
Abstract
WRKY transcription factors play important roles in plant development and responses to various stresses in plants. However, little is known about the evolution of the WRKY genes in the desert poplar species Populus euphratica, which is highly tolerant of salt stress. In this study, we identified 107 PeWRKY genes from the P. euphratica genome and examined their evolutionary relationships with the WRKY genes of the salt-sensitive congener Populus trichocarpa. Ten PeWRKY genes are specific to P. euphratica, and five of these showed altered expression under salt stress. Furthermore, we found that two pairs of orthologs between the two species showed evidence of positive evolution, with dN/dS ratios >1 (nonsynonymous/synonymous substitutions), and both of them altered their expression in response to salinity stress. These findings suggested that both the development of new genes and positive evolution in some orthologs of the WRKY gene family may have played an important role in the acquisition of high salt tolerance by P. euphratica.
Keywords
Introduction
Transcription factors (TFs) regulating gene expression via a modular structure containing at least one DNA-binding domain play pivotal roles in signaling and regulatory networks.1,2 The WRKY TF family in plants comprises numerous members that regulate genes involved in seed germination, seed dormancy, trichome development, lignin biosynthesis, and both biotic and abiotic stress responses.3–8 Since the first WRKY gene was cloned and characterized from sweet potato, numerous WRKY genes have been identified in other plants, including Arabidopsis, rice, cucumber, tomato, grape, and poplar.9–15 Proteins encoded by the WRKY gene family contain two prominent and defining structural characteristics, WRKY domains and zinc-finger motifs, and WRKY proteins are divided into three groups (I–III) on the basis of the numbers and types of these motifs that they contain.16–18 Group I contains two WRKY domains and CX4CX22–23HXH zinc-finger motifs, while the Group II members have the same type of finger motif as Group I, C2H2 (in this case C-X4–5-C-X22–23-H-X1-H) and contain one WRKY domain; the WRKY domain of the third group (III) comprises a C2HC (C-X7-C-X23-H-X1-C) motif. 10 Mutations in either the WRKYGQK motif or the zinc finger of a WRKY domain will result in the loss of DNA-binding ability. 16 WRKY proteins bind to the W-box ((C/T)TGAC(T/C)) elements of their target genes17,18 as well as to a cis-element (SURE). 19
Some WRKY genes have been reported to be activated in response to various abiotic stresses including high salinity.8,18 Salinity is a major abiotic stress, acting primarily by changing osmotic stress and disrupting cellular homeostasis and ion distribution. It also induces oxidative stress, damages membranes and proteins, and activates signaling cascades regulating gene expression. 20 The regulatory networks that underlie salt tolerance are very complex. 21 To date, a small number of WRKY genes have been identified as taking part in this process in plants. For instance, it has been reported that OsWRKY45 plays a part in rice salt tolerance, 22 while GhWRKY39 has a positive role in cotton salt tolerance. 23 Overexpression of a maize WRKY58 gene enhances salt tolerance in transgenic rice. 24 Similarly, in Arabidopsis, overexpression of either AtWRKY25 or AtWRKY33 increases salt tolerance. 25 A recent study also showed that some PeWRKY genes are involved in salt tolerance. 26 However, few studies have been designed in order to examine changes in the number of WRKY genes and adaptive evolution of orthologous genes between species that are closely related but show contrasting degrees of salt tolerances.
In this study, we aimed to examine these issues using Populus euphratica and Populus trichocarpa. The model tree species P. trichocarpa grows in mesophytic habitats, while P. euphratica grows in deserts; it is well adapted to salt stress and has a high level of salt tolerance.27–29 Genome sequences for both species are available30,31 and preliminary analyses suggested that the expression of some WRKY genes is altered in response to salinity stress. 30 The WRKY genes in the P. trichocarpa genome have been identified. 15 In order to better understand the adaptive evolution of the WRKY genes in these two poplars of contrasting salt tolerance, we identified the WRKY genes of P. euphratica and examined the expression under salt stress of both species-specific genes and those that are orthologous in the two species. Our results provide further information about the roles that the WRKY genes may have played in the adaptive history of the desert poplar.
