
Abstract
In humans, taste genes are responsible for perceiving at least 5 different taste qualities. Human taste genes’ evolutionary mechanisms need to be explored. We compiled a list of 69 human taste-related genes and divided them into 7 functional groups. We carried out comparative genomic and evolutionary analyses for these taste genes based on 8 vertebrate species. We found that relative to other groups of human taste genes, human TAS2R genes have a higher proportion of tandem duplicates, suggesting that tandem duplications have contributed significantly to the expansion of the human TAS2R gene family. Human TAS2R genes tend to have fewer collinear genes in outgroup species and evolve faster, suggesting that human TAS2R genes have experienced more gene relocations. Moreover, human TAS2R genes tend to be under more relaxed purifying selection than other genes. Our study sheds new insights into diverse and contrasting evolutionary patterns among human taste genes.
Keywords
Introduction
Taste genes code for proteins that facilitate the sensation of different tastes. The most frequently studied taste genes are sweet, umami, and bitter.1-5 However, the underlying biological processes for salty and sour taste remain poorly understood.6-8 The human taste system also detects noncanonical “tastes” such as water, fat, and complex carbohydrates, but the research on their reception mechanisms is in an early stage. 2
The human TAS1R gene family with 3 members TAS1R1, TAS1R2, and TAS1R3 conducts conserved taste sensation functions in vertebrates. TAS1R1 + TAS1R3 heterodimer receptor functions as an umami receptor, while the TAS1R2 + TAS1R3 heterodimer receptor functions as the sweet receptor.5,9,10 The human TAS2R gene family, with around 25 functional members, functions as bitter taste receptors.11,12 In addition, 11 human TAS2R pseudogenes have been identified. 13 The epithelial sodium channel (ENaC) mediates the sensation of the salty taste.6,14 The ENaC has 4 subunits, α, β, γ, and δ, encoded by the non-voltage-gated sodium channel 1 genes SCNN1A, SCNN1B, SCNN1G, and SCNN1D. The transient receptor potential (TRP) cation channel is a large gene family with diverse sensation roles, including taste sensation. 15 The subfamily V (TRPV1-6) is a promising candidate for transduction of amiloride-insensitive cation-nonselective salty taste.16,17 The classical subfamily (TRPC) can sense membrane lipids. 18 TRPM5, a cation channel, has an essential role in the transduction of bitter, sweet, and umami tastes. 19 The acid-sensing ion channel genes (ASIC1-5) and PKD genes are receptors for acid (sour) taste.8,20 The free fatty acid receptors (FFAR1-4) are G protein-coupled receptors activated by free fatty acids (FFAs), which play essential roles as essential nutritional components. 21
Evolution of taste genes may play an essential role in species adaptations to their specific chemical environments and feeding ecology. 2 Previous evolutionary studies on taste genes were mostly related to TAS1R and TAS2R gene families. TAS2R genes tend to be shorter, which are around 1 kb. 1 TAS1R genes are relatively conserved in evolution, while TAS2R genes are more variable and diverge tremendously among species.22,23 TAS2R genes frequently experienced lineage-specific expansions and losses.13,24,25 TAS2R pseudogenes are differently distributed among species, with some of them (eg, TAS2R38) being polymorphic among human populations.13,26 Copy number variations play an important role in TAS2R genes, especially within the TAS2R43—45s genomic regions.27,28 Moreover, potential positive selection on several TAS1R and TAS2R genes23,29-32 and balancing selection on TAS2R genes32,33 were reported.
It is still unknown what mechanisms have led to the distinct evolutionary patterns of TAS2R genes. Both single gene and whole-genome duplications (WGD) have recurred in vertebrate evolution.34,35 Genes created by different modes often experienced different evolutionary tempos and gene loss rates.36,37 Moreover, gene relocations frequently occur during evolution, which is often associated with increased evolutionary rates.38,39 Genetic polymorphisms and genomic structures may interact to shape taste perception.27,28 The aim of this study is to better understand human taste genes’ evolutionary mechanisms and humans’ taste perceptions and eating behaviors via comparative genomic analysis of taste genes.
