
Abstract
The microbiome plays diverse roles in many diseases and can potentially contribute to cancer development. Breast cancer is the most commonly diagnosed cancer in women worldwide. Thus, we investigated whether the gut microbiota differs between patients with breast carcinoma and those with benign tumors. The DNA of the fecal microbiota community was detected by Illumina sequencing and the taxonomy of 16S rRNA genes. The α-diversity and β-diversity analyses were used to determine richness and evenness of the gut microbiota. Gene function prediction of the microbiota in patients with benign and malignant carcinoma was performed using PICRUSt. There was no significant difference in the α-diversity between patients with benign and malignant tumors (P = 3.15e−1 for the Chao index and P = 3.1e−1 for the ACE index). The microbiota composition was different between the 2 groups, although no statistical difference was observed in β-diversity. Of the 31 different genera compared between the 2 groups, level of only Citrobacter was significantly higher in the malignant tumor group than that in benign tumor group. The metabolic pathways of the gut microbiome in the malignant tumor group were significantly different from those in benign tumor group. Furthermore, the study establishes the distinct richness of the gut microbiome in patients with breast cancer with different clinicopathological factors, including ER, PR, Ki-67 level, Her2 status, and tumor grade. These findings suggest that the gut microbiome may be useful for the diagnosis and treatment of malignant breast carcinoma.
Introduction
Breast cancer is the most commonly diagnosed cancer in women worldwide. 1 Genetic changes and environmental exposures that accumulate in the breast promote development of cancer in only a fraction of all cases. 2 Breast cancer may also be induced or promoted by some other poorly characterized or undescribed factors. Adverse outcomes in breast cancer have also been linked to genetic polymorphisms, obesity, diet, race, and chronic use of antibiotics. 3 Additionally, all these factors are associated with disruption of commensal microbial homeostasis.
The microbiome plays a major role in preserving the human intestinal mucosal barrier, resisting colonization of pathogenic microorganisms, and promoting metabolic balance and immune homeostasis. 4 There is an increasing interest in understanding the role of the altered gut microbiota that may substantially affect human health, such as being a general risk factor for cancer 5 and diabetes, 6 colorectal cancer,7,8 adiposity, 9 inflammatory bowel disease,10,11 lung cancer, 12 cardiovascular diseases, 13 mental diseases,14,15 etc. The microbiota impacts human metabolism and immunity.16,17 The composition of the microbial community can exert negative effects that promote disease occurrence or help in maintaining a healthy status. Studies suggest that pathogenic changes in breast carcinoma may be influenced by microbe-host interactions.18,19 The most influential bacteria in patients with breast cancer were found at different sites, such as the breast tissue, skin, oropharynx, and gastrointestinal tract. 3 Of these, the microbiome of the gastrointestinal tract plays a potential role in steroid hormone metabolism and synthesis of biologically active estrogen mimics during the development of breast cancer. 20 The microbial communities in breast cancer can also act as potential biomarkers for diagnosis, treatment, and prognosis, thereby promoting development of a new era of individualized medicines. 21
Studies have demonstrated a correlation between the gut microbiota and estrogen levels, tumor grade, stage, and tumor type in patients with breast cancer.21-23The microbiome in malignant and benign breast tumor tissues was reported to be dramatically different24,25; but, there is limited research on the diversity of the microbiome in the gastrointestinal tract of patients with malignant and benign breast tumors.
In this study, we collected fecal samples from patients with malignant and benign breast tumors, performed 16S rRNA gene sequencing, and performed functional analysis of the gut microbiota. The results show the distinct richness of the gut microbial communities in patients with malignant or benign breast tumors, and provides an opportunity to explore potential role of the gut microbiome in breast carcinogenesis. Further, we evaluated the association of the gut microbiome with clinicopathological characteristics in patients with breast tumors, which may provide insights in breast cancer treatment and prognosis.
