
Abstract
Recent advances in genetics present unique opportunities for enhancing our understanding of human physiology and disease predisposition through detailed analysis of gene structure, expression, and population variation via examination of data in publicly accessible genome and gene expression repositories. Yet, the vast majority of human genes remain understudied. Here, we show the scope of these genomic and genetic resources by evaluating ZMAT2, a member of a 5-gene family that through May 2020 had been the focus of only 4 peer-reviewed scientific publications. Using analysis of information extracted from public databases, we show that human ZMAT2 is a 6-exon gene and find that it exhibits minimal genetic variation in human populations and in disease states, including cancer. We further demonstrate that the gene and its encoded protein are highly conserved among nonhuman primates and define a cohort of ZMAT2 pseudogenes in the marmoset genome. Collectively, our investigations illustrate how complementary use of genomic, gene expression, and population genetic resources can lead to new insights about human and mammalian biology and evolution, and when coupled with data supporting key roles for ZMAT2 in keratinocyte differentiation and pre-RNA splicing argue that this gene is worthy of further study.
Introduction
The availability of large-scale genomic and gene expression databases 1 makes feasible the study of nearly any human gene, including the ability to fully characterize both gene structure and its chromatin environment, to analyze gene expression patterns at the organ, tissue, developmental stage, and even single-cell levels2-4 and to evaluate genetic variation in populations and in association with different traits and diseases.5-7 Despite these opportunities, 8 the vast majority of human genes remain understudied.9,10 Multiple reasons have been proposed to account for the disparity between a relatively small number of highly analyzed human genes and the remainder, differences that are reflected in the number of publications and in the extent of grant funding.9,10 Some of these discrepancies may be a consequence of the availability of model organisms or of the presence or absence of links to human diseases,9,10 although it has been argued some reasons may be historical or social in origin.9,10
Here, we focus on a gene that has been minimally studied. The gene, ZMAT2, is part of a 5-member family in humans, in which all the encoded proteins contain zinc finger domains, but are otherwise dissimilar to one another. According to a single publication focusing primarily on the functions of human ZMAT2, the protein appears to negatively regulate epidermal cell differentiation. 11 In another context, the yeast ortholog of ZMAT2, termed Snu23, is a component of the spliceosome, 12 the molecular machine responsible for the removal of introns from primary gene transcripts. 13 Human ZMAT2 also has been mapped to the spliceosome. 14 Moreover, it has been postulated based on structural data that Snu23/ZMAT2 may act to facilitate the repositioning of the U6 small ribonucleoprotein (snRNP) at the 5′ splice site during human spliceosome activation. 14
We now use analysis of data obtained from public genomic and gene expression databases to define the organization of the human ZMAT2 gene. We further show that ZMAT2 exhibits very minimal genetic variation in human populations and in disease states, and find that the gene and its encoded protein are highly conserved among primates. Collectively, our studies illustrate how the complementary use of genomic and gene expression resources can lead to new insights about human and mammalian biology and evolution, and in conjunction with data on the human ZMAT2 protein in epidermal cell differentiation, and possibly in spliceosome function, suggest that this gene is worthy of additional investigation.
Materials and Methods
Please see Table 1 for a summary of all publicly accessible data resources used in this article.
Data resources and repositories used in the article.
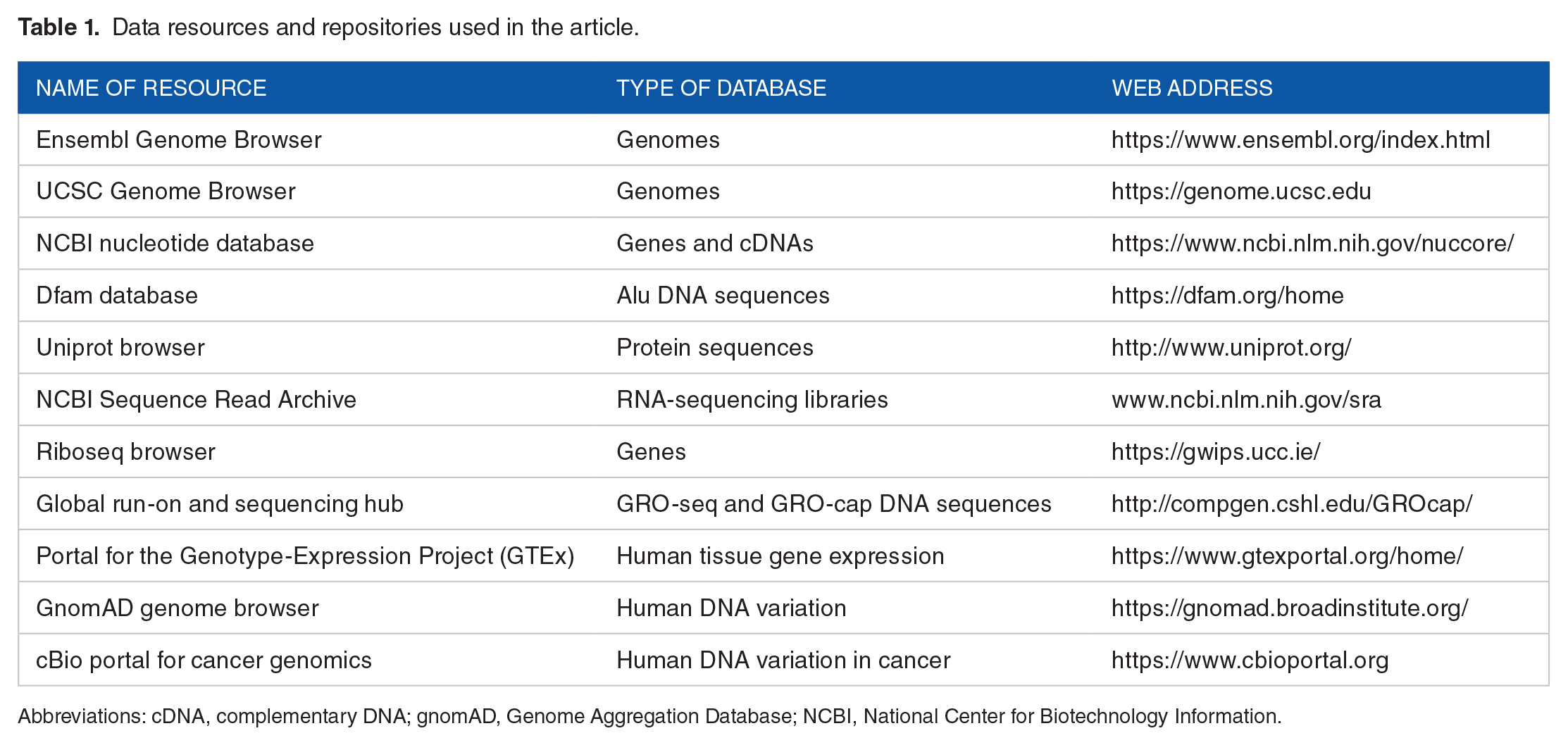
Abbreviations: cDNA, complementary DNA; gnomAD, Genome Aggregation Database; NCBI, National Center for Biotechnology Information.