Methods
Identification of WRKY genes in two poplar genomes
Candidate WRKY proteins in the P. euphratica genome 30 were identified using the BlastP program (P value = le-3). Sequences of Arabidopsis members of the WRKY protein family downloaded from the Arabidopsis genome website TAIR 9.0 (http://www.Arabidopsis.org/index.jsp) were used as query sequences. In addition, the Hidden Markov Model 32 profile for the WRKY domain (PF03106.7) from the Pfam database 33 was applied as a query against the P. euphratica genome using the program HMMER 34 in order to identify WRKY sequences by searching for the WRKY-binding domain. To verify the authenticity of these candidate sequences, the Pfam database was used to confirm that each candidate PeWRKY protein was a member of the WRKY family. The gene model IDs of P. trichocarpa WRKY genes were obtained from the Plant Transcription Factor Database v3.0 (http://planttfdb.cbi.pku.edu.cn/index.php) and confirmed using the same method as above. The PeWRKYs and PtWRKYs were named according to their position on the scaffolds and chromosomes of the respective genomes.
Mapping the PeWRKY genes onto the P. trichocarpa chromosomes
To determine the chromosomal locations of potential poplar WRKY genes, the WRKY orthologs in P. trichocarpa were used as references. Data on gene position, gene length, chromosome size, and position relative to the centromere of PtWRKYs were obtained from the Phytozome v9.1 website (http://www.phytozome.net/poplar). The MCScanX software package 35 was used to complete the chromosomal distribution of PEWRKY genes. The map of the distribution of PeWRKYs on 19 chromosomes was drawn, based on the above data, using custom-designed Perl scripts.
Phylogenetic analyses of WRKY genes
Alignments of WRKY protein and domain sequences were carried out separately using MUSCLE 36 and then adjusted manually before the construction of phylogenetic trees. Phylogenetic trees were constructed based on the aligned WRKY-binding domains and full predicted protein sequences of the PeWRKY genes using CLUSTAL 37 and MEGA 6, 38 respectively. The neighbor-joining (NJ) method was used with the following parameters: Poisson correction, pairwise deletion, and bootstrap (1,000 replicates). Exons and introns were predicted by comparing the coding sequences with the corresponding genomic sequences using custom-designed Perl scripts. The MEME program 39 was used to predict conserved motifs, with the following parameters: maximum number of motifs, 20; minimum motif width, 6; and maximum motif width, 70.
Divergence of orthologs between P. euphratica and P. trichocarpa
Identification of genes orthologous between P. euphratica and P. trichocarpa was performed using the bidirectional best hit method, as described in detail by Zhang et al.40,41 The MCScanX software toolkit and the phylogenetic tree were also used to provide further confirmation. Orthologous genes were aligned by the Probabilistic Alignment Kit software package. 42 To detect divergence of orthologs between P. euphratica and P. trichocarpa, a maximum likelihood method in PAML was applied to calculate nonsynonymous (Ka) and synonymous (Ks) substitution ratios using the YN00 program. 43 The elements in the promoter fragments (from -1500 to +1 bp) were predicted by PlantCARE online (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/).
Gene expression analysis
We collected Illumina RNA-Seq data using the methods described previously by Ma et al. 30 Calli induced from P. euphratica and P. trichocarpa shoots were collected from Tarim Basin desert in Xinjiang and the United States, respectively. RNA-Seq transcriptomic data from P. euphratica and P. trichocarpa calli under salt stress treatments (200 mM NaCl for 0, 6, 12, 24, and 48 hours) were generated using the Illumina HiSeqTM2000 platform. Three biological replicates were carried out. Gene expression levels were calculated as reads per kilobase of exon model per million mapped reads (RPKM). 44 RPKM values were log2-transformed and used for heat map generation using the pheatmap package in the R language.