Materials and Methods
Patient enrollment
The study was performed in accordance with the principles of the Declaration of Helsinki concerning ethical research related to human subjects and was approved by the Institutional Review Board of the Quanzhou First Hospital affiliated to the Fujian Medical University. Informed consent was obtained from all the participants. Eighty-three patients with invasive ductal breast carcinoma who underwent surgery between January 2019 and December 2020 were enrolled in the study. Additionally, 19 patients with benign breast tumors who underwent segmental resection were recruited in the benign group. Segmental mastectomy is the mainstay of therapy for benign breast tumors. Patient data, including age, menopausal status, and tumor characteristics (tumor grade, diameter, lymph node involvement, Ki-67 index, estrogen receptor [ER]/progesterone receptor [PR] expression, and human epidermal growth factor receptor-2 [Her2] status), were obtained from pathology reports. The inclusion criteria for both the groups were as follows: female sex, good physical condition, no history of other cancers, active intestinal dysfunction or gastrointestinal inflammation, no bariatric surgery, celiac disease, pregnancy or nursing within the past 12 months, no special eating history (eg, vegetarian), and no antibiotic treatment prior to fecal specimen collection. Patients with breast cancer were diagnosed with nonspecific invasive carcinoma and did not receive surgery, chemotherapy, endocrine therapy, or radiation prior to sample collection. Subjects were excluded if they received antibiotic treatment within 6 months or other treatment before the onset of the disease, had undefined histological grade in pathological tests, had other pathological types of breast cancer or breast cancer during lactation, or were non-compliant.
Sample collection
We collected fresh stool samples, without undigested food residues or other solid substances, from patients who met the inclusion criteria, before treatment. The DNA preservation solution was used for sample storage at 18°C to 26°C, the main components of the solution were trihydroxy-methyl aminomethane buffer, disodium ethylenediamine tetra-acetic acid, sodium chloride, and sodium dodecyl sulfate.
DNA extraction
DNA extraction was performed using the SDS lysis buffer freeze-thaw method. Genomic DNA was isolated using PowerMax extraction kit (MoBio Laboratories, Carlsbad, CA, USA) and stored at −20°C until further analysis. The quantity of DNA was determined using NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), followed by agarose gel electrophoresis.
16S rRNA gene sequencing and analysis
The 16S rRNA gene amplicons were prepared in a 50 µL reaction. The V4 region of the 16S rRNA gene was amplified by polymerase chain reaction (PCR) with following primers: 515F (5’-GTGCCAGCMGCCGCGGTAA-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’). 26 The forward primer 515F was linked to A-adaptor, a specific 7-bp barcode, while the reverse primer 806R carried B-adapter. The PCR were performed in a 50 μL mixture containing 25 μL of high-fidelity enzyme (Phusion HighFidelity PCR Master Mix with HF Buffer), 2 × 3 µL of each primer (5 μm), 9 µL of double distilled water, and 10 µL of template DNA. The PCR conditions were as follows: 8°C for 30 seconds (one cycle); 98°C for 15 seconds, 58°C for 15 seconds, and 72°C for 15 seconds (25 cycles); and 72°C for 1 minute (1 cycle). Further, the PCR products were purified using AMPure XP Beads (Beckman Coulter, Indianapolis, IN, USA), quantified using PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA), and then pooled amplicons were concentrated using HiSeq platform (Illumina HiSeq 4000).
The 16S rRNA gene sequencing analysis was performed using Quantitative Insights into Microbial Ecology (QIIME, version 1.9.1) software suite 27 and QIIME tutorial (http://qiime.org/) with some modifications. After sequencing, Vsearch (v2.4.4) was used to splice the reads in each sample to obtain the original tag data. Low-quality material or fragments, such as barcode and primer sequence mismatches, sequences sized <150 bp, average mass value <20, sequences with ambiguous characters, single nucleotide repeats of >8 bp, or sequencing errors and chimeras were removed during the pre-processing procedure. The extracted data were saved in the FastQ format. Each sample of the 2-terminal sequencing data contained FastQ1 and FastQ2 files, which were read at both ends of the sequence. The quality of the sequences was controlled and filtered, and splicing was concomitantly performed.
Cluster analysis of operational taxonomic units (OTUs)
Operational taxonomic units (OTUs) are hypothetical taxa in phylogenetic analyses or population genetic research. The OTU in this study represents sequence from the same source. OTU clustering analysis is usually performed at a threshold of 97% identity.
After clustering, the sequences were assigned taxonomy and referenced by GreenGene database. 28
Alpha diversity analysis
Alpha diversity (α-diversity) was determined using several indices to calculate the complexity of species diversity in samples. Differences in α-diversity were computed using the Chao, ACE, and Shannon diversity indices. The Chao and ACE indices reflected species richness of the community. As a reflection of the species diversity, the Shannon value indicates both species richness and evenness—the value considers the abundance of each species.
