
Abstract
Assembled draft genomes usually contain many gaps because of the length limit of next-generation sequencing. Although many gap-closing tools have been developed, most of them still attempt to fill gaps on the basis of next-generation sequencing reads (always < 200 bp). Hence, the gap-filling effect is inferior. Several tools that use long-reads to close gaps have recently been created. However, they require extensive runtimes, which may not be suitable for large genomes. We describe a gap-closing tool called PGcloser, which supports parallel mode and adopts long-reads/contigs to fill gaps in genome sequences. Three tests show that PGcloser is faster than other tools but exhibits similar accuracy. PGcloser is free open-source software that is available at http://software.tobaccodb.org/software/pgcloser.
Keywords
Introduction
Genes are DNA molecular fragments with genetic effects. They are the code of life and record and transmit genetic information. Genetic research has explored biological inheritance and variation. 1 Genetic studies were based on gene sequences that are mainly obtained by genome sequencing. More than 800 kinds of bacteria and more than 100 kinds of eukaryotic organisms’ genome sequences have been released after the introduction of the Sanger sequencing method in 1977. Thousands of genome sequences have been restored in gene databases, and a large number of species are still being sequenced.
The main sequencing technology that has been applied in recent years is called next-generation sequencing (NGS) or second-generation sequencing. Reads generated through this method have short sequences, high coverage rates, and paired-end information. Numerous algorithms have been introduced to assemble reads using the overlap between the sequences of fragments, 2 such as greedy extension algorithm, overlap-layout-consensus algorithm, and De Bruijn graph (DBG) algorithm. And a lot of software including Velvet, 3 SOAPdenovo, 4 AbySS 5 are developed based on these algorithms. However, given the length limitations of next-generation sequencing reads and the high ratio of repeat sequences in genomes, some regions of genome sequences have not been assembled, all these factors make some regions difficult or impossible to assemble, leading to gaps and fragmented genome assemblies. 6 Consequently, the final draft genomes contain many gaps.
The public dataset in National Center for Biotechnology Information (NCBI) (Table 1[June 2018]) showed that only a few species have complete genomes, and most of them are viruses and bacteria. The large genomes of certain plants and animals remain draft genomes at the contig/scaffold level. Thus, they may still contain many gaps.
Genome assembly of various species.

RC: ratio of contig assembly level in the group; RCG: ratio of complete genome assembly level in the group; RChr: ratio of chromosome assembly level in the group; RS: ratio of scaffold assembly level in the group; SNC: species number of contig assembly level; SNCG: species number of complete genome assembly level; SNChr: species number of chromosome assembly level; SNS: species number of scaffold assembly level.
The gaps in scaffolds may contain a considerable amount of useful biological information, such as important genes. Hence, filling the gaps may result in the acquisition of the unknown information, thereby improving the integrity of gene sequences. At present, gaps are filled using five major methods, as follows: (1) assembly by multiple software, such as Velvet 3 and Edena 7 ; (2) use of reference genomes from closely related species, such as software Velvet, 3 Edena, 7 and MUMer 7 ; (3) assembly by multiple types of data, such as ALLPATHS-LG 8 and SSPAKE 9 ; (4) use of polymerase chain reaction amplification at the ends of gaps, such as ABACAS 10 ; and (5) adoption of improved assembly methods based on DBG, such as GapCloser, 11 IMAGE, 12 and GapFiller. 13
Third-generation sequencing technology, including PacBio’s SMRT sequencing technology 14 and Oxford’s single-molecule nanopore sequencing technology, 15 have recently been applied to biological genome sequencing. Compared with the read length of next-generation sequencing, that of third-generation sequencing is considerably longer, possibly exceeding 10 kb 16 ; it is also hundreds of times longer than that of NGS. Several gap-closing tools have been developed using the long-reads instead of short NGS reads, such as LR_Gapcloser, 6 GMcloser, 17 PBJelly, 18 and FGAP 19 . PBJelly and FGAP primarily focus on assembly extension and not gap closing. GMcloser was developed to close gaps by measuring the likelihood ratios of true alignments. Its accuracy is 3-fold to 100-fold higher than that of other available tools that use NGS data. LR_Gapcloser utilizes long reads generated from TGS sequencing platforms. It closes gaps more rapidly with a lower error rate and a considerably lower memory usage than GMcloser. However, LR_Gapcloser and GMcloser have certain limitations. They cannot be run in multiple nodes. Thus, they are slow for large genomes. Moreover, they require the use of a large memory for large genomes and reads.
