
Abstract
Objective:
To construct a competitive endogenous RNA (ceRNA) regulatory network derived from exosomes of human breast cancer (BC) by using the exoRbase database, to explore the possible pathogenesis of BC, and to develop new targets for future diagnosis and treatment.
Methods:
The exosomal gene sequencing data of BC patients and normal controls were downloaded from the exoRbase database, and the expression profiles of exosomal mRNA, long non-coding RNA (lncRNA), and circular RNA (circRNA) were analyzed by using R language. Use Targetscan and miRanda database to jointly predict and differentially express miRNA (microRNA), miRNA combined with mRNA. The miRcode database was used to predict the miRNA combined with differentially expressed lncRNA, and the starBase database was used to predict the miRNA combined with circRNA in the difference table. The related mRNA, circRNA, lncRNA, and their corresponding miRNA prediction data were imported into Cytoscape software to visualize the ceRNA network. Enrichment analysis and visualization of KEGG were carried out using KOBAS. Hub gene was determined by Cytohubba plug-in.
Results:
Forty-two differentially expressed mRNA, 43 differentially expressed circRNA, and 26 differentially expressed lncRNA were screened out. The ceRNA network was constructed by using Cytoscape software, including 19 mRNA nodes, 2 lncRNA nodes, 8 circRNA nodes, and 41 miRNA nodes. KEGG enrichment analysis showed that differentially expressed mRNA in the regulatory network mainly enriched the p53 signaling pathway. Find the key Hub gene PTEN.
Conclusion:
The ceRNA regulatory network in blood exosomes of BC patients has been successfully constructed in this study, which provides an exact target for the diagnosis and treatment of BC.
Introduction
Breast cancer (BC) is the most common malignant tumor in women worldwide, ranking first in tumor incidence and also the leading cause of death from malignant tumors in women. 1 In 2020, there were about 420 000 new cases of BC in China, and the incidence of BC was the highest in the world, with about 117 000 deaths, which has brought a heavy disease burden to Chinese women. 2 In recent years, the clinical diagnosis and treatment of BC have been improving, but the pathogenesis of BC has not yet been fully elucidated. 3 Therefore, it is of great significance to study potential new targets for the diagnosis and treatment of BC.
The exosome is an extracellular disk-shaped vesicle with a diameter of 40 to 100 nm, which is secreted by a variety of active cells specifically and distributed in saliva, milk, serum, plasma, and other body fluids. It contains protein, lipids, DNA, non-coding RNA, and other bioactive substances, thus playing an important role in the regulation of physiological functions.4,5 As an important carrier for intercellular communication and genetic material transfer, exosomes derived from BC cells participate in the proliferation, invasion, metastasis, angiogenesis, immunosuppression, and other development processes of BC by changing the biochemical components, signal transduction pathways, and gene regulation of recipient cells. 6
The competitive endogenous RNA (ceRNA) hypothesis was first proposed by SALMENA et al of Harvard Medical School in the United States. 7 The identified noncoding RNA family members include long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), microRNAs (miRNAs), small nuclear RNAs (snRNAs), small nuclear RNAs (snoRNAs), piwi interacting RNAs (piRNAs), and small interfering RNAs (siRNAs), A large number of miRNA binding sites are present on a wide variety of RNA transcripts. 8 The hypothesis states that non-coding RNAs such as long non-coding RNA (lncRNA), mRNA, and circular RNA (circRNA), can competitively bind to miRNA to reduce the inhibition of mRNA of the target gene, to further regulate a series of biological behaviors such as proliferation, growth, differentiation, and apoptosis of tumor cells. 9 MiRNAs are the center of the ceRNA network, which can bind to the complementary sequence regions of target genes, thereby reducing the stability of target genes or limiting their translation. MiRNAs are generally considered to be active regulatory elements, and when the expression levels of miRNAs at the center of regulatory networks change, they will have effects on epithelial mesenchymal transition (EMT), immune regulation, tumor microvascular formation, cell autophagy, glycolipid metabolism, cell differentiation, and so on. 10 Current studies have shown that ceRNA may be involved in the occurrence and development of BC, 11 but the regulatory mechanism of ceRNA in the blood exosomes of BC patients is still unclear.
