
Abstract
The dysbiosis of the gut microbiome associated with ulcerative colitis (UC) has been extensively studied in recent years. However, the question of whether UC influences the spatial heterogeneity of the human gut mucosal microbiome has not been addressed. Spatial heterogeneity (specifically, the inter-individual heterogeneity in microbial species abundances) is one of the most important characterizations at both population and community scales, and can be assessed and interpreted by Taylor’s power law (TPL) and its community-scale extensions (TPLEs). Due to the high mobility of microbes, it is difficult to investigate their spatial heterogeneity explicitly; however, TPLE offers an effective approach to implicitly analyze the microbial communities. Here, we investigated the influence of UC on the spatial heterogeneity of the gut microbiome with intestinal mucosal microbiome samples collected from 28 UC patients and healthy controls. Specifically, we applied Type-I TPLE for measuring community spatial heterogeneity and Type-III TPLE for measuring mixed-species population heterogeneity to evaluate the heterogeneity changes of the mucosal microbiome induced by UC at both the community and species scales. We further used permutation test to determine the possible differences between UC patients and healthy controls in heterogeneity scaling parameters. Results showed that UC did not significantly influence gut mucosal microbiome heterogeneity at either the community or mixed-species levels. These findings demonstrated significant resilience of the human gut microbiome and confirmed a prediction of TPLE: that the inter-subject heterogeneity scaling parameter of the gut microbiome is an intrinsic property to humans, invariant with UC disease.
Keywords
Introduction
Inflammatory bowel disease (IBD) is a multifactorial disease with probable genetic heterogeneity and is primarily presented as ulcerative colitis (UC) or Crohn’s disease (CD). While CD can affect any part of the digestive system, UC only affects the colon (large intestine)1,2 and thus carries an increased risk of colorectal cancer. Ulcerative colitis involves an imbalance in microbiome-host interactions, with no effective cure currently available. Although the pathogenesis of UC remains unclear, a combination of factors is thought to play a part, including genetics and environmental risk factors (eg, diet, smoking, measles, and appendectomy).3,4
One hypothesis for the etiology of UC is that altered or pathogenic microbiota cause inflammation in genetically susceptible individuals. 5 In the wake of recent advances in high-throughput sequencing, intestinal dysbiosis of microbiota is considered a key factor leading to UC and its complications.6-9 For example, recent studies on fecal and gut mucosa–associated microbiota in UC patients revealed an abnormal microbial composition characterized by low species diversity in gut microbial communities, but high-density mucosal surface colonization and epithelial invasion in areas with active disease. 5 The diversity of mucosa-associated microbiota is also markedly reduced in patients with active CD undergoing surgery. 10 In addition, the abundances of several bacterial species of Firmicutes are lower in patients with CD, 11 with the presence of aggressive bacteria increased in those with UC. 12 Consistently, an imbalance in microbiota composition (dysbiosis) can lead to excessive inflammatory responses and the occurrence and/or progression of UC, irritable bowel syndrome, and functional dyspepsia in humans.6,13 The fecal microbiota of UC patients is dominated by unusual bacterial species. 14 Moreover, UC fecal microbiota exhibits specific discrepancies from the microbiota of healthy subjects. 15 Fecal microbiota transplantation (FMT) is a primary therapeutic method that utilizes target microbiota and could become a potential rescue (or even standardized) therapy for UC. 16
As the detailed pathogenesis of UC remains uncertain, 17 it is difficult to identify how intestinal dysbiosis contributes to UC. In recent years, developments in gene-sequencing technologies, as well as increased availability of powerful bioinformatics tools, have enabled novel insights into the microbial composition of the human gut microbiota and determination of factors important in UC disease progression. For example, to explain longitudinal variations in the intestine, 18 quantitatively assessed the spatial distribution patterns of mucosa-associated microbial flora along the intestinal tract in healthy human individuals.
The objective of this study was to comparatively investigate the spatial heterogeneity of the gut microbiome of UC patients and healthy controls. The spatial heterogeneity can be used to mean the inter-subject variability among individuals in a cohort (or population). Taylor’s power law (TPL) is a classic ecological model for assessing heterogeneity at the population scale19-22 extended TPL from the population to the community level and introduced 4 types of power law extensions (Taylor’s power law extensions [TPLEs]), ie, Type-I to Type-IV TPLEs.22-24 In this study, we aimed to answer the question of whether UC influences the spatial heterogeneity of human gut mucosal microbiome from both community and mixed-species levels by using Ma’s Type-I and Type-III TPLEs, respectively. The results of this study will hopefully generate important insights into the influence of UC on the gut mucosal microbiome. A significant difference between healthy and diseased individuals in spatial heterogeneity parameters (of TPLEs) would indicate the redistribution (or rebalance) of microbes at the community (Type-I TPLE) or species (Type-III TPLE) level. Direct experimental monitoring of the redistribution (rebalance) of microbes in the human gut microbiome is not feasible, but the heterogeneity scaling (change) parameter of the TPLEs offers a powerful theoretic approach for estimating the level of microbial movement, which leads to the redistribution (rebalance) of gut microbes. Although not as important as monitoring the movement of cancer cells, assessment of the redistribution of gut microbes has significance, particularly redistribution induced by diseases such as UC.
Materials and Methods
Subject selection and sampling
In total, 56 volunteers (28 couples) were recruited, including 28 patients with UC and their healthy control partners (see Table 1 for details). All participants were from Kunming, China, and were aged from 18 to 60 years old. The health status of the volunteers was self-reported and confirmed by endoscopy. No healthy volunteers suffered any diseases of the gastrointestinal tract and none were taking medications at the time of their endoscopy nor used antibiotics during the year prior to specimen collection. All participants received information about UC, pathogenicity, risk factors, and the importance of the study in their local language. Verbal and written informed consent was obtained from each participant. This study was approved by the Medical Ethics Board of the First Affiliated Hospital of Kunming Medical University, Yunnan Province, China.
Summary information on datasets.