Results
Functional groups of human taste genes
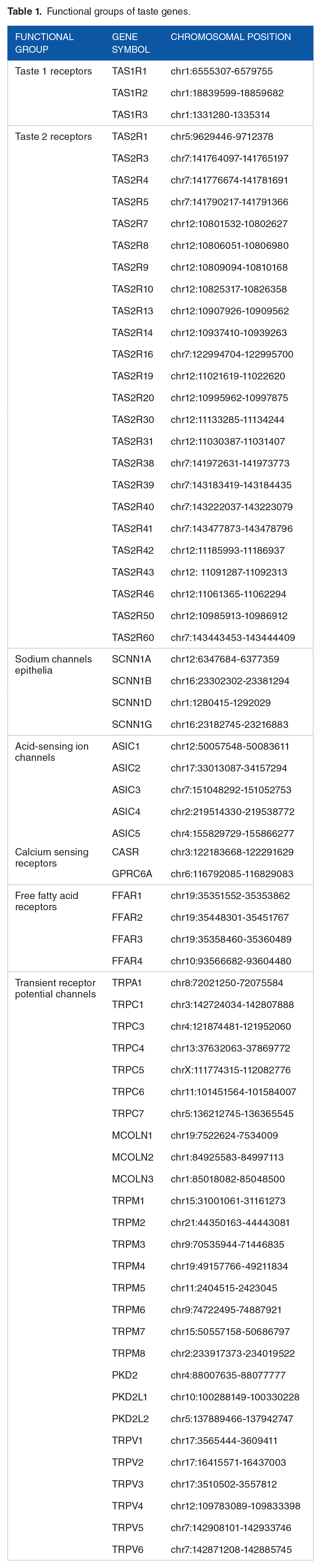
We compiled a list of human taste genes, including 7 functional groups (Table 1). The first group is TAS1R genes, which have 3 members TAS1R1, TAS2R2, and TAS3R3. The second group is TAS2R genes, which are responsible for sensing bitter taste. We collected 24 TAS2R genes, including TAS2R1, TAS2R3, TAS2R4, TAS2R5, TAS2R7, TAS2R8, TAS2R9, TAS2R10, TAS2R13, TAS2R14, TAS2R16, TAS2R19, TAS2R20, TAS2R30, TAS2R31, TAS2R38, TAS2R39, TAS2R40, TAS2R41, TAS2R42, TAS2R43, TAS2R46, TAS2R50, and TAS2R60. The third group of sodium channels epithelia genes are responsible for salt taste perception, including SCNN1A, SCNN1B, SCNN1D, and SCNN1G. The fourth group acid-sensing ion channel genes are candidates for sour taste perception, including ASIC1, ASIC2, ASIC3, ASIC4, and ASIC5. The fifth group is calcium-sensing receptors, which have 2 members, CaSR and GPRC6A. The sixth group is free fatty acid receptors for sensing fatty acid taste, including FFAR1, FFAR2, FFAR3, and FFAR4. The last group is transient receptor potential channels suggested to have essential roles in the sensation of sweet, bitter, and umami tastes. This group includes TRPA1, TRPC1, TRPC3, TRPC4, TRPC5, TRPC6, TRPC7, MCOLN1, MCOLN2, MCOLN3, TRPM1, TRPM2, TRPM3, TRPM4, TRPM5, TRPM6, TRPM7, TRPM8, PKD2, PKD2L1, PKD2L2, TRPV1, TRPV2, TRPV3, TRPV4, TRPV5, and TRPV6. Some genes we involved in the analysis might be just putative taste genes. However, generating an aggressive list of human taste genes will render a comprehensive comparison among different taste genes while not negatively affecting comparative genomic and evolutionary analyses. Detailed information regarding the human taste genes, including various comparative genomics and evolutionary metrics, is included in Supplemental Table S1.
Functional groups of taste genes.
Tandem duplications contribute to the expansions of TAS2R genes
We classified human taste genes into different gene duplication modes, including singletons, whole genome/segmental (ie, collinear genes in collinear blocks), tandem (consecutive repeat), proximal (in the nearby chromosomal region but not adjacent), or dispersed (other modes than segmental, tandem, and proximal) duplications on their copy number and genomic distribution. 38 Among the 24 TAS2R genes, 14 have experienced tandem duplications, while among the 45 other genes, 9 have experienced tandem duplications. The comparison of proportions of tandem duplicates (58.3% vs 20.0%) is significant (P = 3.19 × 10−3, χ2). This observation suggests that tandem duplications are a significant gene duplication mode responsible for expanding human TAS2R genes. Moreover, there are 5 proximal duplicates (20.8%) among human TAS2R genes, while there are only 2 proximal duplicates (4.4%) among other genes. Although the contrast in proportions is obvious, the comparison is not significant due to small numbers. Tandem duplications are believed to be caused by unequal crossing-over and are often associated with inversions. 40 This analysis suggests that human TAS2R genes tend to have experienced different duplication events among human taste genes.
