. (Figure 4). The hydrogen bonding network established between the Syk protein, and its quartet of promising ligands. The intricate diagram elucidates the hydrogen bonding network established between the Syk protein (blue), a pivotal player in cellular signaling, and its quartet of promising ligands. The hydrogen bonds, depicted as dashed yellow lines, delineate the electron-rich donor-acceptor interactions essential for stabilizing the protein-ligand complexes. Such interactions, often characterized by their directionality and strength, orchestrate specific binding events, thereby influencing the protein’s conformation and activity. (A): Syk-118558008; (B): Syk-10344820; (C): Syk-118558000; (D): Syk-118558092.
The investigation into molecular bonds within our compounds involved a comprehensive analysis of various bonding types using BIOVIA Discovery Studio. The examined interaction encompassed a wide spectrum, including conventional hydrogen bonds, carbon-hydrogen bonds, halogen bonds, alkyl bonds, Pi-alkyl bonds, van der Waals interactions, unfavorable donor-donor bonds, Amide-Pi stacked bonds, Pi-sigma bonds, and Pi-sulfur bonds. (Figure 5). The diverse types of interactions between ligands and key residues within the active site, presented through a 2D visualization. A: The 2D interaction between ligand 10344820 and the protein Syk; B: The 2D interaction between ligand 118558008 and the protein Syk; C: The 2D interaction between ligand 118558000 and the protein Syk; D: The 2D interaction between ligand 118557092 and the protein Syk. The analysis of four complexes formed between the SYK protein and specific inhibitors (ID: 10344820, ID: 118558000, ID: 118558008, ID: 118557092) revealed diverse molecular interactions. Notably, several residues exhibited consistent involvement across all complexes, including SER 379, ARG 498, LEU 377, ASN 381, and LYS 533. These residues played pivotal roles in forming essential bonds. Common interaction types observed in all complexes encompassed conventional hydrogen bonds, van der Waals bonds, and, in subsets, alkyl and Pi-Alkyl bonds. Additionally, a halogen (fluorine) bond was shared between complexes 1 and 4. The recurrence of these residues and interaction types suggests a consistent molecular pattern in the binding landscape, providing crucial information for the rational design of inhibitors targeting the SYK protein.
This analysis extended to the study of four complexes (Figure 5(A)–(D)) formed between the SYK (Spleen Tyrosine Kinase) protein and specific inhibitors (ID: 10344820, ID: 118558000, ID: 118558008, ID: 118557092). Molecular docking scores were calculated for each complex, and detailed interactions are reported in Figure 5. The diversity observed in each complex highlights the complexity of molecular interactions, providing a solid foundation for drug design targeting the SYK signaling pathway. Conventional hydrogen bonds, crucial for specificity and stability, are abundant in all complexes. Van der Waals interactions significantly contribute to stability by optimizing protein-ligand contacts. The various types of Pi-interactions enhance the stabilization of complexes, emphasizing the importance of π-π interactions in molecular recognition. Carbon hydrogen bonds add a specific dimension to interactions, while halogen (fluorine) bonds can influence the stability of the complex.
Molecular dynamic analysis
Molecular dynamics simulations were performed to evaluate the behavior of four selected ligands complexed to the SYK protein, alongside a baseline simulation using the R343 ligand in complex with the Syk protein. Analyzes focused on RMSD, Rg, SASA, and hydrogen bonding models to evaluate the stability and interaction dynamics of these complexes. The results are presented in Figures 6 and 7 which presents the means and their standard deviations. Molecular dynamics simulation results of Syk Protein with reference inhibitor R343 and four potential candidate inhibitors, exploring RMSD, Rg, SASA, and H-BOND parameters. Line plot with error bars representing mean and standard deviation values of RMSD, Rg, SASA, and H-BOND parameters from molecular dynamics simulations.