In this study, the exosomal sequencing data of BC patients and normal controls in the exoRBase database were reanalysis, the differential expression profiles of mRNA, lncRNA, and circRNA were found, and the ceRNA network was constructed, which would provide a theoretical basis for exploring new targets for the diagnosis and treatment of BC.
Materials and Methods
Data download and screening of differentially expressed mRNA, lncRNA, circRNA
Exosome gene sequencing data of blood from BC patients and normal control groups were downloaded from the exoRBase 2.0 database (http://www.exorbase.org/). The corresponding gene annotation file was also downloaded. The data deadline was September 10, 2021. A total of 258 sets of sample data were downloaded, including 140 sets of BC exosome gene sequencing data and 118 sets of normal sample exosome gene sequencing data. Differential expression profiles of mRNA, lncRNA, and circRNA in exosomes were analyzed using BC data as the experimental group and normal sample data as the control. The screening condition for differential expression was |log2FC| > 0, and the screening condition after the correction was that P-value < .05.
RNA prediction of interaction and construction of ceRNA network
TargetScan (http://www.targetscan.org/vert_71/) and miRanda (http://www.micro-rna.org/) databases were used to jointly predict and differentially express mRNA-bound miRNAs. The miRcode database was used to predict miRNA binding to differentially expressed lncRNA, and the starBase database was used to predict miRNA binding to differentially expressed circRNA. Finally, the related mRNA, circRNA, lncRNA, and their corresponding miRNA prediction data were imported into Cytoscape (version 3.8.2) software to visualize the ceRNA network.
Functional enrichment analysis of differentially expressed mRNA
Differentially expressed mRNA was ID-converted using the R-package “org.Hs. eg.db” and differentially expressed mRNA was subjected to KEGG enrichment analysis and visualization using Kobas (http://kobas.cbi.pku.edu.cn/) to explore potential roles or potential pathways of influence for differentially expressed mRNA.
Hub gene screening
The CytoHubba plug-in in Cytoscape software was used to calculate the connectivity score of each protein node. The top 10 genes with the score being ranked were identified as Hub genes, and a Hub gene network diagram was constructed.
Statistical analysis
Data were organized using the Perl (version strawbuerry-Perl-5.32.) programming language, and data analysis and drawing were performed using Rstudio (version 4.1.0). Measurement data were expressed as mean standard deviation, and a statistical test was conducted using a T-test or analysis of variance. P < .05 indicated statistically significant.
Results
Data download and difference analysis
The sequencing data of 118 normal groups and the exosomal sequencing data of 140 BC patients were downloaded from the exocrine 2.0 database. The corresponding mRNA expression profile, lncRNA expression profile, and circRNA expression profile matrix were integrated. Differential analysis of mRNA, lncRNA, and circRNA was performed using the R language, and 42 differentially expressed mRNA, 43 differentially expressed circRNA, and 26 differentially expressed lncRNA were screened out. Heat map of the first 20 genes differentially expressed (Figure 1A-C).

(A) Heatmap of differential mRNA expression, (B) heatmap of differential circRNA expression, and (C) heatmap of differential lncRNA expression.
Construction of miRNA-related ceRNA regulatory network
Four hundred sixty-three miRNAs combined with differentially expressed mRNA were predicted using TargetScan and miRanda databases, 65 miRNAs combined with differentially expressed lncRNA were predicted using the miRcode database, and 149 miRNAs combined with differentially expressed circRNA were predicted using the starBase database. The ceRNA network was constructed with Cytoscape software, consisting of 19 mRNA nodes, 2 lncRNA nodes, 8 circRNA nodes, and 41 miRNA nodes (Figure 2).

ceRNA network of differentially expressed genes.