Rarefaction curve
Rarefaction curves were computed from OTU tables using α-diversity, and random sampling of the sequence from each sample indicated community richness.
Beta diversity analysis
Beta diversity (β-diversity), computed using hierarchical cluster dendrograms (Bray-Curtis distance dissimilarities), 29 was used to compare the similarity between samples, which in this study is visibly displayed in the form of principal component analysis (PCA) and principal coordinate analysis (PCoA). The process of PCoA is using OTU sequence and based on UniFrac-weighted distances. The P-values were obtained by the similarities of the analysis of similarities (Anosim). 30
Kyoto encyclopedia of genes and genomes (KEGG) analysis
We performed linear discriminant analysis (LDA) effect size (LEfSe) in the relative abundance of genera to identify influences of biomarkers that were statistically significant. 31 LDA values >2.0 were considered significantly enriched only when P-value <.05. The KEGG 32 was used for abundance and functional annotation. Based on the sequencing data and comparison with the GreenGene database, 28 we used the Phylogenetic Investi-gation of Communities by Reconstruction of Unobserved States (PICRUSt, version 1.1.4) to compare the OTU information and predict the functional composition of the metagenome.
Statistical analysis
Data analysis was performed using the Wilcoxon rank-sum test for binary variables and to explore differences in α-diversity between any 2 groups. The figures are prepared using R (v3.2.0). We used STAMP software (version 2.1.3) 33 to display the graphical representation and predict the KEGG function information. A P-value <.05 was considered statistically significant.
Results
Patient characteristics
To investigate the composition of the gut microbiome in patients with malignant and benign breast tumors, we collected fecal samples from 83 patients with invasive breast cancer and 19 patients with benign breast tumors (clinicopathological information is summarized in Table 1). The median age at diagnosis was 48.07 (range, 29-68) years and 45.14 (range, 31-56) years in the malignant and benign groups, respectively. Of these, there were 30 and 6 postmenopausal patients in the malignant and benign tumor groups, respectively. In the malignant tumor group, 51 (61.44%), and 47 (56.6%) tumors were ER-positive and PR-positive, respectively. Further, 29 (34.9%) and 48 (57.8%) patients had grade II and grade III diseases, respectively, and 37 (54.2%) were Her2 positive.
Demographic information of all subjects used in this study.

Abbreviations: ER, estrogen receptor; HER-2, human epidermal growth factor receptor 2; PR, progesterone receptor.
Wilcoxon rank test between benign vs malignant group for menopausal status.
Microbiota analysis
We first evaluated the richness and evenness of the gut microbiome in the 2 groups (Figure 1). The α-diversity analysis showed that the community richness was not significantly different between benign and malignant groups (P = 3.15e−1 for the Chao index and P = 3.1e−1 for the ACE index; Wilcoxon test; Figure 1a and b). No significant difference in community evenness was observed between the 2 groups using the Shannon index (P = .3901; Wilcoxon test; Figure 1c). The rarefaction curve tended to be flat, suggesting that community richness approached saturation in both groups (Supplemental Figure S1).

Alpha diversity indices boxplot between benign (n = 19) and malignant (n = 83) groups. comparison based on the (a) Chao, (b) ACE, and (c) Shannon.
We investigated the taxonomic profiles of the gut microbiota of patients with malignant and benign tumors at the phylum and genus levels and found that the abundances of major taxa in both the groups were similar (Figure 2a). The gut microbiota of patients with malignant and benign breast tumors is illustrated by a heat map, where an increase in the number of species in the gut microbiota was observed in the malignant group than that in benign group (Figure 2b). The β-diversity analysis was used to compare the similarities between samples, including PCA and PCoA. After visualization, some differences in community membership between the 2 groups were observed in the PCA (Figure 2c), but they were not significant in the PCoA (Anosim P = .953; Figure 2d).