For the SYK-ligand complexes, namely ligand 1 (PubChem ID: 118558000), ligand 2 (PubChem ID: 118558008), ligand 3 (PubChem ID: 118558092) and ligand 4 (PubChem ID: 10344820), l RMSD analysis revealed that ligand 1 had a closer match to the reference structure with an RMSD value of (0.43 ± 0.06). Ligands 2 and 3 exhibited slightly higher RMSD values of (0.55 ± 0.03) and (0.56 ± 0.06), respectively, compared to the reference ligand complex (RMSD = 0.67 ± 0 .05). Ligand 4, however, exhibited a higher RMSD value of (0.78 ± 0.08). (Figure 6(A)).
Analysis of Rg values indicated the compactness or shape of the ligand-protein complex. Ligands 1, 2, and 3 exhibited Rg values of (2.74 ± 0.02), (0.70 ± 0.02), and (0.73 ± 0.030), respectively, reflecting their respective compactness relative to the ligand complex. Reference with an Rg value of (0.68 ± 0.02). Ligand 4 showed an Rg value closer to the reference at 0.66 ± 0.02. (Figure 6-(B)).
Regarding the SASA values, ligands 1, 2 and 3 presented values of (296.17 ± 1.73), (293.70 ± 1.95) and (296.64 ± 1.64), respectively. Which describe the solvent-exposed surface of the ligand-protein complex. While ligand 2 and reference ligand-Syk complex had a SASA value of (294.50 ± 1.63). Ligand 4 displayed a SASA value of (294.32 ± 1.74) which is similar to the reference. (Figure 6(C)).
Hydrogen bond analysis showed that ligand 1 formed a higher number of hydrogen bonds (up to 7) with the SYK protein compared to ligands 2 and 3. This contrasted with ligand 4, which had fewer bonds hydrogen 4 relative to the reference ligand complex. 5 (Figure 6(D)).
3D-pharmacophore model building
In this study, we developed a 3D pharmacophore model from three potential ligands. The inhibitors were analyzed to identify key chemical features essential for their inhibitory activity, leading to the identification of seven key features for interaction with the Syk protein: F1 (Acc&ML), F2 (Don&Acc&ML), F3 (Acc&ML), F4 (ML/Aro/Acc), F5 (Aro), F6 (Hyd), and F7 (Acc&ML). These features include acceptor and metallic sites (F1, F3, F7), a donor, acceptor, and metallic site (F2), as well as sites with metallic, aromatic, and acceptor properties (F4). We also identified an aromatic feature (F5) and a hydrophobic feature (F6). Each feature was characterized to provide information on potential interactions with the Syk protein. Figure 8(A) shows the final pharmacophore model, illustrating the spatial arrangement of the seven identified features (F1–F7) in 3D space. The model highlights key functional groups required for potential ligand-receptor interactions essential for inhibitory activity. Figure 8(B) illustrates the pharmacophore model without the ligands superimposed, providing a clear visualization of the spatial arrangement between features within the pharmacophore model. The inter-feature distances within the pharmacophore model are summarized in Figure 8(C). These distances play a crucial role