Database searches and analyses
Primate genomic databases were accessed in the Ensembl Genome Browser (https://useast.ensembl.org/index.html) and the UCSC Genome Browser (https://genome.ucsc.edu). Searches were performed with BlastN under normal sensitivity (maximum e-value of 10; mismatch scores = 1, −3; gap penalties: opening = 5, extension = 2; filtered low-complexity regions and masked repeat sequences) using human ZMAT2 DNA segments as queries (Homo sapiens genome assembly GRCh38.p13). The following genome assemblies were examined: bonobo (Pan paniscus, Bonobo panpan1.1), chimpanzee (Pan troglodytes, Pan_tro_3.0), gorilla (Gorilla gorilla, gorGor4), macaque (Macaca mulatta, Mmul_8.0.1), marmoset (Callithrix jacchus, ASM275486v1), mouse lemur (Microcebus murinus, Mmur_3.0), olive baboon (Papio anubis, Panu_3.0), and orangutan (Pongo abelii, PPYG2). The highest scoring results in all cases mapped to the ZMAT2 gene, or in marmoset to both ZMAT2 and ZMAT2 pseudogenes. Additional searches were conducted using ZMAT2 complementary DNA (cDNA) sequences as queries to follow up, verify, or extend initial results. The following primate ZMAT2 cDNAs were obtained from the National Center for Biotechnology Information (NCBI) nucleotide database: gorilla (accession number: XM_004042656), human (NM_144723, BC056668), mouse lemur (XM_012748951), and olive baboon (XM_031666488.1). The Dfam database (https://dfam.org/home; release 3.0 from February 2019) was used to identify Alu sequences, and the Uniprot browser (http://www.uniprot.org/) was the source for ZMAT2 protein sequences. When primary protein data were unavailable, DNA sequences from ZMAT2 exons were translated using Serial Cloner 2.6 (see http://serialbasics.free.fr/Serial_Cloner.html).
Mapping 5′ and 3′ ends of human ZMAT2
Inspection of human ZMAT2 and its proposed messenger RNAs (mRNAs) in the Ensembl genome database revealed lack of both an identified termination codon and a 3′ untranslated region (UTR) for the mRNA encoding 1 of the 2 proteins, along with poorly defined 5′ exons for each of the 2 proposed protein-coding transcripts (Figure 2). Because the 2 human ZMAT2 cDNAs did not encode additional DNA, an alternative strategy was used to map these regions of the gene.15,16 RNA-sequencing libraries found in the NCBI Sequence Read Archive (SRA) (www.ncbi.nlm.nih.gov/sra) were queried with adjacent 60 bp probes from genomic DNA corresponding to presumptive 5′ exons 1 and 1a, and from 3′ exons 5 and 6, and read counts were analyzed. These results were then assessed in conjunction with information obtained through the Riboseq browser (https://gwips.ucc.ie/), which provided an overview of the 5′ region of human ZMAT2 exon 1. 17 This segment of human ZMAT2 exon 1 was also examined with data from the global run-on and sequencing (GRO-seq18,19) and 5′-GRO-seq (termed GRO-cap) hub (http://compgen.cshl.edu/GROcap/) and was applied to the 5′ end of human ZMAT2 exon 1 and exon 1a within the UCSC Genome Browser.
Protein alignments and phylogenetic trees
Multiple sequence alignments were performed for ZMAT2 proteins from different species. Amino acid sequences were uploaded into the command line of Clustalw2 (https://www.ebi.ac.uk/Tools/msa/clustalw2/) in FASTA format. This program performs pairwise sequence alignments using a progressive alignment approach and then creates a guide tree using a neighbor-joining algorithm, which is used to complete a multiple sequence alignment. Output files were in GCG MSF (Genetics Computer Group multiple sequence file) format and were used with an .aln extension as input into a command line form of IQ-TREE (http://iqtree.cibiv.univie.ac.at/), which uses maximum likelihood to generate a phylogenetic tree. 20 The output file (with a .filetree extension) became the input file into iterative Tree of Life (iTOL), an online tool for generating pictorial phylogenetic trees (https://itol.embl.de/).