Collinearity analysis of human taste genes
Over the evolutionary course of eukaryotic genomes, genes may remain on corresponding chromosomes (synteny) and corresponding orders (collinearity). 41 Compared with outgroup genomes, we hypothesize that genes without collinear orthologs have undergone relocation, often associated with the reshuffling of chromosomal segments or transposon activities. 40 We generated collinear blocks among 8 vertebrate genomes. We computed the number of cross-species collinear genes for each human taste gene and the number of outgroup species with collinear genes. Comparisons of these 2 indicators between human TAS2R genes and other human taste genes (Figure 1) showed that human TAS2R genes tend to have less collinear genes in outgroup species (P = 1.03 × 10−3, Wilcoxon test) and fewer outgroup species with collinear genes (P = 6.93 × 10−8, Wilcoxon test). They are thus suggesting that human TAS2R genes have more frequently relocated during evolution.

Comparison of gene relocation between TAS2R and other genes in humans. Gene relocation is inversely related to the conservation of collinearity by comparison with multiple outgroup species: (a) comparison of the number of collinear genes and (b) comparison of the number of outgroup species with collinear genes.
Analysis of selection pressure on human taste genes
Relative to TAS1R genes, TAS2R genes are more variable and diverge tremendously among species. 22 Here, we computed nonsynonymous (Ka) and synonymous (Ks) substitution rates for human-mouse orthologous pairs for the taste genes (see Methods). We used Ka/Ks to denote selection pressure. None of human taste genes displayed Ka/Ks > 1 (Supplemental Table S1), indicating no signs of positive selection. We found that human TAS2R genes have significantly higher Ka/Ks than other genes (Figure 2, P = 1.89 × 10−10, Wilcoxon test). TAS2R genes tend to be shorter than other taste genes (P = 2.44 × 10−10, Wilcoxon test) and there was a significant correlation between gene lengths and Ka/Ks (r = −0.307, P = .014) in all taste genes. Thus, we computed gene length-adjusted Ka/Ks and compared it between TAS2R and other genes. A significant p-value (P = 8.88 × 10−8, Wilcoxon test) was remained. This observation suggests that human TAS2R genes are under relaxed purifying selection, while other types of human taste genes tend to be under purifying selection.

Comparison of Ka/Ks between TAS2R and other genes in humans.
We then computed Tajima’D 42 for each taste gene to detect departures from neural selection, based on the African and European populations from the 1000 Genomes Project. 43 TAS2R genes did not show significantly higher Tajima’s D than other genes (P = .701 and .243 for AFR and EUR respectively, Wilcoxon test). This analysis suggests that although individual TAS2R genes may have experienced balancing selection32,33 or positive selection, 30 in general TAS2R genes do not show distinct diviation patterns from neural selection than other taste genes.
Conclusions and Discussion
In this study, we carried out comparative genomic and evolutionary analyses for taste genes across 8 vertebrate species. Human TAS2R genes have a higher proportion of tandem duplicates and tend to have fewer collinear genes in outgroup species. Human TAS2R genes tend to evolve under relaxed purifying selection, while other genes tend to evolve under purifying selection. This study generates new insights into the diverse and contrasting evolutionary patterns of human taste genes.
Tandem duplications occur more frequently within TAS2R genes. Vertebrate genomes have experienced 2 rounds of WGDs during their early evolution. 34 More recent WGDs have also occurred in the teleost fish,44,45 salmonid, 46 and Xenopus laevis 47 lineages, but not the human lineage. Tandem gene duplications, which may frequently occur during evolution, provide a continuous and large amount of genetic materials for species’ evolution and adaptation to specific environments and ecology. 48 Moreover, human-specific gene duplications such as BOLA2 have arisen exclusively in Homo sapiens and have been associated with human diseases. 49 TAS2R genes, under relaxed purifying selection, may quickly evolve new functions which may become fixed because coincidentally fitting to species’ environment. Tandem duplicates have a higher evaporating (ie, gene death) rate. 37 However, many gene duplications offset the high evaporating rate while providing enough raw materials for selection to work.