in defining spatial relationships between features and are important for ligand binding and pharmacophore-based virtual screening studies (Figure 8). The pharmacophore model, derived from the flexible alignment of the top 3 compounds, (A) the various features explored within its structure. (B) A simplified representation of the model. (C) The interfeature distances within the pharmacophore model. Seven significant features were identified and used to construct the pharmacophore model: F1: Acc&ML, F2: Don&Acc&ML, F3: Acc&ML, F4: ML|AroAcc, F5: Aro, F6: Hyd and, F7: Acc&ML.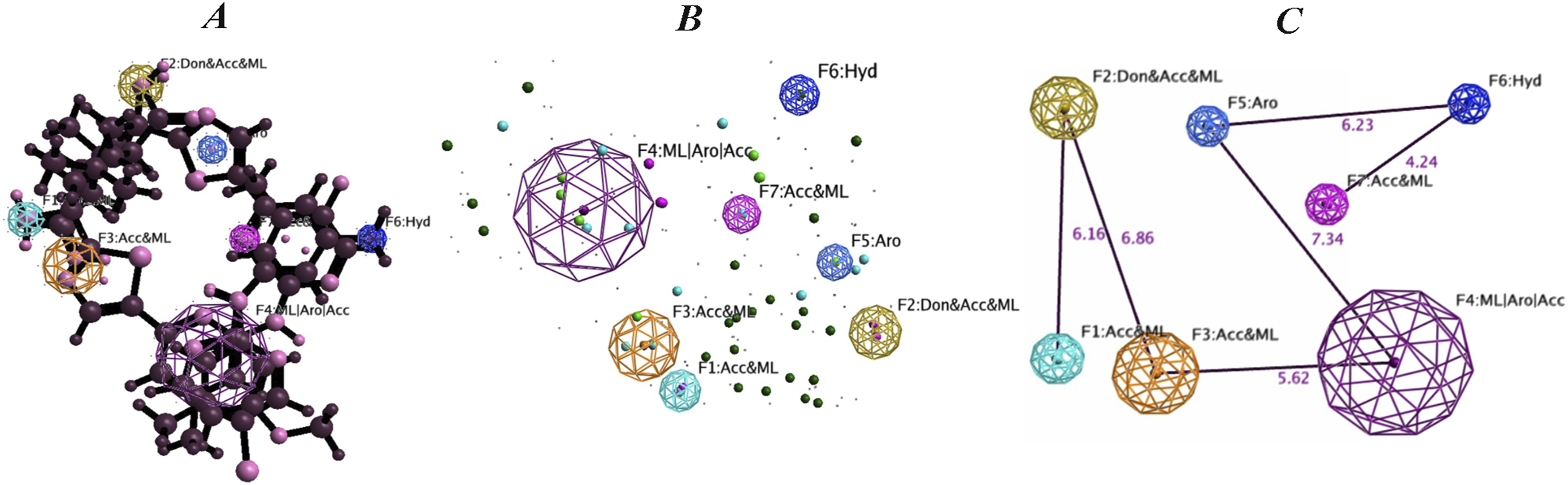
Discussion
In this paper, we were able to gather all reported mutations affecting the Syk protein from the literature to investigate their functional impact. The findings indicated that 60.5% of these mutations were deemed deleterious, meaning they are likely to have a negative impact on the function or structure of the protein. 40 While 39.5% were considered neutral, suggesting that these mutations might not significantly affect the protein's function or could have no discernible impact. 41
Through virtual screening, we identified 3 potential inhibitors (IDs: 118558008, 118558000 and 118558092) that surpass the reference compounds. Additionally, our pharmacophore model provides valuable insights, potentially guiding the development of targeted therapies for AD-HIES and related immunological disorders.
However, it is important to note that while these inhibitors show potential benefits, they are not without drawbacks. Some of these inhibitors have been associated with undesirable side effects. 42 As with many therapeutic interventions, there is a need to strike a balance between the desired efficacy in suppressing mast cell degranulation and the potential risk of adverse effects. Thus, further research and development are essential to refine the specificity and safety profile of SYK inhibitors to maximize their therapeutic potential while minimizing any unintended consequences. 42
Indeed, PRT062607 is an example of an ATP-competitive inhibitor of SYK that has been studied in the context of various inflammatory and autoimmune diseases. While it showed potential as a therapeutic agent for targeting SYK and modulating mast cell degranulation, there have been concerns regarding its selectivity, safety, and efficacy. 43
One of the main issues associated with PRT062607 is its insufficient selectivity, meaning that it not only targets SYK but also affects other kinases. 44 In particular, its inhibition of vascular endothelial growth factor 2 (VEGFR2) has been reported to cause an increase in blood pressure, which can be a significant safety concern. The lack of specificity for SYK and the unintended effects on other kinases can lead to adverse reactions and limit its clinical use. As a result of the initial inhibitor's shortcomings, our focus shifted towards proposing new, more efficient molecules.