Analysis of ZMAT2 gene expression and potential variation
Gene expression analyses were performed by querying the individual RNA-sequencing libraries from the NCBI SRA listed in Additional Table 1 in Supplemental Material. Searches were performed with 60-nucleotide DNA segments comprising parts of different exons (see Additional Table 2 in Supplemental Material). All queries used the Megablast option (optimized for highly similar sequences; maximum target sequences = 10 000 [this parameter may be set from 50 to 20 000]; expect threshold = 10; word size = 11; match/mismatch scores = 2, −3; gap costs: existence = 5, extension = 2; filtered low-complexity regions). Data on human ZMAT2 gene expression were also extracted from the Portal for the Genotype-Expression Project (GTEx v7; https://www.gtexportal.org/home/) using the exon expression module and analyzing variable transcripts, based on the presence of either exon 1a or exon 1. Information on variation in human ZMAT2 was from the Genome Aggregation Database (gnomAD) genome browser (https://gnomad.broadinstitute.org/), which contains results of sequencing of the exons or whole genomes from 141 456 individuals. 21 Data regarding potential ZMAT2 variants in cancer were obtained from the cBio portal for cancer genomics (https://www.cbioportal.org).
Results
ZMAT2 and the human ZMAT gene family are understudied
A recent publication noted that only approximately 10% of human genes had been evaluated in detail. 10 Using the data in that study as a guide, we identified ZMAT2 as among the 4 least-studied human genes (the others are ITFG1, SLC24A3, and DENND5B; see S8 Table in Stoeger et al 10 ). The other 4 members of the human ZMAT gene family are also understudied, and there are very few publications citing them in the scientific literature, with the exception being ZMAT3 (also known as WIG-1, which is a gene regulated by the p53 transcription factor22,23), in which 41 different citations were found in PubMed as of May 2020. The individual ZMAT family genes are located on 5 different human chromosomes, as determined by examining H sapiens genome assembly GRCh38.p13 (Figure 1A). The proteins encoded by these genes range in length from 148 to 638 amino acids. According to information in the Ensembl genome database, ZMAT3 is predicted to produce 4 protein isoforms of 148, 288, 289, and 383 amino acids and ZMAT4 3 protein species of 153, 211, and 229 residues as a result of translation of distinct alternatively spliced mRNAs (Figure 1A). The ZMAT family proteins are dissimilar except for their zinc finger domains (Figure 1C), and even these latter regions are quite variable in terms of amino acid sequence identity or in the number per ZMAT protein, which ranges from 1 to 4 (Figure 1A to C).
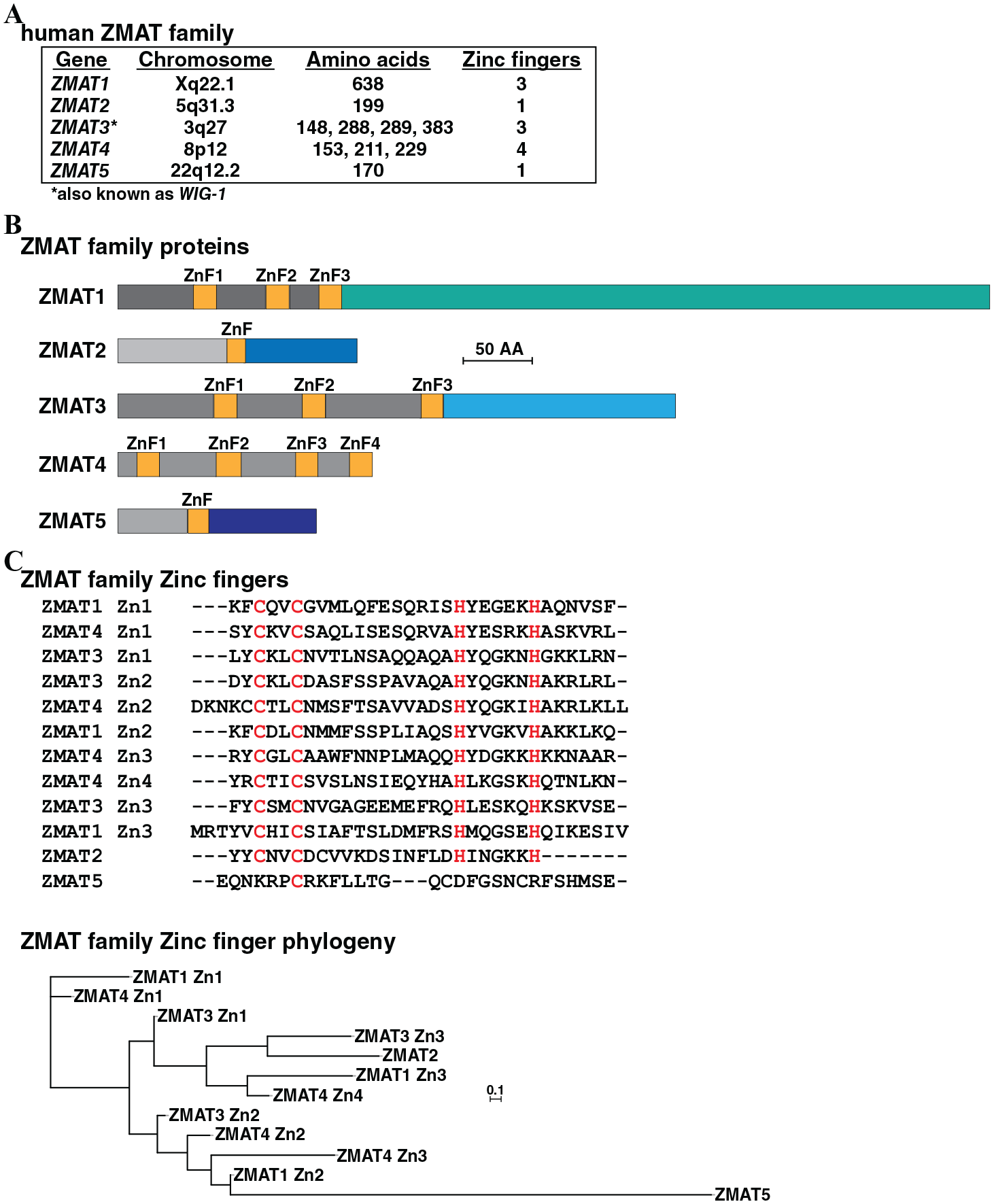
The human ZMAT family. (A) Information on human ZMAT genes 1 through 5, including chromosomal location, the number of amino acids encoded by the respective messenger RNAs, and the number of zinc finger (ZnF) domains per protein. (B) Schematic of human ZMAT proteins, with ZnF regions labeled and colored yellow. Nonsimilar regions are in different colors. Only the longest protein is shown for ZMAT3 and ZMAT4. (C) Upper: alignment of amino acid sequences of 12 human ZMAT ZnF domains, as modeled from the phylogenic tree below. Amino acids that are identical in at least 11 of 12 ZnFs are in red. Zn1 to Zn4 depict the number of ZnF in the specific ZMAT protein, as depicted in (B). Dashes indicating no residue have been placed to maximize alignments. Lower: phylogenetic tree of human ZMAT ZnF domains. The scale bar indicates 0.1 substitutions per site, and the length of each branch approximates the evolutionary distance.