Abbreviations: OTUs, operational taxonomic units; UC, ulcerative colitis.
Microbial samples from the intestinal mucosa of the 56 participants were collected. Intestinal mucosa sampling was undertaken in the morning before the colonoscopy procedure, without bowel cleansing preparation. All samples were immediately frozen in liquid nitrogen and stored at –80°C until transportation on dry ice to Beijing Genomics Institute (BGI), Inc. (China) for sequencing analysis and storage. DNA was extracted from all samples, and the V3-V4 region of the 16S ribosomal RNA gene was amplified, sequenced, and analyzed.
TPL extensions
In 1961, Taylor 19 proposed a widely applied ecological law in population ecology, named Taylor’s power law, to describe the scaling relationship between population mean abundance (m) at a specific site and its corresponding variance (V), which offers an ideal mathematical tool to measure spatial aggregation (heterogeneity). Furthermore, the power law scaling parameter (b) is a species-specific parameter that allows rich ecological and evolutionary insights about species abundance and spatiotemporal distribution across different environments. In contrast, the a parameter is largely influenced by the sampling scheme and is of relatively little biological significance.
In the present study, we used TPL analysis to assess and interpret the “aggregation degree” (heterogeneity level) of intestinal microbial species and communities in UC patients and healthy individuals. The TPL formula is as follows

Since its discovery more than half a century ago, TPL has been applied in numerous field tests and theoretical analyses, especially in macroecological studies of plants and animals.19,20,25 A resurgence in theoretical investigation and extension to wider applications, well beyond ecology, has occurred in recent years. 22 extended the original TPL to the community level by introducing 4 power law extensions (PLEs), with Type-I TPLE proposed to assess community spatial heterogeneity.
Type-I TPLE uses the same mathematical formula as the original TPL, but its parameters are conveyed with different ecological interpretations, ie

where mc is the mean population size (abundance) of all species from the cth community site (sample), Vc is the corresponding variance, and C is the number of communities sampled. Parameter a is primarily influenced by sampling schemes, such as sequencing platform, and contains little biological meaning. Parameter b measures community spatial heterogeneity, with a larger b value indicating higher heterogeneity—a large b value means faster scaling (change) of variance with mean abundance.
Similarly, the Type-III TPLE of the classic TPL can assess spatial heterogeneity of mixed-species populations. It also possesses the same mathematical form as the original TPL, but its parameters are conveyed with different interpretations. Type-III TPLE is defined with the following power function

where ms is the mean population abundance of species s across all communities, V is the corresponding variance, and S is the number of species in the meta-community (consisting of C communities). Parameter b measures the spatial heterogeneity of mixed-species population, with parameter a being of little biological significance.
To fit the PLEs, equations (2) and (3) can be transformed into linear functions on the log scale, ie