Structures of the three potential inhibitors with the best binding affinities shows that the three inhibitors: (1S,4 R)-4-[5-[6-[(5-chloro-4-methylpyridin-2-yl)amino]-4-methylpyridin-2-yl]-1,3-thiazol-2-yl]-4-hydroxy-2,2-dimethylcyclohexane-1-carboxylic acid
In conclusion, this comprehensive screening process identified three promising candidate inhibitors that possess the desired drug-like properties, safety profile and increased affinity which can be explained by the large number of hydrogen bonds that bind the protein with the 3 potential inhibitors whose IDs are 118558008, 118558000 and 118558092;
The results from molecular docking simulations and post-docking interaction visualization between the SYK protein and four specific inhibitors unveil a detailed analysis of the bonds formed in each complex. This molecular investigation provides essential insights for drug design, offering an in-depth understanding of the molecular interactions between the protein and its ligands. This in-depth analysis of molecular interactions between the SYK protein and its inhibitors reveals a diversity of bonds, each contributing specifically to the stability of the complexes. These results offer crucial perspectives for rational drug design, underscoring the need to consider the variety of bonding forces in the development of therapeutic compounds targeting the SYK protein.
Complexes 1 to 4 demonstrate high molecular docking scores, suggesting a strong affinity between the SYK protein and the inhibitors. Examination of the interactions reveals a diversity of bonds, including conventional hydrogen bonds, van der Waals, Pi-interactions (Pi-Sigma, Pi-Alkyl, Amisz Pi-Stacked, Pi-sulfur), carbon hydrogen bonds, and halogen (fluorine) bonds. These bonds contribute specifically to the stability of the complexes. Conventional hydrogen bonds play a crucial role in the specificity and stability of protein-ligand interactions, promoting molecular recognition. Van der Waals interactions provide additional stability, optimizing contacts between the protein and the inhibitor. Various types of Pi-interactions add a specific component to the stabilization of complexes, emphasizing the importance of π-π interactions. Carbon hydrogen bonds introduce another dimension to molecular interactions, contributing to the specificity of bonds. Finally, halogen (fluorine) bonds add extra complexity, potentially influencing the stability of the complex.
In summary, the diversity of observed bond types underscores the complexity of molecular interactions between the SYK protein and its inhibitors. Each type of bond contributes specifically to the stability and selectivity of the complexes, providing opportunities for the rational design of drugs targeting the SYK signaling pathway. These results enrich our understanding of molecular interactions and provide a solid foundation for future studies and the development of therapeutic compounds.
The results of the molecular dynamics simulations provide valuable insights into the behavior of four selected ligands complexed to the SYK protein, as well as a reference simulation using the R343 ligand. The root mean square deviation (RMSD) measures the average deviation of the atomic positions between the starting structure and the snapshots obtained during the simulation. 46 Lower RMSD values indicate more stable ligand binding within the protein binding site. 47 Ligand 1 (PubChem ID: 118558000) had the lowest RMSD of 0.43 ± 0.06 nm, suggesting that it maintains a stable conformation throughout the simulation period, closely resembling the inhibitor reference. This stability implies a strong and coherent interaction with the SYK protein. Ligands 2 (0.55 ± 0.03 nm) and 3 (0.56 ± 0.06 nm) showed slightly higher RMSD values, indicating some flexibility or fluctuation in their binding orientations compared to the reference ligand R343 (RMSD = 0.67 ± 0.05 nm). Ligand 4 (PubChem ID: 10344820), with an RMSD of 0.78 ± 0.08 nm, exhibited the highest deviation among the ligands studied, suggesting less stable binding with the SYK protein and interaction dynamics potentially weaker.