Results and Discussion
Identification of the WRKY genes in P. euphratica
The entire P. euphratica genome was searched by BlastP analysis using 70 Arabidopsis WRKY genes as query sequences. A total of 107 full-length putative WRKY genes were identified. To confirm that these were WRKY genes, the Pfam database was used to search for WRKY domains in each; as a result, all 107 (that were named PeWRKY01 to PeWRKY107) were confirmed as WRKY genes (Table S1). The conserved WRKY domain, which contains approximately 60 amino acid residues, is considered to be a crucial element that usually interacts with the W-box (C/T)TGAC(T/C) to activate a large number of defense-related genes.15,18 Mutations in the WRKYGQK motif will result in the loss of DNA-binding ability. 16 Most WRKY genes contain one WRKY domain except 23 Group I genes, which contain two WRKY domains. Multiple sequence alignments of the 128 WRKY domains showed that 119 contained the conserved amino acid sequence WRKYGQK, while the others had one or two mismatched amino acids in this domain (Fig. S1): WRKYGHK (PeWRKY38), WRKYGRK (PeWRKY71_N), WAKYGQK (PeWRKY94), WRKDGQK (PeWRKY40_C), WKKYGQK (PeWRKY17), WRKYGKK (PeWRKY102, 12, 28), and WRK-(PeWRKY81).
Chromosomal distribution of PeWRKY genes on the reference poplar genome
PeWRKYs were widely distributed within the P. trichocarpa reference genome (Fig. 1). We found that 97 of the 107 PeWRKY genes could be mapped to 18 of the 19 P. trichocarpa chromosomes (Chr) according to the positions of orthologous PtWRKYs or linked genes (Table S2). Thirteen WRKY genes were found on Chr 1, 12 on Chr 2, and 11 on Chr 14. In contrast, Chrs 12, 15, and 19 each possess only two of the genes. We recovered 10 tandemly duplicated gene pairs that contained 20 PeWRKY genes on these chromosomes. We also found that 9 of these 10 duplicated gene pairs were shared with P. trichocarpa (Fig S2), suggesting that they existed before the divergence of the two species. We found one tandemly duplicated gene pair (PeWRKY06 and PeWRKY07) specific to P. euphratica and one pair (PtWRKY31 and PtWRKY32) specific to P. trichocarpa; these probably either formed after the divergence of the two poplars or represent ancestral sequences retained in one species but lost in the other.
Chromosomal distributions of PeWRKY genes in the Populus trichocarpa reference genome and chromosomes. The chromosome number is indicated at the top of each chromosome. Tandemly duplicated genes are indicated with vertical green lines. Gene position and the size of each chromosome can be estimated using the scale on the left of the figure.
Exon–intron organization and motif identification in PeWRKY genes
We next examined the exon–intron structure and motifs of all the PeWRKY genes identified (Fig. 2). Most PeWRKY genes have two to four introns: 56 out of 107 PeWRKYs possess two introns, 14 have three introns, and 15 have four introns. However, 8 PeWRKY genes have only one intron and 13 genes have five introns, while only 1 has six introns. Changes in exon–intron organization may have contributed to evolutionary divergence between the different members of the gene family.
45
Phylogenetic relationship, intron–exon structures, and conserved motif compositions in the PeWRKY family. The unrooted phylogenetic tree was aligned by MUSCLE and constructed using MEGA 6 by the maximum likelihood method with 1,000 bootstrap replicates. The tree was divided into seven phylogenetic subgroups, indicated with different colored backgrounds. The intron–exon structures of 107 PeW/RKY genes are shown in the middle panel. The right panel is a schematic representation of the conserved motifs in the PeWRKY proteins as detected by MEME analysis, represented by different colored boxes. Narrow rectangles representing the E values of motif are not significant as others. The length of each protein can be estimated using the scale at the bottom.