KEGG pathway enrichment analysis
After the differentially expressed mRNA in the constructed ceRNA network was converted from Gene Symbol to Entrez ID, the KOBAS tool was used for KEGG enrichment analysis and visualization of the differentially expressed mRNA, with the first 25 pathways for KEGG enrichment analysis shown in Table 1. Analysis showed that differentially expressed mRNA in the regulatory network was mainly enriched in “p53 signaling pathway,” “Focal adhesion,” “MicroRNAs in cancer,” “Human papillomavirus infection,” “PI3K-Akt signaling pathway,” “Bladder cancer,” “Malaria,” “Pathways in cancer,” “Endometrial cancer,” “Acute myeloid leukemia,” “Central carbon metabolism in cancer,” “RIG-I-like receptor signaling pathway,” “Adherens junction,” “Melanoma,” “Inositol phosphate metabolism,” “Glioma,” “Chronic myeloid leukemia,” “EGFR tyrosine kinase inhibitor resistance,” “ECM-receptor interaction,” “PD-L1 expression and PD-1 checkpoint pathway in cancer,” “Small cell lung cancer,” “TGF-beta signaling pathway,” “Glycerophospholipid metabolism,” “Prostate cancer,” “Phosphatidylinositol signaling system” (Figure 3) . The most important difference between PTEN and THBS gene enrichment in the p53 signaling pathway (Figure 4). These results indicate that the ceRNA regulatory network constructed by blood exosomes of BC patients plays an important role in the occurrence and development of BC.
KEGG enrichment analysis of mRNA in ceRNA regulatory network of blood exosomes in breast cancer patients.

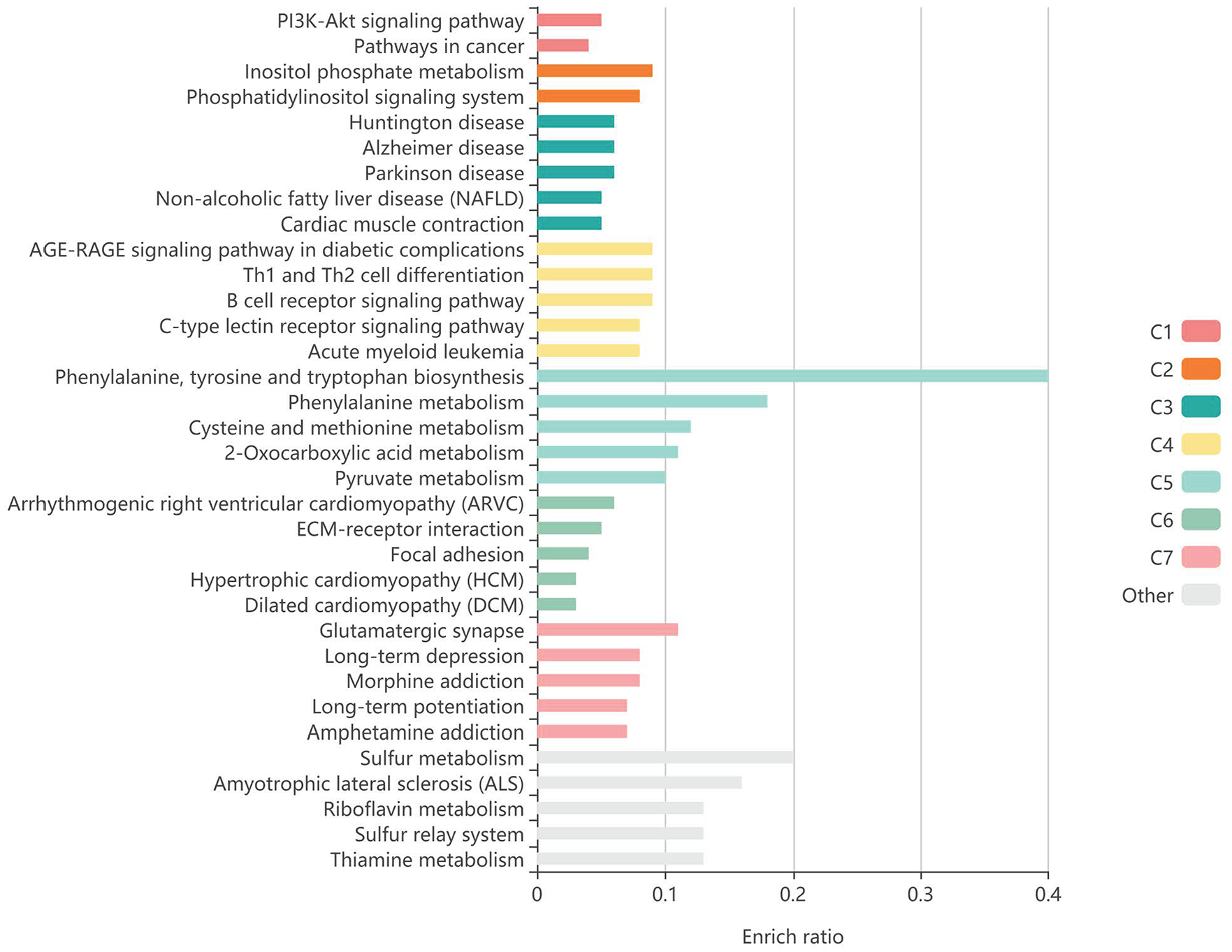