Defining the human ZMAT2 gene
According to Ensembl, human ZMAT2 is a 7-exon gene on chromosome 5q31.3, where it resides adjacent to and overlapping with HARS2 in the same transcriptional orientation. The 3 proposed ZMAT2 transcripts in Ensembl are stated to encode proteins of 199 or 53 amino acids (Figure 2A and B), along with a third mRNA that is predicted to undergo nonsense-mediated decay. Of note, inspection of the gene reveals that the shorter coding transcript lacks a stop codon and a 3′ UTR, and thus must not be fully characterized. In addition, each of the 2 proposed protein-coding transcripts have poorly defined 5′ exons (Figure 2B). In contrast, in the UCSC Genome Browser, a single major ZMAT2 transcript is listed that resembles the Ensembl mRNA containing exons 1 to 6 (Figure 2B). Moreover, there are no published data available about either identification of a ZMAT2 gene promoter or promoters, or regulation of gene expression.

Human ZMAT2 gene in the Ensembl genome database. (A) Map of the human HARS-HARS2-ZMAT2 locus on chromosome 5, as presented in Ensembl. Boxes depict exons (red for HARS, blue for HARS2, black for ZMAT2), with coding regions being solid and noncoding regions white. The direction of transcription of each gene is indicated and a scale bar is shown. (B) Human ZMAT2 protein-coding messenger RNAs (mRNAs) as found in Ensembl. Coding segments are in black and noncoding regions in white (note the absence of a translational stop codon for the smaller mRNA, which lacks additional DNA information in Ensembl). (C) Diagram of human ZMAT2 exon 1, and gene expression data from the National Center for Biotechnology Information Sequence Read Archive RNA-sequencing library, SRX5281080 (Additional Table 1 in Supplemental Material), using as probes 60 bp genomic segments a to d (each letter marks the center of each probe). A scale bar is shown. The DNA sequence below the graph depicts putative 5′ end for exon 1, with location of the 5′ end of the longest RNA-sequencing clone indicated by a vertical arrow. (D) Diagram of human ZMAT2 exons 5 and 6. Illustrated below map are locations of 60 bp DNA probes that were used to screen RNA-sequencing library, SRX4654287, and a graph of the number of full-length transcripts that matched each probe. A scale bar is shown. (E) Diagram of human ZMAT2 exon 6, along with gene expression data from SRX4654287, using as probes 60 bp genomic segments a to f (each letter marks the center of each probe). A scale bar is shown. Also depicted below the map is the DNA sequence of the putative 3′ end of exon 6. A potential polyadenylation signal is underlined, and a vertical arrow denotes the possible 3′ end of ZMAT2 transcripts.
We thus performed a series of investigations to better characterize human ZMAT2. As the 2 human ZMAT2 cDNAs in the NCBI nucleotide database (NM_144723.2 and BC056668.1) did not contain any information beyond what was found in genome data, an alternative approach was used to map the beginnings and ends of the gene. This analysis took advantage of the availability of searchable RNA-sequencing libraries.15,16 Specifically, we constructed a series of adjacent 60 bp probes from genomic DNA corresponding to the 5′ end of presumptive exon 1a and exon 1, and used them to query the RNA-sequencing library SRX5281080 from the NCBI SRA (Additional Table 1 in Supplemental Material). Based on the number of hits, our results showed that exon 1 was ~136 bp in length, rather than the 32 bp stated in Ensembl (Figure 2C). In contrast, a 5′ end of presumptive exon 1a could not be mapped, as this DNA region completely overlapped the most 3′ exon of HARS2 (see Figure 2A). No potential TATA box, which helps position RNA polymerase II at the start of transcription, 24 and no initiator element, which performs a similar role, 25 were found adjacent to the 5′ end of the longest ZMAT2 transcripts for exon 1 detected in these RNA-sequencing libraries (Figure 2C). Further confirmation regarding different 5′ ends for human ZMAT2 exon 1 came from the analysis of GRO-seq and GRO-cap data and the Riboseq Web site, as applied to information in the UCSC Genome Browser about human ZMAT2 (see Methods). Each of these resources showed that a range of 5′ ends for ZMAT2 exon 1 had been identified in different human cell lines using sequencing-based methods. Taken together, these results defined longer 5′ ends of exon 1 for ZMAT2 than had been recorded in Ensembl. Although our observations did not definitively identify the location of a gene promoter, the presence of several binding sites for transcription factors adjacent to the range of 5′ ends for ZMAT2 exon 1 is highly suggestive, as is evidence of an area of DNAse-I hypersensitivity and acetylation of histone H3 lysine 27 in this same region, although other supportive information, such as the presence of CpG islands, is lacking (see http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chr5%3A140079562%2D140080497&hgsid=769183249_79TJJsqNJdMb3UJWbEQaKe2fPdWf). In contrast, no GRO-seq or GRO-cap data were observed adjacent to presumptive exon 1a of Ensembl, and there was no evidence of accumulation of transcription factor binding sites either.
