
Abstract
Trehalose-6-phosphate synthase (TPS) is a key enzyme in the biosynthesis of trehalose, with its direct product, trehalose-6-phosphate, playing important roles in regulating whole-plant carbohydrate allocation and utilization. Genes encoding TPS constitute a multigene family in which functional divergence appears to have occurred repeatedly. To identify the crucial evolutionary amino acid sites of TPS in higher plants, a series of bioinformatics tools were applied to investigate the phylogenetic relationships, functional divergence, positive selection, and co-evolution of TPS proteins. First, we identified 150 TPS genes from 13 higher plant species. Phylogenetic analysis placed these TPS proteins into 2 clades: clades A and B, of which clade B could be further divided into 4 subclades (B1-B4). This classification was supported by the intron-exon structures, with more introns present in clade A. Next, detection of the critical functionally divergent amino acid sites resulted in the isolation of a total of 286 sites reflecting nonredundant radical shifts in amino acid properties with a high posterior probability cutoff among subclades. In addition, positively selected sites were identified using a codon substitution model, from which 46 amino acid sites were isolated as exhibiting positive selection at a significant level. Moreover, 18 amino acid sites were highlighted both for functional divergence and positive selection; these may thus potentially represent crucial evolutionary sites in the TPS family. Further co-evolutionary analysis revealed 3 pairs of sites: 11S and 12H, 33S and 34N, and 109G and 110E as demonstrating co-evolution. Finally, the 18 crucial evolutionary amino acid sites were mapped in the 3-dimensional structure. A total of 77 sites harboring functionally and structurally important residues of TPS proteins were found by using the CLIPS-4D online tool; notably, no overlap was observed with the identified crucial evolutionary sites, providing positive evidence supporting their designation. A total of 18 sites were isolated as key amino acids by using multiple bioinformatics tools based on their concomitant functional divergence and positive selection. Almost all these key sites are located in 2 domains of this protein family where they exhibit no overlap with the structurally and functionally conserved sites. These results will provide an improved understanding of the complexity of the TPS gene family and of its function and evolution in higher plants. Moreover, this knowledge may facilitate the exploitation of these sites for protein engineering applications.
Keywords
Introduction
Trehalose, a nonreducing disaccharide in which the 2 glucose units are linked in an α,α-1,1-glycosidic linkage, is present in a wide variety of organisms, including bacteria, yeast, fungi, insects, invertebrates, and plants. 1 The ubiquitous presence of trehalose is accompanied by a wide range of different functions. In plants, a clear role of trehalose in stress tolerance, to drought in particular, has been demonstrated for cryptobiotic species, such as the desiccation-tolerant Selaginella lepidophylla. 2 Trehalose has been shown to protect the integrity of cells against environmental injuries and nutritional 3 limitations and can also be used as the sole carbon source at low and high osmolality in Escherichia coli. 4 Moreover, bacteria both appropriate exogenous trehalose as an energy source and synthesize enormous amounts as a compatible solute. Some mycobacteria also contain petrified trehalose as a structural component of the cell wall. In contrast, yeast cells are largely unable to grow on trehalose as carbon source 3 although trehalose reserves in dormant yeast spores can be used as an energy supply. 5
Trehalose is the principal sugar circulating in the blood or hemolymph of most insects as an energy store, cryoprotectant, protein stabilizer during osmotic and thermal stress, and component of a feedback mechanism regulating feeding behavior and nutrient intake. 6 In Arabidopsis, trehalose is present at almost undetectable levels; however, overexpression of the AtTPS1 gene, encoding trehalose-6-phosphate synthase (TPS), resulted in induction of stress-associated genes, including those involved in abscisic acid (a plant hormone) and glucose signaling pathways. 7 Moreover, commercial uses of trehalose include application as a stabilizing agent for preserving biomolecules such as enzymes and to preserve the freshness characteristics of dried or frozen foodstuffs. 8