Here, we developed PGcloser to efficiently and rapidly close gaps in assemblies using long reads or contigs. Compared with the abovementioned gap-closing tools, PGcloser has advantages in runtime, average memory usage, and efficiency.
Methods
The main idea of PGcloser is to reduce the amount of computing data and increase running speed. Thus, we split a genome into small sub-files for parallel computation and then used the error-corrected and repeats-removed long reads or contigs to minimize the number of sequences in the reference reads. The pipeline for PGcloser is shown in Figure 1.
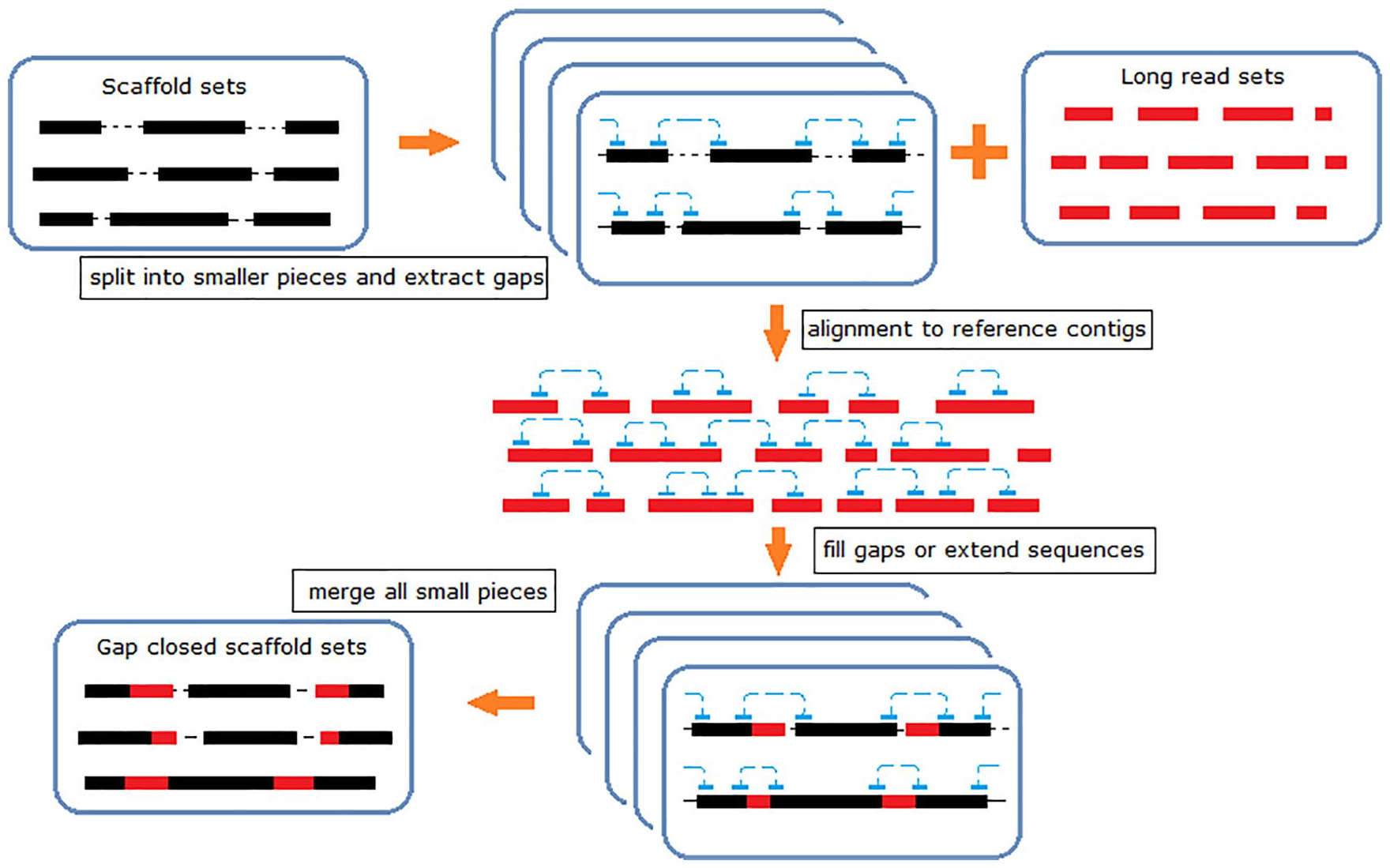
Pipeline of PGcloser.