This linear function can be easily fitted with simple linear regression analysis. Either a P value or linear correlation coefficient (R) can be used to determine the goodness of fit.
We used the permutation test to test the differences in Type-I and Type-III parameters between UC patients and healthy controls. The null hypothesis (H0) of permutation test is that the difference of values of TPLE parameters (ie, b and ln(a)) between UC patients and healthy controls is no less than that between 2 random groups; in other words, the difference of parameters is affected on random effect alone. We will take parameter b as an example to introduce the specific steps of the permutation test as follows:
(i) First, compute the absolute difference of b between UC and healthy (H) treatments,
(ii) Pool together all samples from the UC and healthy treatments, and evenly divide them randomly into 2 groups. Fit the TPLE models to these 2 new groups, respectively, and record their b values as bp1 and bp2. Compute absolute difference between bp1 and bp2, and record it as Δ pb

In this step, we generate a permutation.
(iii) Repeat step (ii) for 1000 times, generating 1000 TPLE models and corresponding 1000 Δ pb .
(iv) If the number of permutations with Δ pb > Δ b is D, the P value for permutation test can be defined as

If P ⩽ .05, we have strong evidence to reject the null hypothesis and receive the alternative hypothesis, ie, the parameter (b) is different between UC patients and healthy controls.
Results and Discussion
We recruited 28 couples for this study, divided into 2 groups (UC patients and healthy controls; see Table 1 for characteristics). A mucosal sample was collected from each subject. Details on sequencing and operational taxonomic units (OTUs) for each group generated from bioinformatics analysis are also provided in Table 1. We built Type-I and Type-III TPLEs for the mucosal microbiomes of the healthy and UC subjects, including b values, ln(a), their standard errors, P values, correlation coefficients (R), and sample/OTUs (n) (see Table 2 and Figure 1). The P value was used to determine whether the model was fitted successfully, and R was used to validate the goodness of fit. As shown in Table 2, all 4 TPLE models were successfully fitted to the mucosal microbiome data (P < .05), in which the 2 Type-III TPLE models performed well (R > 0.98). For the Type-I TPLE, the b values for the healthy and UC groups were 9.212 and 6.504, respectively, suggesting that the mucosal microbiome of healthy individuals had higher community spatial aggregation/heterogeneity than that of UC patients. For the Type-III TPLE, the b values for the healthy and UC groups were 1.728 and 1.784, respectively, indicating that the variations in mixed-species population spatial aggregations/heterogeneities of the healthy and UC groups were very similar. Table 3 shows the permutation test results for differences in the parameters of the 4 models. For the Type-I and Type-III TPLE models, there were no significant differences in b values between healthy individuals and UC patients.
Parameters of PLE-I for community spatial heterogeneity and PLE-III for mixed-species population spatial aggregation.

Abbreviations: PLE-I, Type-I power law extension; PLE-III, Type-III power law extension; UC, ulcerative colitis.