Overall, 5 naturally occurring routes of trehalose biosynthesis have been identified: the OtsA-OtsB, TreP, TreS, TreY-TreZ, and Tre-T pathways. The OtsA-OtsB pathway, which is the only pathway to involve the intermediate T6P, is the most widespread, being found in all prokaryotic and eukaryotic organisms that synthesize trehalose, and is the only trehalose pathway found in plants. 9 This pathway involves 2 enzymatic steps catalyzed by TPS (EC 2.4.1.15) and trehalose-phosphatase (TPP; EC 3.1.3.12). TPS catalyzes the transfer of glucose from uridine diphosphate (UDP)-glucose to glucose 6-phosphate (G6P), forming trehalose 6-phosphate (T6P) and UDP. Subsequently, TPP dephosphorylates T6P to trehalose and inorganic phosphate.1,10 Plant TPS proteins have been shown to contain 2 essential domains: Glyco_transf_20 (Pfam: PF00982) and Trehalose_PPase (Pfam: PF02358), whereas TPP proteins contain only the PF02358 domain. 11 Also, plant TPP proteins exhibit TPP activities; however, many studies have not detected the TPP activity of plant TPS proteins.6,12,13
T6P, the direct product of TPS, had been extensively studied as a signaling metabolite for regulating carbohydrate allocation and utilization.14,15 The interaction between T6P and SNF1-Related Kinase 1/AMP-activated protein kinase (SnRK1) significantly affects source-sink relationships in plants.16-18 Increasing T6P levels in response to high sucrose levels in a cell inhibits SnRK1 activity, thus promoting anabolic processes associated with growth and yield. When T6P levels are decreased, active SnRK1 promotes catabolic processes to relocate and alter sucrose allocation in response to abiotic stress, enabling better performance.15,16 Thus, T6P targeting serves as a strategy to improve yield potential and resilience through genetic modification, 19 gene discovery via quantitative trait locus (QTL) mapping, 16 and chemical intervention 15 approaches.
Accordingly, trehalose plays an important role in metabolic regulation and abiotic stress tolerance in plants. Trehalose contents are potentially modulated by TPS, which not only constitutes a key enzyme in the trehalose biosynthetic pathway but also participates in stress signal transduction in higher plants.20,21 In yeast, the TPS enzyme can increase the efficiency of T6P control on glucose influx into yeast glycolysis. 22 In higher vascular plants, some TPS genes encode active proteins that also play important roles in plant development. 23 Specifically, higher plants contain a TPS multigene family comprising 11, 11, 12, and 28 members in Arabidopsis, rice, poplar, and Pigeon pea genomes, respectively. 24 Mutations in class I genes (AtTPS1-AtTPS4) indicate a role in regulating starch storage, resistance to drought, and inflorescence architecture. Class II genes (AtTPS5-AtTPS11) encode multifunctional enzymes exhibiting synthase and phosphatase activity. 25 The tps1 mutant in Arabidopsis, disrupting the TPS1 protein, plays a major role in coordinating cell wall biosynthesis and cell division along with cellular metabolism during embryo development. 26 Rice TPS family members may form TPS complexes and therefore potentially modify T6P levels to regulate plant development. 13 Moreover, rice TPS1 overexpression improves rice seedling tolerance to cold, high salinity, and drought treatments without other significant phenotypic changes. 27 SlTPS1, isolated from the resurrection plant S lepidophylla, encodes the functional plant homolog SlTPS1 that could sustain trehalose biosynthesis and plays a major role in stress tolerance in this plant. 28 Furthermore, a TPS gene cloned from maize demonstrated upregulated expression in response to both salt and cold stresses, 29 and 3 TPS genes were identified as being involved in maize domestication, 30 supporting that TPS plays important roles in plant growth and development.
Considering the significant functions of TPS, we conducted a comparative genome study to improve the understanding of the evolution and functions of the TPS family. In this study, we isolated TPS members from 13 higher plant species representative of the 2 major higher plant lineages. A phylogenetic tree was constructed to evaluate evolutionary relationships. Subsequently, functional divergence, positive selection, co-evolution, and conserved amino acids crucial for TPS evolution and functions were identified using bioinformatics tools. The results provide useful information for further studies regarding TPS family molecular evolution and protein engineering.