PGcloser is a Linux-based integrative analysis workflow. This tool contains the following steps:
It extracts the gaps for each scaffold in the genome file. Then, two ends of each gap are extracted as anchors for each scaffold.
The long reads are regarded as reference, and an index file is built for this reference. Then, anchor sequences are mapped to the long reads.
PGcloser analyzes the mapping results and obtains a specified mapping position for each anchor sequence. If two anchors map to the same sequence in the reference with reasonable distance, close this gap. If one or two anchors map to different sequences in the reference, then the corresponding gap is extended.
All gap-closing results are combined, and the final gap-closing genome is produced.
Step 1: the genome is split into small sub-files, and the gaps are extracted
PGcloser splits the genome file into sub-files by the number of threads provided by the user. Then, it extracts the gaps for each scaffold, and two ends of each gap are used as anchors. The corresponding parameters, including the minimum gap length, anchor length, and thread number, are provided by the user.
Step 2: the anchor sequence is aligned to the long reads
PGcloser regards the long reads/contigs as the reference and builds an index for it. Then, the anchor sequences generated in Step 1 are mapped to the index file. The mapping result is then stored in SAM/BAM format.
Step 3: the gaps are filled or extended
The mapping result generated in Step 2 can be divided into three categories, as follows:
Two anchors mapped to the same read. In this mapping process, we defined two filter criteria. First, two anchors map to the same read in the reference. And another is the ratio of the gap length in the reference sequence to the gap length in the genome (error ratio) is within a reasonable range (this parameter can be submitted by the user). By these two filter criteria, the anchors mapped to repetitive regions will be greatly reduced after filtering. For the last repetitive regions, we will keep all the possible mapping reads.
Two anchors mapped to two different reads or just one anchor mapped to one read. If two anchors map to different reads or just one anchor maps to one read in the reference, PGcloser will extend the corresponding gap related to these anchors.
Two anchors are not mapped to any read. If two anchors do not map to any reference read, PGcloser will continue to keep this gap.
Step 4: all sub-files are merged
In this last step, PGcloser combines the processed gaps and forms the final result.
Implementation
In accordance with the abovementioned algorithm, we developed the gap-closing tool PGcloser, which contains the following modules: SplitFa, ExtrGap, BwtBuilt, CompGap, ClsGap, MergFa, and GetCls.
SplitFa: This module splits the genomic file into small sub-files in accordance with the number of threads and nodes provided by the user.
ExtrGap: This module extracts all the gaps of the sub-files in multiple threads and nodes. Then, it generates a fasta file of the anchor sequences.
BwtBuilt: This module constructs the index file for the input long reads set.
CompGap: This module aligns the anchor sequences of the gaps to the index file and generates SAM files for the next module.
ClsGap: This module extracts the specified position sequence for the anchor sequences and deals with gaps in accordance with their mapping result.
MergFa: This module merges the closed and extended gaps to form the final result.
GetCls: This module executes all the modules in PGcloser and outputs the GapCloseed.fa file.
Result
Datasets
To test the performance of PGcloser, we selected three datasets with different genome sizes from Arabidopsis thaliana, Oryza sativa, and Homo sapiens (Table 2), more data description can be visited and downloaded from the website http://software.tobaccodb.org/software/pgcloser.
Information for three datasets.

Benchmark
We compared the performance, efficiency, and genome evaluation of PGcloser, LR_Gapcloser, GMcloser, GapFiller, and FGAP on a 48-core server with Intel(R) Xeon(R) Gold 6126 CPU @ 2.60 GHz and 512 GB RAM.