KEGG enrichment analysis of differential mRNA.
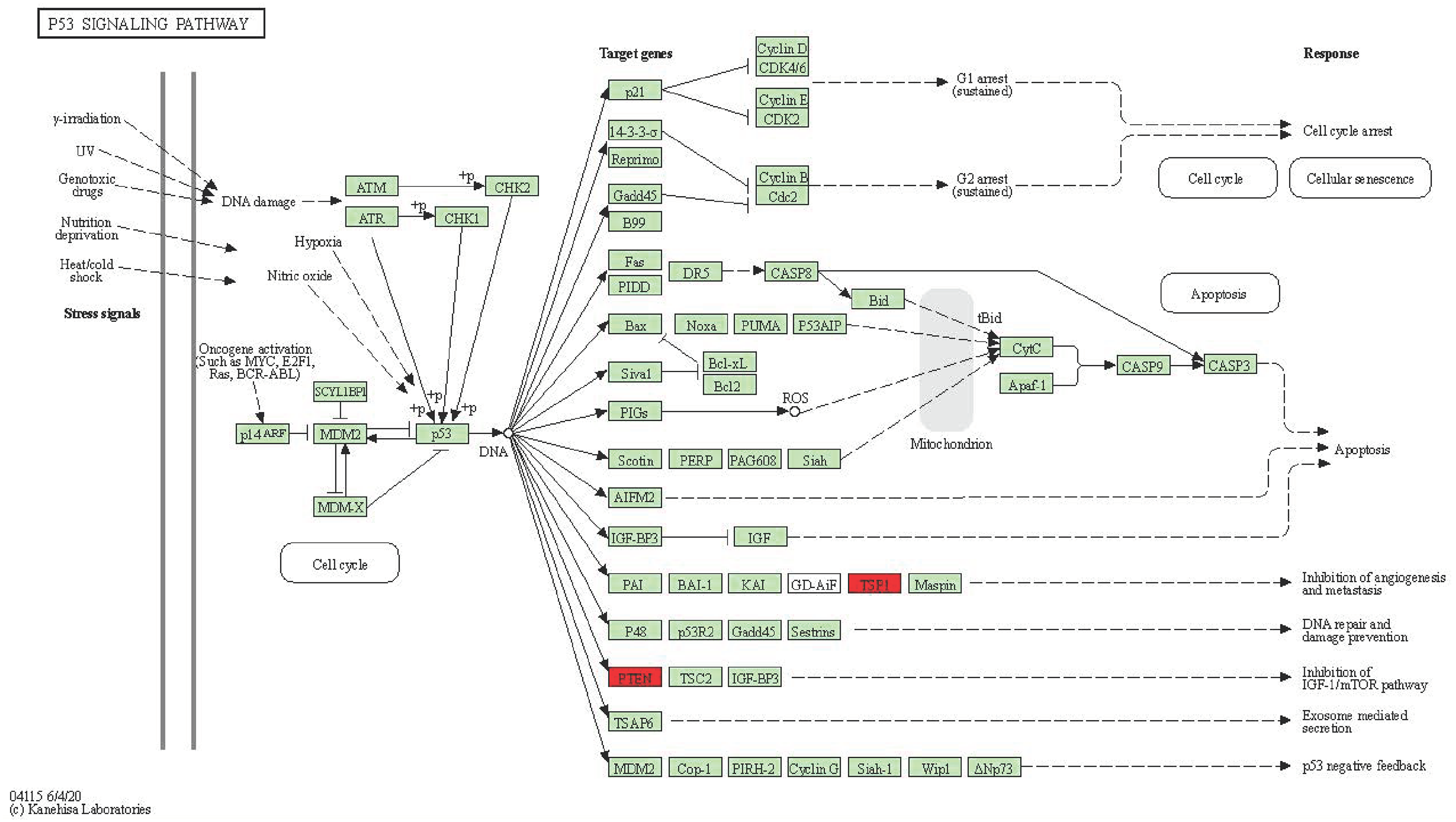
p53 signaling pathway mechanism diagram.
Hub gene screening
The Cytoscape software screened out the Hub gene with the top 10 scores, including 5 mRNA levels: PTEN, DDX3X, SERBP1, THBS1, and SMC1A; 1 miRNA: hsa-miR-429; 4 circRNAs: hsc_circ_0000437, hsa_circ_0000652, hsa_circ_0001472, and hsa_circ_0001009. The corresponding Hub gene network diagram was constructed, indicating that the genes closely related to the occurrence and development of BC were PTEN, DDX3X, and SERBP1, as shown in Figure 5.

Hub gene network diagram.
Discussion
Breast cancer (BC) is one of the most common malignant tumors in female patients. In recent years, some progress has been made in the research on the molecular mechanism and treatment of BC, but it is still the most important cause of death from malignant tumors in women. The occurrence and development of BC involve a series of complex biological processes, including multi-gene mutation and multi-protein interaction. 12 Therefore, it is of great significance to deeply explore the molecular mechanism of BC and identify specific therapeutic targets. Exosomes are small lipid bilayer particles released by cells with multi-functional properties, which regulate complex cellular pathways and participate in all stages of the occurrence and development of BC. 13 In recent years, competitive endogenous RNA (ceRNA), as a new mechanism to explain the interaction between RNAs, is gradually entering our field of