Materials and Methods
Identification of TPS members
TPS genes were identified from 13 completely sequenced plant genomes. The 11 nonredundant TPS protein sequences downloaded from the TAIR database (http://www.arabidopsis.org) were used as queries for BLASTP searches against the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html). Sequences were obtained from the following groups and species: the dicot Arabidopsis thaliana, Brassica rapa, Citrus clementine, Glycine max, Gossypium raimondii, Populus trichocarpa, Solanum lycopersicum, and Prunus persica, and monocot Brachypodium distachyon, Oryza sativa, Setaria italica, Sorghum bicolor, and Zea mays. Sequences with complete open reading frames and E values ⩽ 1e-5 were selected as candidate proteins. Redundant genes were removed manually. All candidate proteins were then verified using online tools Pfam (http://pfam.xfam.org/) and SMART (http://smart.embl-heidelberg.de/) to detect the typical domains of the TPS protein. We finally identified 150 genes and submitted all to the ExPASY database (https://www.expasy.org/) to predict the isoelectric point (pI) and molecular weight (Mw).
Phylogenetic tree construction and structure analysis
TPS protein sequences were aligned using the MUSCLE program with default parameters. 31 The phylogenetic tree was generated using the neighbor-joining and maximum likelihood methods with MEGA6.06. 32 To confirm the tree topology, Bayesian tree was constructed using MrBayes. 33 Finally, the Bayesian tree was used for further analysis. The intron-exon gene structures of these genes were obtained using the Gene Structure Display Server (GSDS: http://gsds.cbi.pku.edu.cn).
Positive selection and functional divergence
DIVERGE was applied to calculate coefficients of Type I and Type II functional divergence (θI and θII) between any 2 clusters. Also, we used posterior probability (Qk) to predict critical amino acid residues that were responsible for functional divergence (Qk > 0.9).34-36 Values of θI and θII that were significantly greater than 0 implied site-specific altered selective constraints or radical shifts in amino acid physiochemical properties following gene duplication and/or speciation.34,37 The large Qk values indicated a high probability that evolutionary rates, or site-level physiochemical amino acid properties, differed between 2 clusters. 34
Positive selection was identified using a maximum likelihood method in PAML v4.4.38,39 Two pairs of models were contrasted to test the selective pressures at codon sites. First, models M0 (one ratio) and M3 (discrete) were compared, using a test for heterogeneity between codon sites based on the dN/dS ratio value, ω. The second comparison involved M7 (beta) versus M8 (beta & ω > 1). In addition, we introduced the likelihood ratio test (LRT) to compare the 2 extreme models. When the LRT suggested positive selection, the Bayes empirical Bayes method was used to calculate the posterior probabilities that each codon was from the site class of positive selection under models M3 and M8. 40
Co-evolution of TPS amino acid sites
To identify co-evolution among amino acid sites, Co-evolution Analysis using Protein Sequences (CAPS) was performed with PERL-based software, which provides a mathematically simple and computationally feasible method of comparing the correlated variance of evolutionary rates at 2 amino acid sites corrected by time since divergence of the protein sequences to which they belong. Blosum-corrected amino acid distance was used to identify amino acid covariation. The phylogenetic sequence relationships were used to remove phylogenetic and stochastic dependencies between sites. 41
Identification of critical structural and functional sites and 3-dimensional structure prediction
The CLIPS-4D online tool 42 was used to distinguish structurally and functionally important residue positions based on sequence and 3-dimensional (3D) data. The multiple sequence alignment and 3D structure of AT1G78580 were uploaded as input information for prediction. Each prediction was assigned a P-value, which enables the statistical assessment and selection of predictions with similar quality. This program uses the 3D structure of a single protein chain to deduce the local environment of each residue and does not use the position of ligands. 42
To better study the relevance of amino acid sites based on their structure and function, both PHYRE2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) 43 and I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER) 44 were used to construct the 3D structure. Then, PyMol was used to flag the critical sites on the 3D structure. 45
Results
Collection of TPS genes
To obtain TPS members in higher plants, 11 TPS proteins from A thaliana were used as query sequences for BLASTP searches against 13 species representing 2 major plant lineages: dicot and monocot. All retrieved sequences were screened according to E value and open reading frame. The remaining sequences were further examined using SMART and Pfam databases to confirm the presence of the conserved TPS domains PF00982 and PF02358. Finally, we identified 150 TPS genes from the queried 13 species representing the 2 major lineages. The names of the TPS genes, along with their amino acid sequence length, pI, and Mw are supplied in Online Additional File 1. As indicated in the table, the protein lengths and pI values were largely variable, which suggested that various TPS proteins might exhibit a wide range of functions to accommodate different environments.