For performance, we compared speed and memory usage. For efficiency, we compared the gap-closing ratio and gap length reduction. For genome evaluation after gap-closing, we compared BUSCO and reads mapping rate.
With m as the gap number before gap-closing and n as the gap number after gap-closing, the close rate is defined as follows:

With x as the gap length before gap-closing and y as the gap length after gap-closing, the gap length reduction is defined as follows:

Artificial data test
In order to initially evaluate the gap-closing quality of PGcloser, we have created 2000 artificial gaps with length from 100 to 300 bp in three genomes. Half of them were inserted in gene regions, and others were inserted into repetitive elements. Then, we compared the filling sequence of the gaps by the gap-closing tools with the original sequence. The results are as follows.
Tables 3 and 4 show the results of artificial gap-closing. For the closed gaps, the accuracy rate of PGcloser in genes is around 95% and in repetitive elements is more than 50% on three test datasets, which are similar to other tools.
Gap-closing results in gene regions.

Accurate closed—sequence difference before and after gap closing < 5%.
Gap-closing results in repetitive elements regions.

The results of the artificial gap test showed, that the quality of gap-closing using PGcloser is reliable.
Main content
Performance comparison
LR_Gapcloser, GMcloser, FGAP, and GapFiller tools cannot run in multiple nodes. Thus, we tested them with different thread numbers (from 1 to 48) in one node. We also performed an additional test using PGcloser in multiple nodes. The performance results between these tools are as follows:
The time consumption of LR_Gapcloser, GMcloser, GapFiller, and FGAP is too long. Hence, we stopped the processes after 10 days. GapFiller and FGAP failed to obtain gap-closing results on the H. sapiens dataset with all threads. Figures 2 and 3 show that PGcloser outperforms the other tools in terms of running time and memory usage. Compared with the other tools, PGcloser achieved a 10-fold to 100-fold faster running time, especially in the multiple thread mode. Memory usage decreased by more than 90% using A. thaliana and O. sativa.

Running time (min).

Average memory usage (Mb).
Efficiency comparison
The efficiency for gap closing is as follows. Table 5 shows the results for the three datasets. PGcloser has a similar gap-closing number compared with most of the other tools, including the relatively stable tools LR_Gapcloser and GMcloser. FGAP only has superior performance for A. thaliana, which has a small genome. As for the closing ratio for the gaps, PGcloser, GMcloser, and LR_Gapcloser outperform GapFiller and FGAP, which respectively can achieve 60% and 90% for H. sapiens, which has a large genome.