Variations in the TAS1R genes were previously reported to be under positive selection.23,29 Among TAS2R genes, TAS2R16 and TAS2R38 were previously reported to be under recent positive selection.30-32 TAS2R38 might have also experienced balancing selection. 26 Variations within the TAS1R3 promoter were found to be associated with human taste sensitivity to sucrose and show signs of departure from neutral selection. 50 We did not find signatures of positive selection in human taste genes, which is consistent with a recent study. 51 Our finding that human TAS2R genes tend to be under relaxed purifying selection aligns with the neural evolution hypothesis.13,52
A limitation is that it remains unclear when the relaxation of purifying selection on TAS2R genes started. Although alignments of ancient human genomes such as Neanderthal, Denisovan, archaic sapiens were available, the sequencing depths did not allow us to accurately predict their taste genes.
Methods
Identification of taste genes in humans
We collected the best-characterized families of human taste genes for sweet and umami, bitter, salty, sour, ENaC-independent salt, noncanonical tastes, fat, and complex carbohydrates from HGNC at https://www.genenames.org.
Gene sequences and homology search
Whole-genome protein sequences, CDS sequences in FASTA format, and gene positions for human, gorilla, macaque, mouse, chicken, lizard, frog, and zebrafish were retrieved Ensembl at https://uswest.ensembl.org/info/data/ftp/index.html.
We only selected the longest transcript was selected in the annotation for any genes that had more than 1 transcript. To search for nomology between humans with 2 other primates and 5 non-primate genomes, we conducted an all-vs-all BLASTP for each above genome against human and human against each genome. Respectively, we used an e-value cut-off of 1e − 10 and reported the best 5 non-self-hits in each target genome. We also performed BLASTP for the human genome against itself with the same setting but kept the best 6 self-hits.
We searched the Ensembl database for each human taste gene to identify human-mouse orthologous pairs and kept 1-1 ortholog. We treated Tas2r136 as the mouse ortholog for TAS2R19, TAS2R20, TAS2R30, and TAS2R50 because of its highest gene order conservation score. TAS2R5, TAS2R8, TAS2R9, TAS2R45, and SCNN1D had no mouse ortholog.
Detection of syntenic blocks and collinear genes
To identify syntenic blocks and collinear genes between multiple species genomes, we concatenated all above inter-/intra-species m6 BLASTP outputs into a .blast concatenated all gene positions of different species into a .gff file. Then, we analyzed the homologous genes by scanning syntenic blocks using the software MCScanX 53 with all default parameters among 8 genomes (a match score of 50, gap penalty of −1, E-value of 1e − 5, maximum gap size between any 2 consecutive protein pairs of 25, and at least 5 consecutive proteins to define a syntenic region). We also generated self-syntenic blocks within the human genome using MCScanX with the same setting.
Classification of duplicate gene origins
We uploaded the output of Human self-genome BLASTP to MCScanX. Then we used duplicate_gene_classifier from MCScanX to detect duplicate genes and classify them into different origins.
Ka and Ks calculation
We computed Ka and Ks using the Yang and Nielsen method, 54 available in the yn00 module of the PAML package. 55
Neutral mutation test
We collected all taste receptor genes’ variant calls of Europeans and Africans for Human (GRCh38.p13) with “Data Slicer” from 1000 Genomes Project at https://uswest.ensembl.org/Homo_sapiens/Tools/DataSlicer. Then Tajima’s D 42 was calculated at the population and gene levels using vcftools v0.1.13. 56
Supplemental Material
sj-xlsx-1-evb-10.1177_11769343211035141 – Supplemental material for Contrasting Patterns of Gene Duplication, Relocation, and Selection Among Human Taste Genes
Supplemental material, sj-xlsx-1-evb-10.1177_11769343211035141 for Contrasting Patterns of Gene Duplication, Relocation, and Selection Among Human Taste Genes by Yupeng Wang, Ying Sun and Paule Valery Joseph in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of Alcohol Abuse and Alcoholism under award number, Z01AA000135 and the Office of Workforce Diversity, National Institutes of Health Distinguished Scholar, and the Rockefeller University Heilbrunn Nurse Scholar Award to PVJ.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Y.W. and P.V.J conceived the study. Y.W. and Y.S. made the programs. Y.W. and Y.S. performed the analyses. Y.W. and P.V.J wrote the manuscript.
Data Availability
The data underlying this article are available in the article and its Supplemental Material.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.