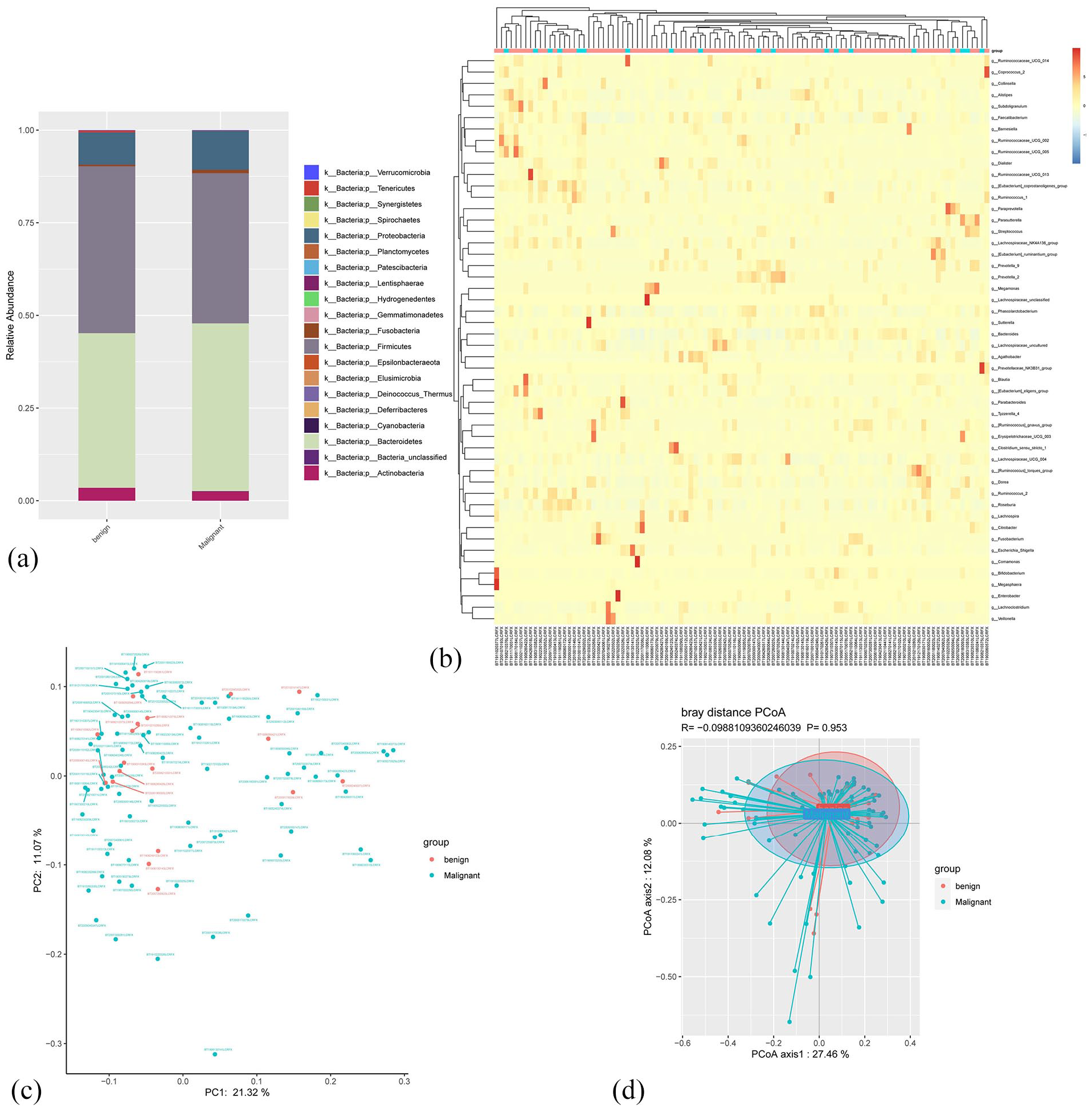
Comparation of the gut microbiota taxonomic profiles from patients with benign (n = 19) and malignant (n = 83) tumors. (a) Barplots of the gut microbiota taxonomic profiles of the benign and malignant cases at phylum level. (b) Heatmap shows the OTU presence and absence of the top 50 fecal samples at genus level. (c) The analysis of principal component analysis (PCA) based on OTU between groups. (d) Principal co-ordinates analysis (PCoA) based on OTU between groups (P-value from Anosim analysis are shown).
To further assess the differences in the microbiome between the benign and malignant groups, an LEfSe analysis was performed to identify differences in the microbiota and detect significant biomarkers. The relative distribution of 31 genera was found to be significantly different between the benign and malignant groups (P < .05), including Faecalibacterium, Lachnospira, Clostridium, Brachybacterium, Butyrivibrio, and Mobiluncus. Furthermore, LEfSe showed that Clostridium, Faecalibacterium, Lachnospira, Erysipelotrichaceae, Romboutsia, Fusicatenibacter, Xylophilus, and Arcanobacterium were significantly more abundant in the benign tumor group than that in malignant tumor group. The amount of Citrobacter was significantly higher only in the malignant tumor group (LDA score >3; Figure 3a). Differential analysis of taxa in the gut microbiota of the malignant and benign groups based on a permutation test is shown in Figure 3b.