An analogous strategy was used to map the 3′ end of human ZMAT2. We found that exon 5, which was proposed in Ensembl to contain the 3′ terminus of the transcript encoding the 53-amino-acid ZMAT2 protein, instead appeared to end in an exon-intron junction. In fact, by searching the RNA-sequencing library SRX4654287, we determined that exons 5 and 6 formed 1 continuous transcript (see Figure 2D). Thus, in contrast to what is shown in Ensembl, exon 5 is not the final exon for any ZMAT2 mRNA. We did find that exon 6 contained an “AATAAA” presumptive poly A recognition sequence, and we mapped a poly A addition site 26 beginning at 43 bp in the further 3′ direction (Figure 2E). Thus, in total, exon 6 was 1071 bp in length and included a 3′ UTR of 927 bp. Taken together, these results define a 6-exon human ZMAT2 gene that spans 6341 bp (Figure 3A, Table 2) and that is transcribed and processed into a single coding mRNA of 1646 nucleotides (Figure 3B). This mRNA contains exons 1 to 6 and is predicted to encode a protein of 199 amino acids.

Human ZMAT2 gene and gene expression. (A) Structure of the human ZMAT2 gene, incorporating mapping studies shown in Figure 2. Labeling is as in Figure 2. P indicates a possible promoter. (B) Human ZMAT2 mRNA is based on gene characterization in Figure 2. Coding segments are in black and noncoding regions in white. (C) Transcript levels were analyzed for ZMAT2 (exons 1a + 2 and exons 1 + 2) and MRPS17 (top and bottom panels, respectively) in liver, fat, and adrenal gland using RNA-sequencing libraries from the NCBI SRA (left) and data from GTEx (right). Data are presented as hits/106 reads (NCBI SRA; see Additional Table 1 in Supplemental Material for characteristics of RNA-sequencing libraries and Additional Table 2 in Supplemental Material for DNA probes) or as sequence reads/base (GTEx). GTEx indicates Genotype-Expression Project; mRNA, messenger RNA; NCBI, National Center for Biotechnology Information; SRA, Sequence Read Archive.
Organization of primate ZMAT2 genes (in base pairs).

Approximate, because exon 1 has not been characterized fully.
Human ZMAT2 gene expression
Gene expression studies for mRNAs containing either Ensembl-defined exons 1a and 2 or exons 1 and 2 showed that the former transcript was minimally expressed in human RNA-sequencing libraries from liver, white fat, and adrenal gland, in contrast with a control transcript MRPS17, a gene encoding a mitochondrial ribosomal protein that is expressed in nearly all cell and tissue types (see: https://www.ncbi.nlm.nih.gov/gene/51373) (Figure 3C). Collectively with observations noted above, these results indicate that ZMAT2 mRNAs containing exon 1a are at best a very minor species.
The initial publication focusing on human ZMAT2 showed that silencing of ZMAT2 mRNA enhanced the differentiation of primary human foreskin keratinocytes, 11 implying that ZMAT2 somehow prevented differentiation. We thus interrogated human keratinocyte RNA-sequencing libraries (Additional Table 1 in Supplemental Material) to determine whether concentrations of ZMAT2 transcripts changed during a 6-day differentiation time course. Levels of ZMAT2 mRNA remained essentially constant during keratinocyte differentiation, as did a control transcript for MRPS17 (variation of ⩽35%, Figure 4). In contrast, steady-state levels of mRNAs of 2 epidermal terminal differentiation markers, envoplakin (EVPL) and periplakin (PPL), 27 rose by ~7-fold and ~12-fold, respectively, during 6 days of treatment of keratinocytes with differentiation-inducing medium, indicating that differentiation had occurred. 27 Thus, based on these results, the mechanisms by which the actions of ZMAT2 might decline during human keratinocyte differentiation 11 do not appear to be secondary to a major reduction in ZMAT2 gene expression.

ZMAT2 gene and gene expression during human keratinocyte differentiation. Transcript levels were measured for ZMAT2, EVPL, and PPL, markers of keratinocyte differentiation, 27 and MRPS17 (top, 2 middle, and bottom panels, respectively) in RNA-sequencing libraries from the NCBI SRA (see Additional Table 1 in Supplemental Material for characteristics of the libraries and Additional Table 2 in Supplemental Material for DNA probes). Data represent the mean of 2 experiments and are presented as hits/106 reads. NCBI indicates National Center for Biotechnology Information; SRA, Sequence Read Archive.
The ZMAT2 gene in other primates
By examination of the Ensembl Genome Browser and by searching genome databases with human exons, ZMAT2 was mapped in 8 nonhuman primate species. The single-copy primate ZMAT2 genes also appeared to consist of 6 exons (Figure 5, Table 2), and their overall structures closely resembled human ZMAT2 (Figure 5). However, for 3 species, orangutan, olive baboon, and marmoset, the structure of ZMAT2 was incomplete, as exon 1 lacked a 5′ UTR (and consisted of only 18 bp). When their 5′ ends were mapped using species-homologous RNA-sequencing libraries (Additional Table 1 and Figure 1 in Supplemental Material), exon 1 and their overall gene structures closely resembled human ZMAT2, including reasonable congruence in the lengths of all exons and introns among these primates (Figure 5, Table 2). Total gene sizes ranged from 6309 bp in chimpanzee to 7079 bp in marmoset and 7980 bp in rhesus macaque, with variation in the 2 latter species being secondary to a longer intron 4 and longer exon 1 for macaque (Table 2). DNA conservation among ZMAT2 exons was high among the primate species studied, with nucleotide sequence identity with the human gene for all 6 exons in chimpanzee, gorilla, orangutan, macaque, bonobo, and olive baboon being >95% and for exons 2 to 5 in marmoset and mouse lemur (Table 3). As might be expected, these analyses also showed that DNA identity with human ZMAT2 was highest in primate species evolutionarily closest to humans. For example, in chimpanzees and gorillas, in which the overall match with the human genome is >98.5%,28,29 DNA sequence identity ranged from 97.8% to 100% for all 6 exons. These parameters were lower in rhesus macaque, where identity with the human genome was ~93.5% 29 (95.7%-100% for exons 1-6, Table 3), and were less in the more distantly related marmoset and mouse lemur (86.6%-99.3%; Table 3).