In addition, the online Multiple Expectation Maximization for Motif Elicitation program was used to find conserved motifs in the PeWRKY proteins (Table S4). As illustrated in Figure 2, other than motifs 1, 2, 3, and 5, which characterize the WRKY domains, PeWRKY members of the same subgroup were generally found to share a common motif composition. For instance, motif 17 is specific to 8 out of 12 genes in Group III while motif 10 and motif 15 are only present in Subgroup IId. Both Subgroup IIa and Subgroup IIb contain motif 7; Subgroup IIc could be divided into two sections, of which one contains motif 14, which is specific to Subgroup IIc and the other only contains motifs 1, 2, and 19, which comprise WRKY domains. Members of Subgroup IId mostly shared a conserved CaM-binding domain (motif 10), which is associated with calmodulin in Arabidopsis, 46 implying that these WRKY proteins may play pivotal roles in signal transduction by regulating the activity of target proteins. In general, the motifs within each group were relatively consistent and are probably associated with the similar functions carried out by members of that subgroup.
Phylogenetic analyses
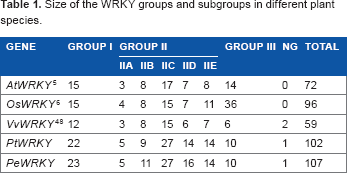
To examine the evolutionary relationships among the WRKY domains, phylogenetic relationships were constructed using the amino acid sequences of the conserved domains of AtWRKYs, PtWRKYs, and PeWRKYs (Fig. 3) and the protein sequences encoded by PeWRKYs (Fig. 2). Based on AtWRKY classification and PeWRKY sequences, all the PeWRKY genes identified could be divided into three tentative groups. The first group (Group I) comprised 23 members, most of which contained two WRKY domains. Group II, which had one WRKY domain could be divided into five subgroups (Subgroups IIa–e), which contained 5, 11, 27, 16, and 14 members, respectively. The third group contained the remaining 10 members, which contained the WRKY domains and C2HC zinc-finger structure (C-X4-C-X23-H-X1-C).
47
One gene (PeWRKY17), which lacked this structure, clustered weakly with the third group. The three groups recovered here were consistent with the previous classification of WRKYs in Arabidopsis
10
and grape.
48
The ortholog of PeWRKY17 in P. trichocarpa was found to have lost the characteristic structure of Group III WRKYs.
49
The small group of IIC genes in Figure 3 (orthologs of AtWRKY49) that separated from the larger group were mainly the result of domain sequences and also agreed with the result in grape.
48
In addition, two genes (PeWRK71 and PeWRKY98) of Group I were found to have lost the WRKYGQK motif at the C-terminal, according to the WRKY domains predicted by the Pfam database (Fig. S1). Because genes of Group I have been found to exist in all plants and in some organisms with ancient origins, they may be ancestral to the other two groups and subgroups.
47
Phylogenetic tree of WRKY domains of Arabidopsis, Populus trichocarpa, and Populus euphratica. Multiple sequence alignment of WRKY domains was done using MUSCLE, and the phylogenetic tree was constructed using CLUSTAL with 1,000 bootstrap replicates by the NJ method. Group I proteins with the suffix N or C indicate N-terminal WRKY domains or C-terminal WRKY domains, respectively. The tree was divided into eight phylogenetic subgroups, designated as IC, IN, I IIa–e, and III. Groups and subgroups are shown in different colors.
Through phylogenetic analysis, we found 10 PeWRKY genes specific to P. euphratica while P. trichocarpa has five species-specific genes. One P. euphratica–specific gene was formed by tandem duplication, while the other nine genes in this species, although located in different scaffolds, show high sequence similarity to the other PeWRKY genes and cluster together with them in the phylogenetic tree. The latter nine genes probably either originated by segmental duplication after the two species diverged or were retained in P. euphratica but lost in P. trichocarpa after genome duplication in the ancestor of the two species. Comparison of the number of WRKY genes in the different groups in other species with those in P. euphratica shows that among the PeWRKYs, duplication occurred mainly in Group I and Subgroups IIb and IId. We also found five unique genes (PtWRKYs) specific to P. trichocarpa. These results indicated that either the WRKY gene family expanded separately in the two species or some members were selectively retained in each poplar species after genome duplication in their common ancestor.