Comparison of gut microbiota using LDA and KEGG. (a) Differential taxa between benign and malignant gut microbiota based on a Linear Discriminant Analysis (LDA). Taxa with LDA score >2 at the family and genus level are defined as statistical differences. (b) Differential taxa between the gut microbiota of the 2 groups based on a permutation test. (c and d) Histogram of Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathway of gut microbiota.
We used PICRUSt with STAMP software to calculate the KEGG pathways in the gut microbiome of patients with benign and malignant tumors. The gut microbiota in the malignant tumor group showed an increase in poorly characterized glycan biosynthesis and metabolism, whereas there was a reduction in transcription than that in benign group (P < .05, Figure 3c). A more detailed analysis of KEGG pathways indicated significant differences in 26 pathways (P < .05), including lipopolysaccharide biosynthesis, glycolysis/gluconeogenesis, folate biosynthesis, glycerophospholipid metabolism, and sporulation between the 2 groups (Figure 3d).
Microbial profiles associated with clinicopathological factors in breast cancer
Further, we evaluated the association between the gut microbial communities and clinicopathological factors in breast cancer. Patients with PR-positive breast cancer displayed an enrichment of Prevotellaceae and Tyzzerella, whereas those with PR-negative breast cancer had an abundance of Barnesiellaceae, Lactobacilliaceae, Lactobacillus, Prevotellaceae, Cloacibacillus, Acinetobacter, Hydrogenophilus, Rhodobacteriae, and Hydrogenophilaceae (Figure 4a). Microbial species of Megasphaera, Roseburia, and Prevotellaceae were found to be of distinct richness in patients with ER-positive breast cancer, while Bacteroides, Bacteroidaceae, Puniceicocceae, Opitutales, Hydrogenophilus, and Hydrogenophilaceae were significantly enriched in those with ER-negative breast cancer (LDA score > 3) (Figure 4b). The gut microbes, such as Megasphaera, Barnesiellacea, Alloprevotella, Lachnospiraceae, Moraxellaceae, Acinetobacter, Enorma, Flavonifractor, Burkholderiaceae, and Eubacterium were more abundant in patients with Her2-positive cancer than in those with Her2-negative cancer (Figure 4c). In patients with low Ki-67 expression (Ki-67 < 30%), the gut microbiomes were enriched for Lactobacillus, Clostridium, Clostridiaceae, Megasphaera, Proteus, and Burkholderiaceae. However, Ruminiclostridium, Tenericetes, Mollicutes, Ruminococcaceae_UCG, Izimaplasmatales, Sporobacter, Syntrophomonadaceae, and Clostridiales_vadinBB60 were enriched in the Ki-67 high expression group (Ki67 ⩾ 30%) than in Ki-67 low expression group (Figure 4e). Enrichment of Coriobacteriaceae, Collinella, and Faecalitalea was found in patients with histologic grade III disease (Figure 4f). Additionally, an increase in numbers of microbial species, such as Enterobacter, Erysipelotrichaceae, Romboutsia, Anaerostipes, Granulicatella, and Carnobacteriaceae was particularly observed in premenopausal patients; whereas, numbers of Herbinix, Ruminococcaceae, and Epulopiscium were increased in postmenopausal women (Figure 4d).

LEfSe identified the most differential gut microbiota in 83 breast cancer patients. Analysis performed based on clinicopathologic groups in (a) PR+ (n = 47) and PR+ (n = 36), (b) ER+(n = 51) and ER-(n = 32), (c) Her2+ (n = 37) and Her2- (n = 45), (d) pre-menopause(n = 53), post-menopause (n = 30), (e) Ki67 < 30% (n = 21), Ki67 ⩾ 30% (n = 62), and (f) tumor grade I (n = 3), grade II (n = 29), grade III (n = 48). Taxa with LDA score > 2 are defined as statistical differences.
Discussion
We investigated the gut microbiome of patients with benign and malignant breast tumors using 16S rRNA gene sequencing and compared the microbial communities based on clinicopathological factors in patients with breast cancer. Microbiome community richness was higher in benign group than in malignant group. The metabolic pathways in patients with malignant tumors were significantly different from those in patients with benign tumors. The study also established the distinct abundance of the gut microbiome in patients with breast cancer with different clinicopathological factors, including ER, PR, and Ki-67 levels, Her2 status, and tumor grade.