As for the radius of gyration (Rg), it evaluates the compactness or overall shape of the protein-ligand complex. Ligands 1, 2, and 3 displayed Rg values of 2.74 ± 0.02 nm, 2.70 ± 0.02 nm, and 2.73 ± 0.03 nm, respectively, indicating their different degrees of compactness within the protein binding site relative to the reference (Rg = 2.68 ± 0.02 nm). These Rg values suggest that while all ligands retain a generally compact structure, ligands 1 and 3 exhibit slightly larger Rg values, potentially indicating a more extended or differently shaped binding conformation compared to ligands 2 and the ligand of reference. 48
Concerning Solvent Accessible Surface Area (SASA), this measurement parameter measures the surface area of the ligand-protein complex accessible to solvent molecules. Ligands 1, 2, and 3 showed SASA values of 296.17 ± 1.73 nm2, 293.70 ± 1.95 nm2, and 296.64 ± 1.64 nm2, respectively, indicating their different degrees of exposure to the solvent environment compared to the reference (SASA = 294.50 ± 1.63 nm2). Higher SASA values suggest that ligands 1 and 3 may interact more broadly with the solvent, potentially influencing their binding kinetics and accessibility within the protein pocket. 49
Lately hydrogen bonds play an essential role in stabilizing ligand-protein interactions. Ligand 1 formed a higher number of hydrogen bonds (up to 7) with the SYK protein, indicating strong and specific interactions that contribute to its stability within the binding site. In contrast, ligands 2 and 3 formed fewer hydrogen bonds than ligand 1, suggesting potentially weaker or less specific interactions. Ligand 4 formed 4 hydrogen bonds, fewer than the reference ligand R343 (5 hydrogen bonds), which may impact its binding affinity and stability. 47
In summary, these results collectively provide insight into the dynamic behavior and interaction characteristics of SYK-ligand complexes. Ligands 1, 2, and 3 exhibit different degrees of stability, compactness, solvent exposure, and hydrogen bonding patterns compared to the reference ligand R343. These results are crucial and served as a matrix for the design of a 3D pharmacophore model from the 3 potential ligands 1, 2 and 3.
The constructed pharmacophore model appears to identify seven distinct features that are likely essential for the inhibitors' interaction with the SYK protein and contribute to their potential effectiveness; (F1) is a Metal Acceptor and Ligand (Acc&ML), this feature represents a functional group that acts as a hydrogen bond acceptor and has the potential to coordinate with metal ions. 48 Hydrogen bond acceptors play a crucial role in stabilizing ligand-receptor complexes through hydrogen bond interactions with hydrogen donors on the receptor. Metal ligand functionality enhances ligand binding specificity and affinity by coordinating with metal ions in the active site of the protein. 50 (F2) is a Donor, Acceptor, and Metal Ligand (Don&Acc&ML), this complex feature includes Hydrogen Donor Capability (Don), Hydrogen Acceptor Capability (Acc), and Ligand Feature metallic (ML). Hydrogen donor and acceptor capabilities contribute to potential hydrogen bonding interactions with the target receptor, thereby enhancing ligand-receptor binding. The property of the metal ligand further strengthens the interaction by coordinating with the metal ions present in the active site.51,52 (F3) is an Acceptor and Metal Ligand (Acc&ML) which is similar to Feature 1, this feature includes a hydrogen bond acceptor and metal ligand functionality. Hydrogen bond acceptors participate in stabilizing hydrogen bond interactions with the target receptor, while the metal ligand property contributes to binding specificity through coordination with metal ions.53-55 (F4) is a Metal Ligand/Aromatic/Acceptor (ML/Aro/Acc), this feature combines the functionalities of a metal ligand (ML), an aromatic (Aro) and an acceptor hydrogen (Acc). The aromatic nature suggests a potential for π–π interactions with aromatic residues on the receptor, providing additional stabilizing forces for the ligand-receptor complex. 56 The hydrogen acceptor ability allows this functionality to form hydrogen bonds with appropriate donors on the receptor, thereby contributing to ligand-receptor interactions. The metal ligand property enhances binding affinity through coordination with metal ions. 52 (F5) is an Aromatic (Aro), this feature represents an aromatic moiety that can engage in aromatic stacking or π–π interactions with complementary aromatic residues on the receptor. 57 These interactions provide stabilizing forces to the ligand-receptor complex, contributing to overall binding affinity. (F6) is a Hydrophobic (Hyd), this feature represents a hydrophobic group that contributes to binding affinity by engaging in hydrophobic interactions with non-polar residues on the receptor.58,59 These interactions are essential to stabilize the ligand-receptor complex in a non-aqueous environment. (F7) is an Acceptor and Metal Ligand (Acc&ML), this feature is similar to Features 1 and 3, including the Hydrogen Bond Acceptor and Metal Ligand features. It participates in the stabilization of hydrogen bonding interactions with the target receptor and improves binding specificity through coordination with metal ions.53,60
In conclusion, the constructed pharmacophore model identifies seven distinct features that are likely essential for the inhibitors' interaction with the SYK protein and contribute to their potential efficacy. These features include various functional groups such as hydrogen bond acceptors, donors, metal ligands, aromatic moieties, and hydrophobic groups. Each feature plays a crucial role in stabilizing ligand-receptor complexes through specific interactions such as hydrogen bonding, coordination with metal ions, π–π interactions, and hydrophobic interactions. Comprehensive identification of these features shows key functional groups required for potential ligand-receptor interactions, which is essential for the design and optimization of effective SYK inhibitors.61,62
These findings offer promising prospects for future research and potential clinical benefits for affected individuals. To advance research on autosomal dominant hyper-IgE syndrome (AD-HIES) and its potential therapeutic approaches, several perspectives can be considered.