Evolutionary divergence of orthologs between P. euphratica and P. trichocarpa
To examine the divergence of orthologs between PeWRKYs and PtWRKYs, nonsynonymous substitutions (dN) and synonymous substitutions (dS) per site between orthologous genes were calculated: dN/dS = 1, <1, and >1 indicate, respectively, neutral selection, purifying selection, and accelerated evolution with positive selection. 50 A total of 97 pairs of orthologous genes were identified, and the dN/dS value of each was determined (Table S1). The results indicated that most of the gene pairs (95/97) showed dN/dS ratios <1, showing that purifying selection predominated (Table S3) as is the case for other gene families. However, we found that two pairs of genes (PeWRKY45 versus PtWRKY6 and PeWRKY102 versus PtWRKY52) had dN/dS ratios >1, suggesting that positive evolution was likely for these two genes.
Expression data showed that the positive evolved gene pairs showed different expression levels under salt stress in two poplar species (Fig. S3, Fig. 4). We examined the functions of the Arabidopsis orthologs of PeWRKY45 and PeWRKY102 and AtWRKY17 and AtWRKY50. Both of them negatively regulate jasmonic acid (JA) signaling.51,52 JA concentration have also been found to be associated with salt tolerance in P. euphratica.
53
We further found that the promoter regions of these pairs of genes varied slightly (Table S5–S6). Taken together, these results suggest that PeWRKY45 and PeWRKY102 probably mediate the response to salt stress by increasing the level of expression of JA-related genes, and this may shed light on how P. euphratica tolerates salt stress.
Heat map representation of expression of orthologous genes between PeWRKYs and PtWRKYs under salt stress. The Illumina RNA-Seq data were analyzed, and the RPKM values were log2-transformed and a heat map generated. The left and right panels represent RNA-Seq data from Populus euphratica and Populus trichocarpa, respectively. Blue and red colors are used to represent low and high levels of expression, respectively. Adaptive evolved genes were marked by red asterisk. Size of the WRKY groups and subgroups in different plant species.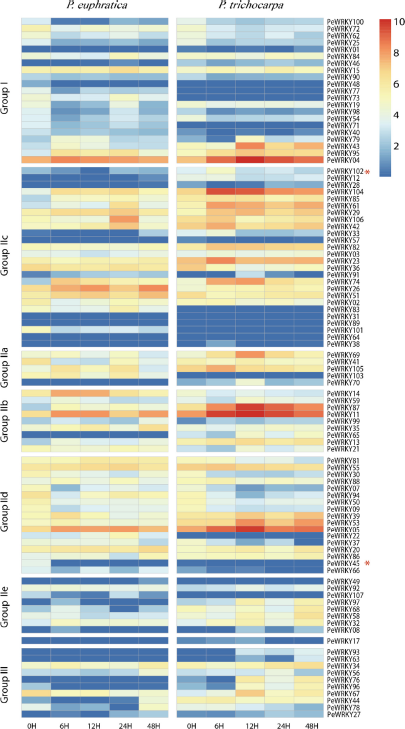
Expression profiling of PeWRKYs and PtWRKYs under salt stress
A total of 92/107 PeWRKY genes and 84/102 PtWRKY genes were detected as being expressed in calli with or without salt stress (Table S7). Of 92 PeWRKY genes, 23 showed changes in expression in response to salt stress: most (16/23) were downregulated while seven were upregulated. However, only five of the PtWRKY genes altered expression in response to salt stress. Further analysis showed that a total of 11 PeWRKY genes, among which are included two pairs of orthologs (PeWRKY45 versus PtWRKY6 and PeWRKY102 versus PtWRKY52) that show evidence of adaptive evolution (Fig. S3), underwent different changes in expressions in response to salt stress compared to their P. trichocarpa orthologs (Fig. 4). Of the 23 PeWRKY genes differentially expressed under salt stress, nine belong to Group I and five to Subgroup IId while the others are members of the remaining groups or subgroups. Members of Subgroup IId mostly share the conserved CaM-binding domain that is associated with calmodulin in Arabidopsis. 46 The upregulated genes include the one reported to mediate salt stress in P. euphratica. 26 These findings suggest that members of Group I and Subgroup IId may play more roles in response to salt stress than members of the other groups of genes.