view, including long non-coding RNA (lncRNA), mRNA, and circular RNA (circRNA) et al. 14 A large number of frontier studies show that the ceRNA network regulates the target genes of BC, thus affecting the proliferation, migration, invasion, apoptosis, treatment, angiogenesis, and drug resistance of tumor cells. 15
Chi et al 16 showed that lncRNA SNHG5 is an oncogene in breast cancer and a potential predictor of breast cancer. As a “molecular sponge” of mir-154-5p, it weakened the inhibitory effect of mir-154-5p on the target gene PCNA, enhanced the proliferation of breast cancer cells, and suggested the role of ceRNA in breast cancer proliferation. Dong et al 17 found that lncRNA TINCR was highly expressed in trastuzumab-resistant breast cancer patients and associated with patient prognosis, which interacted with HER-2 as a ceRNA and co competed for the binding site on miR-125b to silence miR-125b expression to promote HER-2 expression, resulting in resistance to trastuzumab in breast cancer patients and simultaneously promoted EMT in cancer cells; MiR-125b expression increased and HER-2 expression decreased after TINCR knockdown. Studies have also found that mRNA Snail-1 is also a downstream target gene of miR-125b, and overexpression of miR-125b significantly inhibited Snail expression in breast cancer cells; This suggests that the ceRNA network helps to newly type breast cancer and further elucidates the pathogenesis of unique to various subtypes. However, the ceRNA network of exosome blood in BC patients needs further study.