ZMAT2 gene in primates. Diagrams of human, chimpanzee, gorilla, orangutan, macaque, bonobo, olive baboon, marmoset, and mouse lemur ZMAT2. Exons are depicted as boxes (black coding, white noncoding). The locations of ATG and TGA codons are indicated, and a vertical arrow defines the location of the putative polyadenylation site at the 3′ end of exon 6 for each gene. A scale bar is shown. Also see Tables 2 and 3.
Nucleotide identity with human ZMAT2 exons.

Coding and noncoding DNA.
ZMAT2 gene expression in primates
Gene expression studies showed that ZMAT2 mRNAs were present in liver RNA-sequencing libraries from different primate species. However, steady-state levels varied by a factor of ~15 among different primates, as did the abundance of a control transcript for MRPS17 (Figure 6).
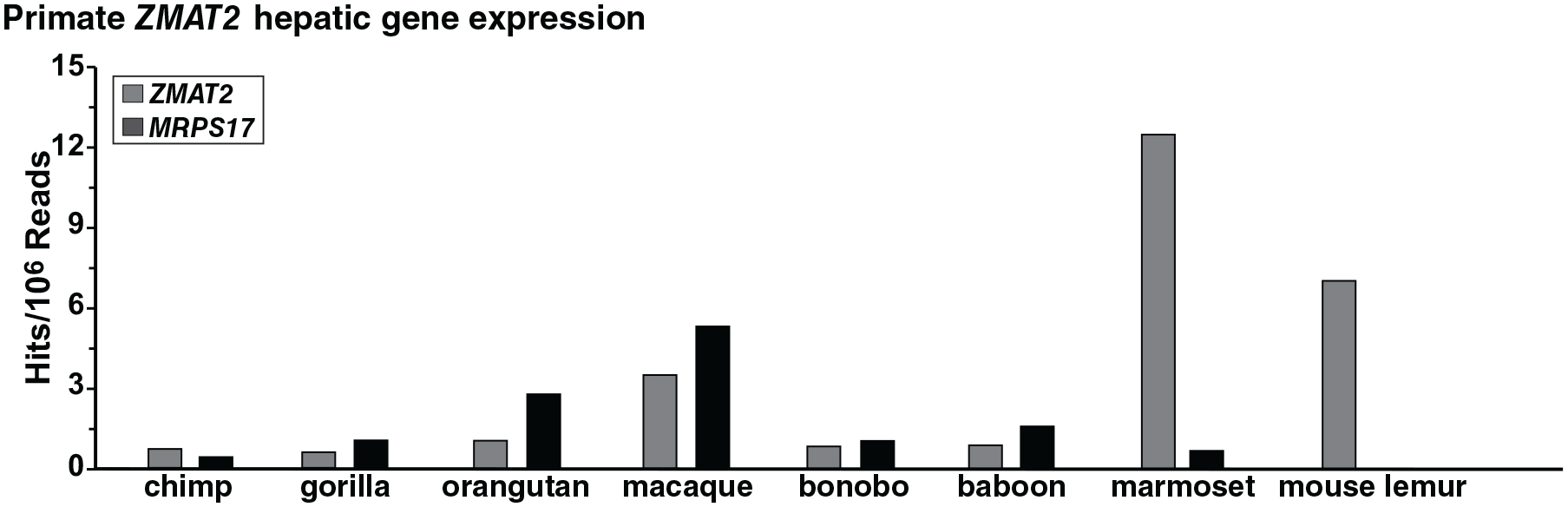
ZMAT2 gene expression in primates. Transcript levels were examined for ZMAT2 and MRPS17 in liver for different primates by querying RNA-sequencing libraries using specific 60 bp genomic DNA segments from each species. Results are plotted as hits/106 reads. See Additional Table 1 in Supplemental Material for the libraries and Additional Table 2 in Supplemental Material for DNA probes.
Three ZMAT2 pseudogenes are found in the marmoset genome
Initial screening of the marmoset genome revealed 4 sets of DNA sequences with similar levels of identity with human ZMAT2 exons 1 through 6 (90%-99.3%). These DNA segments were distributed to 4 different locations in the marmoset genome (Figure 7A). One contained ZMAT2, but 2 of the other 3 consisted of continuous DNA sequences, and thus resembled processed mRNAs that were retro-transposed as DNA copies back into the marmoset genome. 30 For the other DNA sequence, a putative “intron” of 302 bp separated copies of “exons 1 to 3” from “exons 4 to 6,” which is located in the single intron of marmoset protein-coding gene, ENSCJAT00000066532.1 (Figure 7A), but its junctions did not resemble normal exon-intron or intron-exon boundaries. 31 Moreover, the DNA within this “intron” appeared to be an Alu repeat element32,33 and was identified as such using the Dfam database.