Graphs fitting Type-I PLE and Type-III PLE with mucosa microbial samples of healthy and UC groups. (A) Type-I PLE for community spatial heterogeneity and (B) Type-III PLE for mixed species. IBD indicates inflammatory bowel disease; PLE, power law extension; UC, ulcerative colitis.
Permutation test for differences in parameters of PLE-I for community spatial heterogeneity and PLE-III for mixed-species population spatial aggregation.
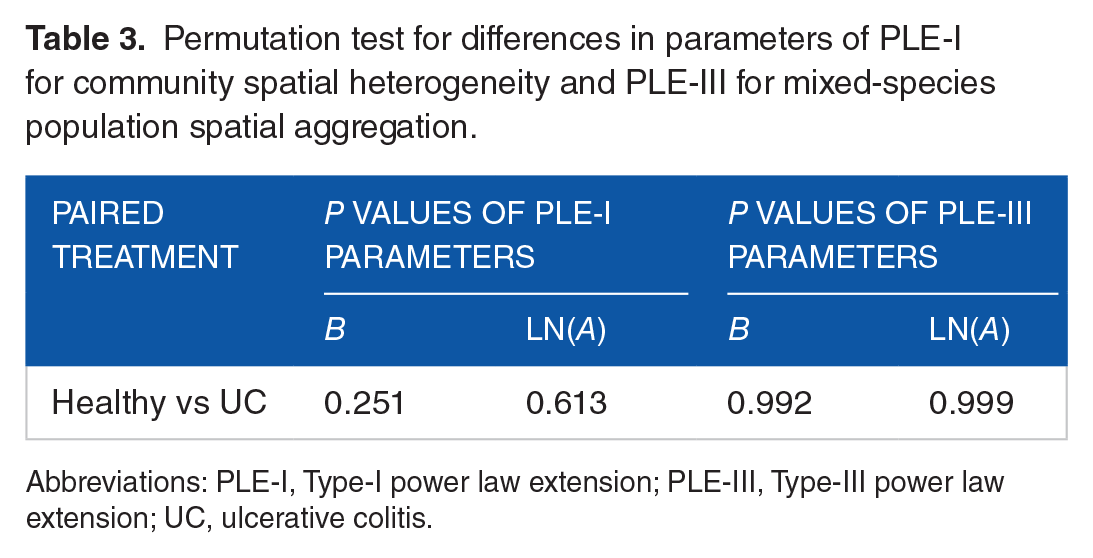
Abbreviations: PLE-I, Type-I power law extension; PLE-III, Type-III power law extension; UC, ulcerative colitis.
Previous studies have demonstrated that UC can significantly change the ecological characteristics of gut microbiota; however, few studies have focused on heterogeneity. 19 Power law model provides a useful tool to measure heterogeneity and TPLEs make it possible to assess community-level and mixed-species-level heterogeneity of microbiota. 22 For example, Oh et al 26 applied TPLE to human skin microbiome research. Community-level heterogeneity can indicate interspecies abundance variations exhibited at the community scale, whereas mixed-species-level heterogeneity can indicate population abundance variations among spatial sites in terms of mixed species at the population level. 7 In our previous study, 27 we found that IBD may influence the community spatial heterogeneity of the gut microbiota but not the spatial heterogeneity of mixed populations (ie, IBD may influence community-level spatial heterogeneity but not mixed-species-level heterogeneity). However, we did not adopt a robust statistical test to prove this finding in that study.
In the current study, we designed a more sophisticated experiment to investigate UC-related changes in the heterogeneity of the gut microbiota. We recruited couples consisting of 1 UC patient and 1 healthy individual. We studied couples who live together and share similar living and eating habits and environments, which should help eliminate the effects of external environmental factors to a large extent. In addition, a permutation test was adopted for pairwise statistical testing, with a P value of less than .05, indicating a significant difference.
Dysbiosis of the gut mucosal microbiome has been reported in UC patients previously, including a decrease in species of the phyla Firmicutes and Bacteroidetes, an increase in the abundance of facultative anaerobes, and a reduction in bacteria producing short-chain fatty acids.28-31 However, based on the permutation test, we found that UC did not influence community-level or mixed-species-level heterogeneity of the gut mucosal microbiome. This indicates that, although changes in species composition were found in UC patients, UC did not influence the aggregations/heterogeneities of the gut mucosal microbiome. This may be because the effects of species loss can be compensated for by the acquisition of other species. These results also emphasize the high resilience (or low variability) of the gut mucosal microbiome against UC.
It is worth noting that, at the community level, spatial scaling of the heterogeneity quantifies the degree of inter-individual variations of gut mucosal microbiome across subjects. In effect, the community spatial heterogeneity can be considered as a proxy of diversity. Our results revealed that the scaling parameter (b) was not affected by UC, which also further verified that parameter b is intrinsic to the human gut microbiome.22,25
Our quest for a specific pathogen in UC has been hindered by a lack of understanding of the host conditions required for pathogenesis. However, in the wake of recent advances in high-throughput sequencing, it is becoming increasingly evident that intestinal microbes are essential for the development of UC. Our study adopted a pairwise comparison with robust statistical testing to confirm that UC did not influence either community-level or mixed-species-level microbiome heterogeneity. Although this research is novel, as few studies have examined UC-related changes in gut microbiota heterogeneity, there are some study limitations. We only provided an overall (or general) view of the ecological changes in the gut microbiota caused by UC and did not identify precisely which species were significantly different in the gut microbiota of UC patients and healthy individuals. In future work, we will identify and investigate the significantly different species found in paired UC patients and healthy individuals.
Footnotes
Acknowledgements
The thank Prof. Zhanshan (Sam) Ma for designing the study and revising the manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (U1802282, 81660100, 81670501) to Yinglei Miao and Open Grants (#GREKF19-07) from State Key Laboratory of Genetic Resources and Evolution.
Declaration of conflicting interests:
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
YS, JL, and LW collected the human specimens and data; LL, HC, WX, and LD performed data analysis and interpretation; AL, KW, and JN participated in the discussion and interpretation of the results; LL and YS drafted the manuscript; and YM and LD designed the study and revised the manuscript. All authors approved the submission.
Data Availability
Sequence data are available from Sequence Read Archive (SRA) BioProject PRJNA600852.