Zhu et al 18 found that the gut microbial communities differed between patients with breast cancer and healthy controls both in pre- and post-menopausal women using the Chao and Shannon indices. Goedert et al 19 suggested that postmenopausal patients with breast cancer had a less diverse and compositionally different fecal microbiota than postmenopausal healthy women. Moreover, the present study also showed that the fecal microbiota of patients with malignant tumor than those with benign tumor had a lower α-diversity with the Chao and ACE indices, but not with Shannon index. Therefore, patients with malignant tumors have less species richness, but indistinctive evenness of the gut microbial community than those with benign tumors.
Antibiotic use can cause disorder of host immune system, change the composition of gut microbiota34,35 and possibly aggravate chronic inflammation and carcinoma.36,37 However, the debate on the role of antibiotics remains inconclusive. Several observational studies indicate that antibiotic use won’t increase the risk of breast cancer.38-42 Elkrief et al concluded that the effect of broad-spectrum antibiotics on the intestinal flora of cancer patients receiving immunotherapy is also controversial. Other studies show that antibiotic-induced turbulence of gut microbiota can aggravate breast tumor and metabolic disorder.43,44 A systematic review demonstrates that the risk of breast cancer modestly increases for individuals who have ever used antibiotics. 45 The causality between antibiotics and breast cancer remains unclear, so further explanation and mechanistic studies still need to be done.
Further, the study revealed that patients with malignant tumors possessed elevated levels of Citrobacter, whereas a great majority of the microbiota elevated in those with benign tumors included Clostridium, Faecalibacterium, Lachnospira, Erysipelotrichaceae, Romboutsia, Fusicatenibacter, Xylophilus, and Arcanobacterium. Zhu et al 18 found that 38 species were enriched in the postmenopausal group, including Citrobacter, Escherichia, and Klebsiella. In contrast, a previous analysis demonstrated that the levels of Clostridaceae, Faecalibacterium, and Ruminococcaceae were higher, while those of Dorea and Lachnospiraceae were lower in postmenopausal patients with breast cancer than levels in healthy controls. 19 Preclinical studies in a mouse model showed that infection with Citrobacter increased epithelial cell proliferation and promoted growth of chemically-induced colon tumors. 46 In contrast to previous studies,18,47,48 Clostridium richness was found in patients with benign breast tumor than in those with malignant tumor. Diversity in the gut microbiota composition of patients with breast carcinoma suggests that microbial metabolism or dysbiosis may play an important role in breast cancer development.
Dysbiosis in the microbial community can regulate systemic immune function and increase production of inflammatory mediators that correlate with poor outcome in many diseases, including malignant tumors.3,49,50 It is established that estrogen metabolism is associated with some species of the gastrointestinal microbes.51,52 However, the metabolic output of the microbiota often reflects the condition of the commensal ecosystem. 3 Microorganisms and their metabolites have variable effects on breast cancer through different metabolic pathways. For instance, bacterial metabolites, such as lithocholic acid and cadaverine, have been shown to exert an inhibitory effect on breast cancer.53-55 Kovács et al showed that lithocholic acid increases oxidative stress in breast cancer. Based on the present analysis, the gut microbial metabolism in patients with malignant and benign tumors was significantly different. We observed an increase in a majority of metabolic pathways in patients with malignant tumors, especially the lipopolysaccharide biosynthesis pathways, than that in patients with benign tumors. In contrast, a significant increase in sporulation in patients with benign tumors was found by KEGG analysis. A previous study has shown that bacterial lipopolysaccharides are frequently present in many human solid tumors. 56 Moreover, high-fat diets may increase the serum levels of bacterial lipopolysaccharide, thereby impacting breast cancer risk in mouse models. 57
Patients with breast cancer with different clinical characteristics show diverse compositions of the gut microbiota.21,58 A study conducted by Bard et al 59 showed that the number of Blautia sp. was higher in patients with grade III disease than in those with grade I disease, and the enrichment or proportions of some bacteria was significantly different according to the clinical stage and body mass index (BMI). Moreover, Luu et al 22 suggested that the microbiome in patients with breast cancer significantly varied with different clinical stages, histoprognostic grades, and BMI. Similar to a previous study, the present study found that the number and abundance of the gut microbial communities were extraordinarily higher in patients with grade III than in those with grade I/II breast cancer. Furthermore, an abundance of Herbinix, Ruminococcaceae, and Epulopiscium was detected in postmenopausal patients.