It is believed that Group III of the WRKY gene family evolved more recently than the other two groups. 47 Many members of this group in Arabidopsis, including AtWRKY30, 53, 54, and 70, have been identified as regulators of senescence. 54 They are involved in a number of signaling pathways in response to different stresses. We calculated the dN/dS ratios for WRKY genes in the two Populus species; the average ratio in Group III was found to be 0.42 while the average in the other groups was 0.37, a finding consistent with the above assertion. In addition, no difference in expression was detected for Group III members when P. trichocarpa was subjected to salt stress. 15 However, in the present study we found that PeWRKY78, a member of this group, was downregulated, whereas its P. trichocarpa ortholog PtWRKY44 was slightly upregulated under salt stress.
Of the 10 PeWRKY genes unique to P. euphratica, three were downregulated and two were upregulated in response to salt stress (Fig. 5); this supports the notion that the origin or retention of these species-specific genes may have been driven by the need for high salt tolerance in this desert poplar. Overall, the results suggest that WRKY genes play an important role in salt stress tolerance in P. euphratica.
Expression levels of 10 PeWRKY genes specific to Populus euphratica under salt stress. The y-axis shows the log2-transformed RPKM values of genes expressed in callus after different periods of salt stress, and the x-axis shows the duplicated WRKY genes.
Conclusion
A total of 107 WRKY genes were identified in P. euphratica with 10 of them being species-specific genes that had originated through tandem and segmental duplication. When members of the WRKY family were compared between P. euphratica and P. trichocarpa, two pairs of genes exhibited evidence of positive selection (dN/dS > 1); they are probably significant in the adaptation of this poplar species to salt stress. Expression profiling of the PeWRKY genes revealed that five genes specific to P. euphatica and two that showed evidence of adaptive evolution showed alterations in expression in response to salt stress. These findings suggested that the formation of new genes and the occurrence of adaptive evolution in other genes may have played an important role in the evolution of high salt tolerance in P. euphratica.
Author Contributions
Conceived and designed the experiments: JLiu. Analyzed the data: JM. Wrote the first draft of the manuscript: JM. Contributed to the writing of the manuscript: JLu, JX, XH. Agree with manuscript results and conclusions: JLiu, JM. Jointly developed the structure and arguments for the paper: JLiu, XH. Made critical revisions and approved final version: JM, XH, BD. All authors reviewed and approved of the final manuscript.
Supplementary Material
Supplementary Table S1
Identification of WRKY TF genes in Populus euphratica.
Supplementary Table S2
WRKY TF genes in Populus trichocarpa.
Supplementary Table S3
Divergent selection between PeWRKY and PtWRKY genes.
Supplementary Table S4
The details of the motif sequences.
Supplementary Tables S5–S6
Cis-acting elements analysis of positive evolved genes.
Supplementary Table S7
RNA-seq data of PeWRKYs and PtWRKYs in calli.
Supplementary Figure S1
Multiple sequence alignment of WRKY domains using DNAMAN.
Supplementary Figure S2
Chromosome distribution of PeWRKYs and PtWRKYs.
Supplementary Figure S3
Expression levels of positive evolutional orthologous genes.