Efficiency results.
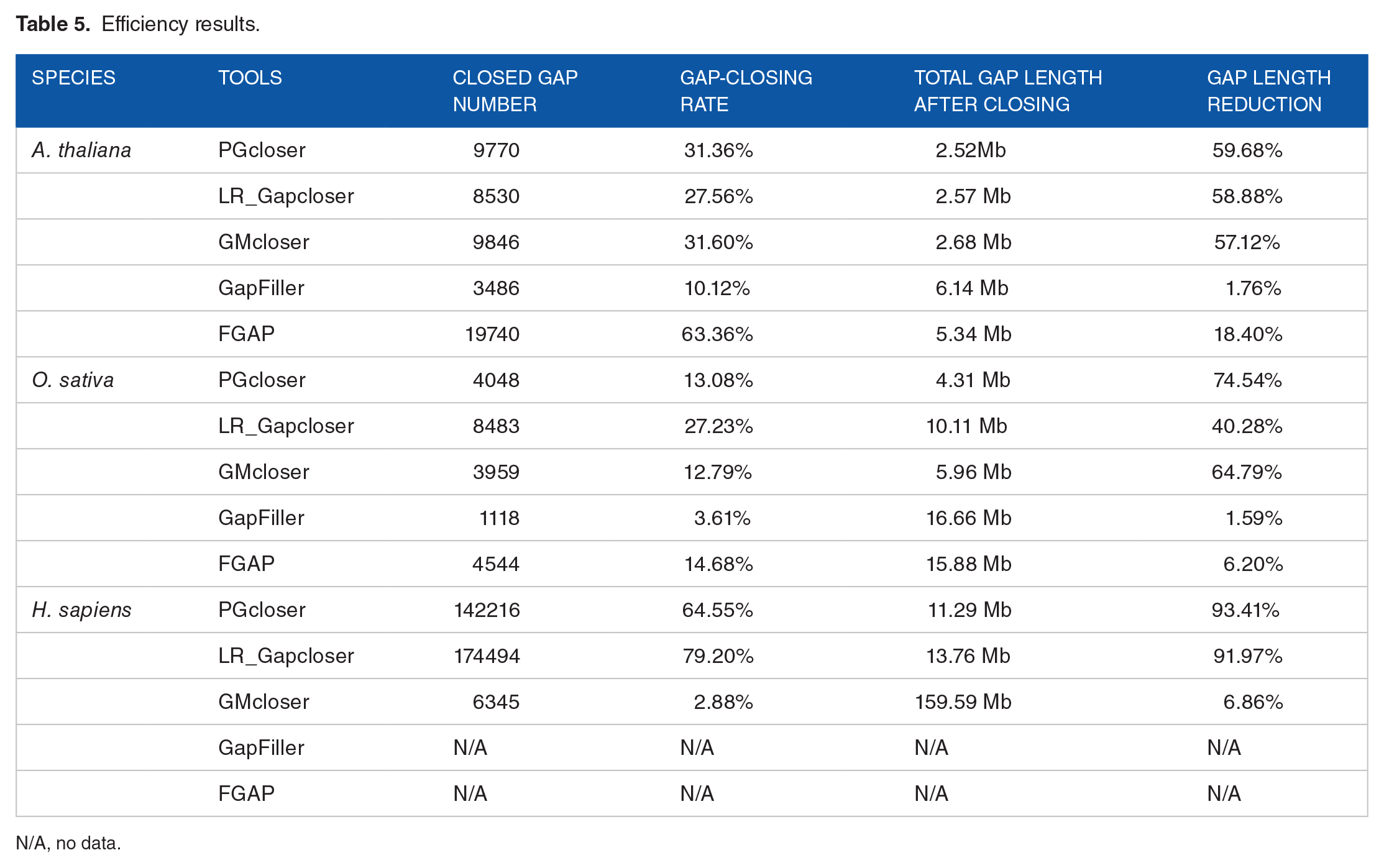
N/A, no data.
Genome evaluation
We used two methods to evaluate the quality of genomes after gap-closing. One is showing BUSCOs before and after gap-closing by running BUSCO. 20 Another is showing the long reads mapping rates before and after gap-closing by running Minimap2. 21 More details are as follows.
Table 6 shows the results of BUSCOs before and after genome gap-closing. The BUSCO result of PGcloser is similar to other tools on the three datasets.
BUSCO results.
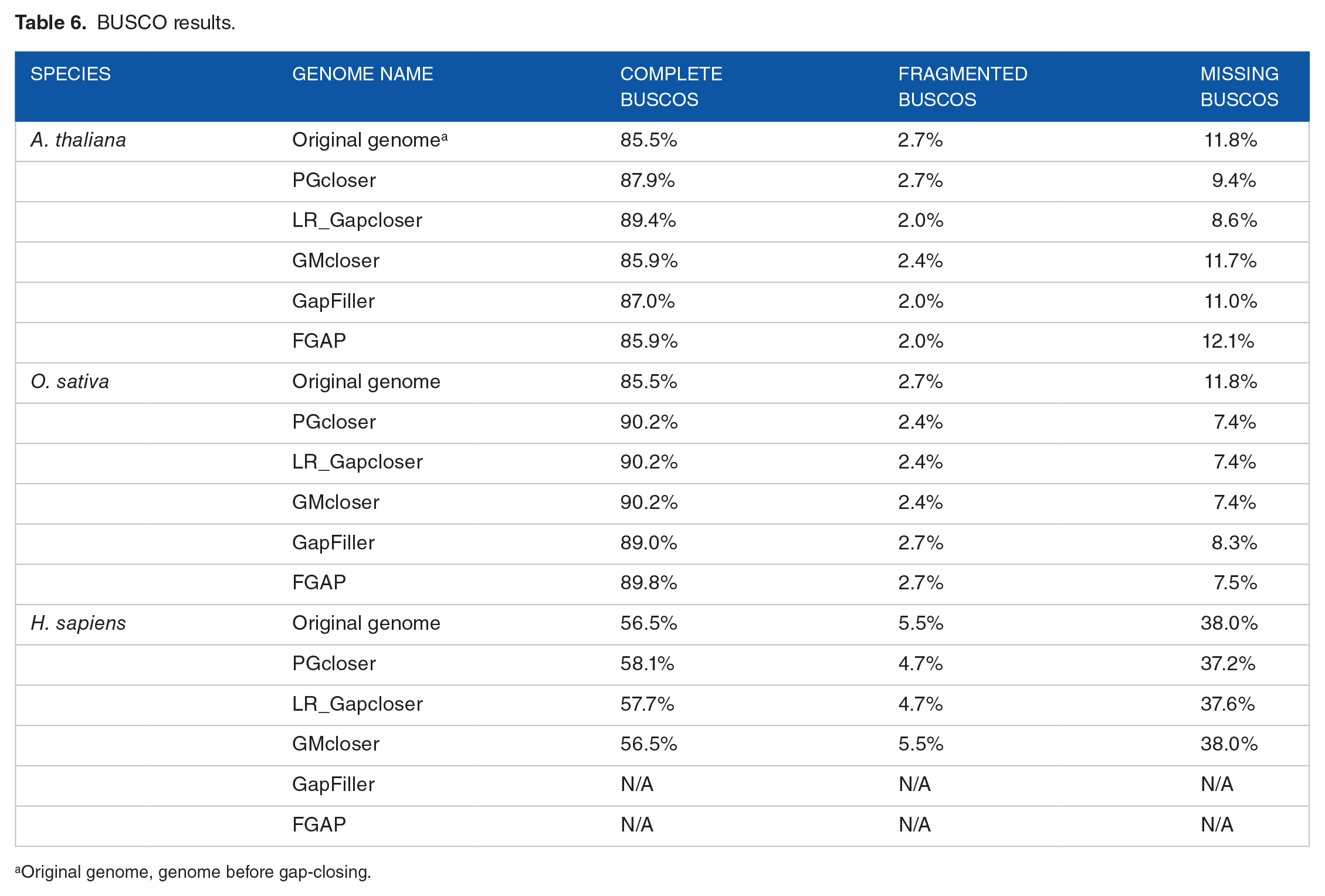
Original genome, genome before gap-closing.
Table 7 shows the results of reads mapping rate before and after gap-closing. The mapped rates had increased after gap-closing for A. thaliana. Although reads mapping rate is same for O. sativa and H. sapiens, the number of mapped reads had increased after gap-closing. The reads mapping result of PGcloser is similar to other tools on the three datasets.
Reads mapping rate results.
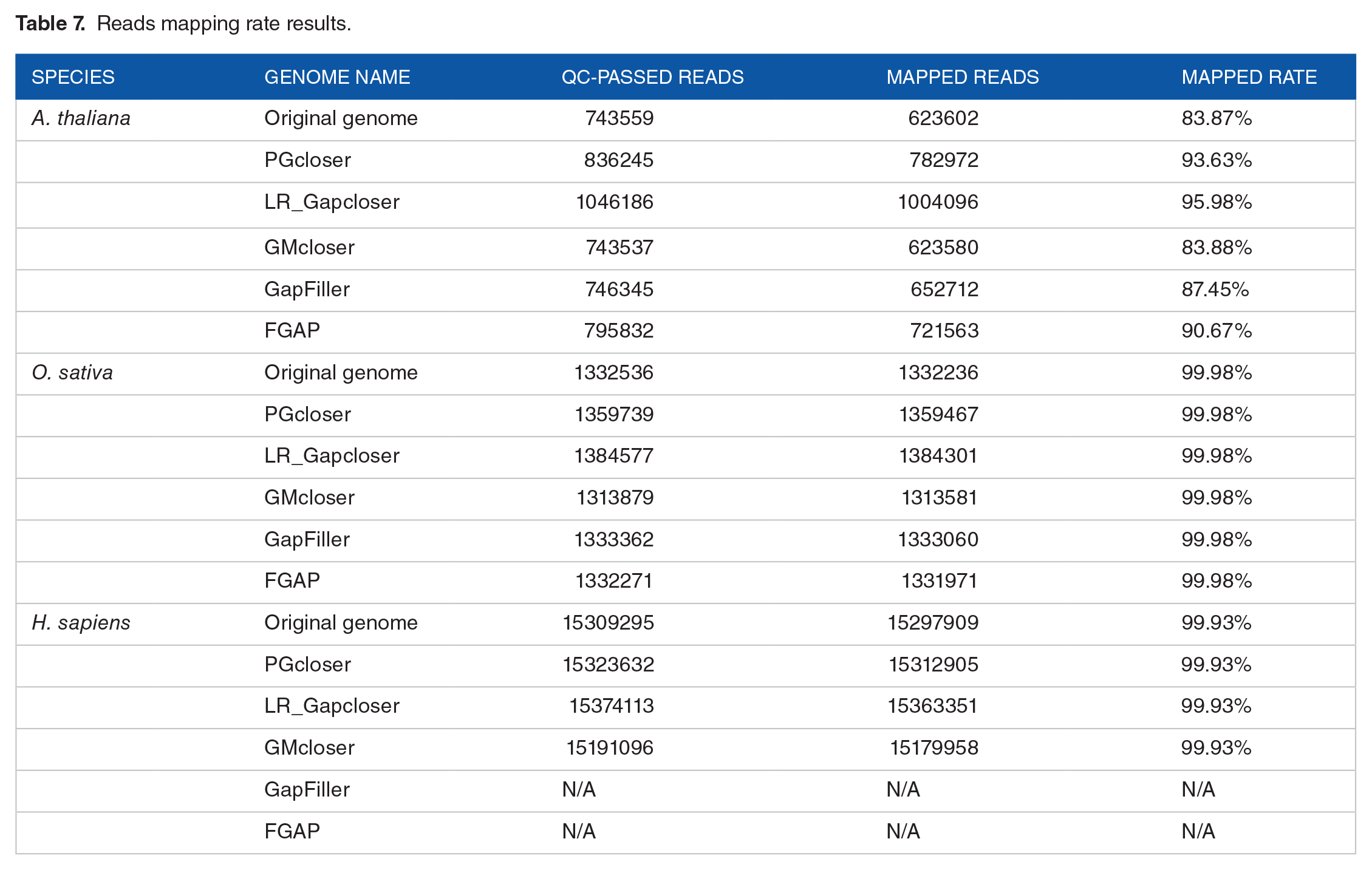
The evaluation results of the above two methods showed that the quality of three genomes has improved after gap closing. And the quality of the genome after gap-closing by PGcloser is similar as the quality obtained with other tools.
Conclusion