First, rigorous experimental validation of potential inhibitors identified by virtual screening is essential to confirm their efficacy and safety. Following successful validation, clinical trials should be initiated to evaluate these inhibitors in real-world conditions in patients with AD-HIES. Additionally, exploring combination therapies with multiple existing treatments could potentially improve outcomes for individuals suffering from allergic symptoms related to AD-HIES.
Among the identified inhibitors, ligand 1 with ID: 118558000 stands out for its most stable binding (lowest RMSD), potentially favorable compactness (Rg), and extensive interaction with solvents (SASA), supported by a higher number of hydrogen bonds. These results provide an in-depth understanding of the molecular mechanisms underlying ligand binding to SYK and guide the future optimization of potential inhibitors targeting the Syk protein, particularly in the context of allergies characteristic of hyper-IgE syndrome.
In conclusion, these advances not only deepen our understanding of AD-HIES mechanisms but also guide the development of new targeted therapies, offering tangible hope for patients suffering from this rare pathology.
Although this study provides valuable information on Syk mutations and potential inhibitors, several limitations should be considered. First, the accuracy of virtual screening and molecular docking is highly dependent on the computational models and assumptions used, which may not fully replicate real-world conditions. Variability in protein conformations and dynamics could impact the reliability of binding affinity predictions.
As well, the pharmacokinetic properties and toxicity profiles of the identified inhibitors were evaluated computationally and may not accurately reflect their in vivo behavior. Experimental validation using in vitro assays and animal models is essential to confirm efficacy and safety.
Conclusion
In conclusion, this study gathered all reported mutations affecting the Syk protein and identified that 60.5% of them are deleterious, impacting the function or structure of the protein. Through virtual screening, three potential inhibitors (ID: 118558008, 118558000, and 118558092) were found to be superior to the reference compounds. Ligand 1 (ID: 118558000) demonstrated the most stable binding, favorable compactness, and extensive interaction with solvents, supported by a higher number of hydrogen bonds. The pharmacophore model provides information for the development of targeted therapies for AD-HIES and related disorders. These advances are guiding the development of new targeted therapies, offering hope to patients suffering from allergies in the context of Hyper IgE syndrome.
Supplemental Material
Supplemental Material - Syk protein inhibitors treatment for the allergic symptoms associated with hyper immunoglobulin E syndromes: A focused on a computational approach
Supplemental Material for Syk protein inhibitors treatment for the allergic symptoms associated with hyper immunoglobulin E syndromes: A focused on a computational approach by Mariam Mansouri, Ghyzlane ElHaddoumi, Ilham Kandoussi, Lahcen Belyamani, Azeddine Ibrahimi and Naima El Hafidi in International Journal of Immunopathology and Pharmacology.
Footnotes
Acknowledgements
This work was carried out under national funding from the Moroccan Ministry of Higher Education and Scientific Research (COVID-19 program) to A.I. This work was also supported by a grant from the Moroccan Institute of Cancer Research and the PPR-1 program to A.I. and Biocodex grant to AI. The CNRST-PPR2 program provided a informatic station to support bioinformatic analyses for this study. The authors express their gratitude toward these funding sources. We would like to express our sincere gratitude to Professor Rachid Eljaoudi for his expertise and significant contributions to the integration of new results and the overall improvement of the manuscript. We also extend our thanks to Dr Nasma Boumajdi for her invaluable assistance in generating the molecular dynamics graphs as well as for her critical insights. Their contributions were essential to the success of this work.
Author contributions
⁃
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.