The marmoset genome contains 3 ZMAT2 pseudogenes. (A) Top to bottom: schematics of marmoset ZMAT2 and pseudogenes 1, 2, and 3. The color coding indicates regions of each pseudogene that are similar in DNA sequence to individual exons of marmoset ZMAT2. (B) Alignment of amino acid sequences of marmoset ZMAT2 and predicted pseudogene proteins 1 and 3 (Z1 and Z3, respectively). The open reading frame for Z3 starts at amino acid 77 of marmoset ZMAT2. Similarities and differences are shown, with identities being indicated by asterisks. Differences also are marked by blue or red text. (C) Gene expression of marmoset ZMAT2 and the 3 pseudogenes in liver. Data were obtained by querying NCBI SRA library SRX347666 (Additional Table 1 in Supplemental Material) with probes listed in Additional Table 2 in Supplemental Material. Only transcripts from authentic marmoset ZMAT2 could be detected. NCBI indicates National Center for Biotechnology Information; SRA, Sequence Read Archive.
Conceptual translation of the RNAs predicted from the 2 DNA sequences that formed a continuous open reading frame (pseudogenes Z1 and Z3, Figure 7B) revealed marked similarity with the marmoset ZMAT2 protein. Pseudogene Z1 was identical with marmoset ZMAT2 in 196 of 199 residues (98.5% identity), and pseudogene Z3 matched ZMAT2 in 120 of 123 amino acids (Figure 7B). However, analysis of gene expression of these variant ZMATs revealed no transcripts encoding any of them in an RNA-sequencing library from marmoset liver RNA, although authentic ZMAT2 mRNA was detected readily (Figure 7C). Thus, all 3 of these variant versions of marmoset ZMAT2 appear to be pseudogenes. As no potential ZMAT2 pseudogenes were detected either in the human or in any of the other primate genomes studied here, these presumably arose in marmoset subsequent to the divergence of its progenitors from other primates, such as mouse lemur and macaque, and thus entered the marmoset genome more recently than approximately 25 to 30 million years ago. 29
Limited predicted population variation in the human ZMAT2 protein
Human ZMAT2 appears to be remarkably nonpolymorphic, as very few missense or other variants could be detected in human populations, at least as judged by analysis of the data from gnomAD, which contains results of whole exon and whole genome sequencing from 141 456 different individuals. 21 Only 41 different missense modifications were identified, and collectively they were found in 0.014% of alleles in this study population, with the most frequent variant (Glu154 to Gly) being present in less than 1 in 50 000 alleles (Figure 8A, Table 4). In addition, no alterations were detected that caused loss of protein expression or errors in gene splicing (Table 4). A few other different ZMAT2 coding changes appeared to be present in a range of human cancers, with 32 of 36 encoding single predicted amino acid substitutions (in addition, there was 1 stop codon, 2 splicing alterations, and 1 frameshift) and with nearly all of the alterations being detected uncommonly in individual cancer types (Table 5; see the cBio portal for cancer genomics—https://www.cbioportal.org).

Primate ZMAT2 proteins. (A) Schematic of the human ZMAT2 protein, with NH2 (N) and COOH (C) terminal (term), and zinc finger (ZnF) regions labeled and color-coded. The overall population prevalence of variant alleles for each segment of the protein is listed above the map, and the number of missense mutations in various cancers is found below. Also see Tables 4 and 5. (B) Alignments of amino acid sequences of ZMAT2 from human, chimpanzee, gorilla, orangutan, macaque, bonobo, marmoset, mouse lemur, and olive baboon are shown in single-letter code. The amino acid sequences are identical, as depicted by the asterisks. An “I” followed by a number indicates the location of each intron.
Human population variation in ZMAT2. a

Data are from the gnomAD genome browser (https://gnomad.broadinstitute.org/).
Cancer-associated predicted mutations in ZMAT2. a

Data are from the cBio portal for cancer genomics (https://www.cbioportal.org).
Identical ZMAT2 protein sequences among primates
ZMAT2 was identical to the human protein in all 8 of the nonhuman primates evaluated here (Figure 8B). However, for olive baboon, this conclusion is based only on data from cDNA XM_031666488.1, as it could not be validated in the genomic DNA sequence in Ensembl because of a stretch of nucleotides in exon 6 that could not be determined.
Discussion
The major goals of the investigations presented here were to characterize the nearly unstudied human ZMAT2 gene by mining the resources of public databases and to place these findings in an evolutionary context with ZMAT2 homologues from other nonhuman primates. Our main observations include defining the structure of a 6-exon single-copy human ZMAT2 gene, showing that ZMAT2 exhibits very limited genetic variation in human populations and in disease states, finding that the gene and its encoded protein are highly conserved among primates, and identifying ZMAT pseudogenes in a single species, marmoset. More importantly, our study demonstrates how a strategy involving the focused and complementary examination of publicly accessible genomic, gene expression, and population genetic databases can lead to new insights about human and mammalian biology and evolution, and illustrates the value of investigating understudied genes as a means of generating new experimentally testable hypotheses.
The ZMAT2 gene in humans and other primates
The genomic and gene expression data described and analyzed here show that ZMAT2 is a 6-exon gene in humans and in at least 8 other nonhuman primates (Figures 3 and 5). Our results thus appear to contradict information from Ensembl, which states that a seventh ZMAT2 exon is located further 5′ within the most 3′ exon of HARS2 (Figure 2A). Our experimental data obtained by querying human RNA-sequencing libraries and the GTEx gene expression database show that transcripts containing this additional exon fused to ZMAT2 exon 2 are minimally expressed (Figure 3), and moreover that data derived from GRO-seq and GRO-cap analysis do not support the presence of an additional 5′ exon for human ZMAT2.