We compared the gap-closure performance of PGcloser and four currently available tools: LR_Gapcloser, 6 GMcloser, 17 GapFiller, 13 and FGAP. 19 We ran each tool with 1, 10, 20, and 48 threads on the same machine. We estimated the performance of each tool using runtime and average memory usage, approximated the efficiency using the gap closing rate and gap length reduction, and then evaluated the quality of genome after gap-closing by BUSCOs and reads mapped rates.
The results of the three datasets showed that PGcloser reduced the running time and memory usage compared with the other tools. PGcloser was considerably faster and had similar or even better efficiency than the other tools. PGcloser showed a bigger advantage than the other approaches, especially for large genomes.
Footnotes
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Projects of Gap-Closing in the Tobacco Genome (902016CA0170), Tobacco Breeding Big Data (110201901024(SJ-03)), ENCODE of Tobacco Genome (110201601033(JY-07)), and the Young Elite Scientists Sponsorship Program by CAST (2016QNRC001).
Author Contributions
PL, JJ, and PC designed the project. PL, ZL, and YX collected sample data used in this project. PL, JJ, DH, and JL designed the algorithm. PL and JJ wrote the program codes and performed the bioinformatic analyses. PL, JJ, and PC wrote the manuscript. PL and JJ contributed equally to the study.