Additionally, patients with breast cancer with different expression of Ki-67 (<30%/⩾30%), ER (+/−), PR (+/−), and Her2 (+/−) showed microbiome diversity in the present study. A study by Banerjee et al 23 demonstrated similar microbial signatures in ER+ and Her2+ samples, while the triple negative and triple positive samples presented with distinct patterns of microbiomes. Similarly, Nejman et al 56 revealed that the prevalence of multiple bacterial taxa was various in different breast cancer subtypes based on their ER, PR, and Her2 status. It is suggested that each breast cancer type might have type-specific communities. The connection of breast cancer type and gut microbiome might via enterohepatic circulation, which were thought to affect circulating and excretory estrogen levels in breast cancer.
According to analysis of different clinicopathological grouping, Wu et al 60 found that Her2 status and age at menarche had significant association with α-diversity measures of the gut microbiome and specific microbial composition. He et al 48 found that the composition and symbiosis of the gut microbiota in patients with premenopausal breast cancer changed significantly in comparison with that in healthy premenopausal women. However, further studies are needed to confirm the characteristics of the human microbiome and intrinsic connection between the pathological and clinical features of breast cancer and established risk factors.
Enrichment of Megasphaera was identified by 16S rRNA sequencing analysis in patients with ER+ and Her2+ tumors in the present study. It is known that estrogen levels are influenced by the diversity of the gut microbiome, and that increased levels of endogenous and circulating estrogen are related to a high risk of breast cancer in postmenopausal women. 61 Interestingly, in the present study, members of only the Prevotellaceae family were abundant in both patients with PR+ and ER+ tumors, while microbes such as Hydrogenophilus, Lactobacillus, and Acinetobacter were highly abundant in patients with PR- and ER- tumors. Therefore, the function and connection of the gut microbiome in estrogen production warrants further investigation. Moreover, it will be interesting to investigate the role of the gut microbiome in the treatment response, including chemotherapy, endocrine therapy, and targeted therapy.
Previous studies have attempted exploration of similar topics. Terrisse et al 62 found that chemotherapy drastically shifted the microbiome composition, by reducing abundance of microflora associated with side effects and increasing abundance of favorable commensals after adjuvant or neoadjuvant treatment with anthracylines and taxanes. A study by Di Modica et al 63 revealed the existence of specific microbiota in patients with Her2+ breast cancer that can influence their response to targeted therapy. This suggests that manipulation of the fecal microbiota is an optimal future regimen to achieve effective treatment or to explore its potential as a biomarker for therapeutic response. Moreover, anticancer properties of the intestinal flora should be closely studied. It is possible that the gut microbiome community in patients with breast cancer may contribute to the prognosis and survival outcome. However, a lack of a healthy control group and a low number of samples were limitations in this study. Thus, further analysis of a healthy cohort and a larger sample size is imperative for whole-genome sequencing.
In summary, the study demonstrates that the proportion and metabolic pathways of the gut microbiome in patients with malignant breast tumors are significantly different from those in patients with benign tumors. Furthermore, we observed a distinct enrichment of the gut microbiome in patients with breast cancer with different clinicopathological factors, including ER, PR, Ki-67 levels, Her2 status, and tumor grade. These findings suggest that the gut microbiome may be useful for the diagnosis and treatment of malignant breast tumors.
Supplemental Material
sj-jpg-1-evb-10.1177_11769343211057573 – Supplemental material for Comparison of the Gut Microbiota in Patients with Benign and Malignant Breast Tumors: A Pilot Study
Supplemental material, sj-jpg-1-evb-10.1177_11769343211057573 for Comparison of the Gut Microbiota in Patients with Benign and Malignant Breast Tumors: A Pilot Study by Peidong Yang, Zhitang Wang, Qingqin Peng, Weibin Lian and Debo Chen in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this study was provided by Quanzhou Science and Technology (No. 2019N033S).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors Contributions
D.B.C. contributed to the conception and design of the study; P.D.Y., Z.T.W., and W.B.L. contributed to the acquisition of data and analysis; P.D.Y. and Q.Q.P. contributed to drafting the article, and/or revised the manuscript. D.B.C. did study supervision.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.