In this study, the exoRbase database was used to analyze the genetic data of exosomes in the normal population and peripheral blood of patients with BC. Forty-two differentially expressed mRNA, 26 differentially expressed lncRNA, and 43 differentially expressed circRNA were screened out. A ceRNA network consisting of 19 mRNA nodes, 2 lncRNA nodes, 8 circRNA nodes, and 41 miRNA nodes was constructed using Cytoscape software. KOBAS tool was used for KEGG enrichment analysis and visualization of differentially expressed mRNA. The most abundant differentially expressed mRNA was in the p53 signaling pathway. The obtained network information was imported into Cytospace software, and 10 gene Hub genes, including 5 mRNA, 1 miRNA, and 4 circRNA, were obtained by using plug-in CytoHubba. The corresponding Hub gene network diagram was constructed, indicating that the genes closely related to the occurrence and development of BC were PTEN, DDX3X, and SERBP1.
Literature retrieval of the Hub gene revealed that the PTEN gene was closely related to the occurrence and development of BC. PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a deletion on chromosome 10, which encodes a phosphatase and a tumor suppressor gene homologous to tensin and helper proteins, and is usually found missing on human chromosome 10. 18 PTEN gene mutation is often found in a variety of malignant tumors, such as glioblastoma, malignant melanoma, endometrial cancer, prostate cancer, breast cancer, colorectal cancer, pancreatic cancer, etc. Li et al 19 have found that the PTEN gene is closely related to the proliferation, metastasis, infiltration, apoptosis, prognosis, and treatment of BC. Ji et al 20 selected cell line MDA- with normal expression of PTEN-MB-231(M231) were used to silence the expression of PTEN by using the shRNA method, and M231 cell line (M231-3001) with low expression of PTEN and control group cell (M231-SCR) were constructed. The expression inhibition of the target gene was analyzed by using RT-PCR and Western-blot methods. By using the CCK-8 method, cell scratch test, and Transwell cell migration test, it was found that the PTEN gene had a cancer-inhibiting effect, and the reduced expression could promote the proliferation, migration, and invasion of cancer cells. Liu et al 21 studied human BC MDA-MB-231 cells using the CCK-8 method, double-staining method, and Western-blot method and concluded that overexpression of the PTEN gene could inhibit the Akt-mTOR signaling pathway, and increase the apoptosis index of BC cells, and reduce cell proliferative activity, thereby exerting the anti-cancer effect. Ye et al 22 found that the expression of SpALT-like transcription factor 2 (SALL2) was significantly reduced during tamoxifen treatment. Silencing of SALL2 can induce the down-regulation of estrogen receptors and PTEN, leading to estrogen-independent growth of positive BC and tamoxifen resistance, which has provided a direction for the treatment of tamoxifen-resistant BC.
Conclusion
In this study, we successfully identified the exosome RNA related to the occurrence of BC, screened out the differentially expressed mRNA, lncRNA, and circRNA genes, and constructed the corresponding ceRNA network to identify the Hub gene PTEN, which is the key to the occurrence and development of BC. The P53 signaling pathway is an important pathway for the occurrence and development of tumors. KEGG enrichment analysis showed that PTEN was also involved in the signaling of the p53 signaling pathway, which provided an exact theoretical target for the p53 signaling pathway and the occurrence and development of BC. Because the ceRNA regulatory system is complex and cross-through, most of the studies done so far suffer from the problems of small samples and insightful mechanistic studies. In the next step, clinical verification will be performed to further confirm the value and significance of this target in the biological behavior of BC through exosomal cell experiments and vector experiments.
Footnotes
Acknowledgements
Not applicable.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The current study was supported by grants from Jiangsu college students’ innovation and entrepreneurship training plan [202113993016y], Nantong Science and technology planning project [MSZ19217]
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Contributions
All authors participated in the design, interpretation of the studies and analysis of the data and review of the manuscript; Kangle Zhu and Qingqing Wang participated in the statistical analysis. Lian Wang conceived the research, participated in the research design and coordination.
Ethical Approval
Not applicable.