Remarkably, the marmoset genome contains 3 distinct ZMAT2 pseudogenes that are highly similar to the authentic gene, but do not appear to function, as they are not expressed (Figure 7). Two of these pseudogenes resemble fully processed mRNAs that were retro-transposed as individual DNA copies back into the marmoset genome. 28 The other appears to be the copy of a partially spliced mRNA, although analysis of its single “intron” reveals that it contains an Alu element32,33 and lacks appropriate splicing signals at its junctions, 29 and thus that it must have been extensively modified during its residence time in the marmoset genome. As we did not find any other ZMAT2 pseudogenes in other primate genomes, these must have entered the marmoset genome more recently than ~25 to 30 million years ago, at a time after the divergence of the progenitor of this species from other primate precursors. 29 Other recently published studies from our group have demonstrated that Zmat2 pseudogenes are present in at least 9 other mammalian species. 34 As the DNA sequence of each of these pseudogenes was more similar to the paralog from the homologous mammalian species than to other Zmat2 pseudogenes, it seems likely that each Zmat2 pseudogene arose independently subsequent to the divergence of each mammal from its closest ancestors. 34
ZMAT2 proteins
Our results show that the human and primate ZMAT2 proteins are identical to each other (Figure 8). Moreover, ZMAT2 is remarkably nonpolymorphic in humans, as judged by the fact that of more than 280 000 alleles studied in the gnomAD project, only 31 different potential codon changes that predict amino acid substitutions were identified, and these occurred collectively in only 0.014% of the alleles in the study population (Figure 8, Table 4), a percentage substantially lower than that had been described previously for the prevalence of variant alleles in at least 19 other human genes (eg, 0.08% [AKT3 35 ], 31% [IGFBP1 36 ], 86% [RGMA 37 ], and 121% [IGF2R 36 ]) in the Human Exome Consortium (ExAC38,39). Moreover, and unlike these other genes,35-37 no frameshift alterations or splicing site changes were found in human ZMAT2, and in addition, very few modifications were identified in different human cancers (Figure 8, Tables 4 and 5). A potential reason for this lack of variation could be that ZMAT2 plays a critical structural and functional role in pre-mRNA splicing in the nucleus. This statement is based on the identification of ZMAT2 as a component of the yeast 12 and human spliceosome, as determined in the latter recently by cryo-electron microscopy. 14 As defined by that study, the α-helical region of ZMAT2, along with the protein Prp38, contacts the U6 snRNP at the 5′ splice site of the intron and may facilitate its activation 14 and step 1 of splicing, which leads to a cleaved 5′ exon and the development of a lariat intermediate between the intron and 3′ exon. 13 Remarkably, ZMAT2 also appears to have a specialized function as a negative regulator of human keratinocyte differentiation, potentially via selective inhibitory effects on pre-mRNA splicing of certain genes. 11 It is unknown whether ZMAT2 might act similarly in other organs or tissues in which epithelial cell differentiation is critical for normal development or response to disease (eg, bronchi or alveoli in the lungs 40 ) or regeneration (eg, the intestines,41,42 wound healing 43 ), or whether it is dysfunctional in skin diseases in which terminal differentiation could be altered.44,45 Moreover, while this manuscript was in review, a novel mutation was described in human ZMAT2 in a child with a bone disorder termed congenital radioulnar synostosis. This mutation, predicting amino acid substitution F142I, 46 had not been identified previously in humans (see Table 5). Thus, there are several potentially important topics for future investigation into ZMAT2 gene regulation and protein function.
The ZMAT family and other understudied human genes
Despite advances in access to information through public genomic and gene expression databases and other resources,2,5-7 only a small fraction of human genes has been evaluated.8-10 In fact, according to a recent report, approximately 90% of human genes are understudied. 10 Among these are all 5 members of the ZMAT family, as collectively they have been the main topic of analysis in ~50 publications to date, with the vast majority being devoted to ZMAT3 (also known as the p53 target WIG-1 [wild-type p53-induced gene], Figure 1).22,23
Genes and databases
Publicly available genomic repositories contain extensive data on different genes from many species, yet as shown here, the information about ZMAT2 in humans and in at least a cohort of primates had not been annotated completely or correctly. This problem does not appear to be uncommon, as similar deficiencies have been shown by us for several other genes in mammals and in nonmammalian vertebrates as well.15,47 It is clear that a substantial effort is needed to improve the accuracy of the data in these resources to enhance the opportunity for future discoveries, and more broadly for the general benefit of the scientific community.
Final comments
The genetics of modern humans represents the distillation of extensive interactions over multiple generations with many different ancestral groups. These interactions have resulted in the presence of various amounts of chromosomal DNA in current human populations, which were derived from extinct groups such as Neanderthals, Denisovans, and others.48-51 Modern humans have also been shaped by a variety of genetic roadblocks, founder effects (eg, see Belbin et al 52 and other interactions53,54 that collectively have influenced and continue to influence both human physiology and disease susceptibility55,56). It is thus conceivable that further analysis of ZMAT2 and other understudied human genes may lead to new insights of potentially high genomic, biological, and biomedical significance.
Supplemental Material
Suppl_Table_1-RNA-seq_libraries_xyz427519184a252 – Supplemental material for ZMAT2 in Humans and Other Primates: A Highly Conserved and Understudied Gene
Supplemental material, Suppl_Table_1-RNA-seq_libraries_xyz427519184a252 for ZMAT2 in Humans and Other Primates: A Highly Conserved and Understudied Gene by Kabita Baral and Peter Rotwein in Evolutionary Bioinformatics
Supplemental Material
Suppl_Table_2-probes-revised_xyz427511641c6ec – Supplemental material for ZMAT2 in Humans and Other Primates: A Highly Conserved and Understudied Gene
Supplemental material, Suppl_Table_2-probes-revised_xyz427511641c6ec for ZMAT2 in Humans and Other Primates: A Highly Conserved and Understudied Gene by Kabita Baral and Peter Rotwein in Evolutionary Bioinformatics
Footnotes
Acknowledgements
There are no human or animal studies in this manuscript. All data generated and analyzed during this study are included in this published article and in its supplementary information files.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported in part by National Institutes of Health research grant R01 DK042748-28 (to P.R.).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
P.R. conceived the study, performed the research, and wrote and edited the manuscript. K.B. performed the research and edited the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.