Phylogenetic tree reconstruction
To access the evolutionary relationships of TPS genes among a wide variety of plant species, the 150 full-length protein sequences were subjected to multiple sequence alignment using MUSCLE. Based on the alignment, distance-based neighbor-joining and character-based maximum likelihood phylogenetic trees were constructed using MEGA6. 32 MrBayes3.2 was used to further confirm the topology of phylogenetic tree. 5 The results revealed that all 3 phylogenetic trees shared similar topologies with only minor modifications in the terminal clades.
According to the topology of the Bayesian tree (Figure 1), the plant TPS genes could be distinctly divided into 2 major clades: clades A and B. The clade B subfamily could be further divided into 4 subclades: B1, B2, B3, and B4. This result was consistent with the previous studies.12,13,46 The topology of the phylogenetic tree was also supported by the gene structures among clades. The numbers of introns and exons in 150 TPS genes were shown using the GSDS online tool. 47 Gene structures (Online Additional File 1) in clade A were more complicated than those in clade B. Almost all clade A members contained 15 to 17 introns with 2 exceptions (Bra035049 and Bra008366, containing 9 introns), whereas all clade B members contained 1 to 3 introns; moreover, the length of TPS genes and the number of introns were strictly restrained in clade B. The gene structures revealed in our study were consistent with those from the previous studies. 46

Phylogenetic tree of 150 TPS genes from 13 species.
As indicated in Figure 1, with a minor exception, genes from eudicot species clustered together more closely than genes from monocot species, supporting the ancestral origin of the genes as reflecting divergent evolution following the monocot-eudicot separation. This result is consistent with that of a previous study. 15 All sequences from Arabidopsis tightly clustered with sequences from B rapa. Clade A included 4 Arabidopsis TPSs among which AtTPS2 (At1G16980), AtTPS3 (At1G17000), and AtTPS4 (At4G27550) clustered in the same subgroup, whereas AtTPS1 (At1G78580) was much closer to the monocot sequences. Clade B constituted 7 Arabidopsis TPSs that were distributed across the 4 subclades. The 2 maize genes Zm00001d043468 (GRMZM2G304274) and Zm00001d043469 (GRMZM2G123277), which are associated with domestication improvement, 30 located closest to AtTPS7, attributed to subclade B4. Another gene involved in maize domestication, Zm00001d032311 (GRMZM2G008226), 30 was closest to AtTPS9, attributed to subclade B3.
Identification of the functionally divergent residues in the TPS family
Two types of functional divergence (Type I and Type II) between the 5 gene subclades in the TPS family were inferred by posterior analysis using DIVERGEv3.0. 34 - 36 Type I functional divergence constitutes the evolutionary process resulting in site-specific shifts in evolutionary rates following gene duplication. Type II functional divergence represents the process resulting in site-specific amino acid physiochemical property shifts. 48 The results of Type I functional divergence were statistically significant (θ > 0, P < .01; LRT statistic > 43.526), thereby supporting the hypothesis of highly different site-specific altered selective constraint between subclades (Table 1). The results also revealed that there was evidence of Type II functional divergence between 8 subclade pairs (A/B1, A/B2, A/B3, A/B4, B1/B2, B1/B3, B1/B4, and B2/B4), indicating a radical shift in amino acid property changes.
Functional divergence between subclades of the TPS gene family.

Abbreviations: LRT, likelihood ratio test; Qk, posterior probability; TPS, trehalose-6-phosphate synthase; θI and θII, the coefficients of Type I and Type II functional divergence between any 2 gene clades.
P < .01.
We used the posterior probability to predict whether critical amino acid sites were relevant to the functional divergence between TPS subclades. A large Qk value indicates a high possibility that the evolutionary rates or physiochemical amino acid properties differ between 2 clusters. 36 To reduce the false positives, values of Qk > 0.9, P < .01 were set as a cutoff to identify Type I and Type II functional divergence–related residues between the 5 subclades (Table 1). A total of 138 nonredundant Type I functional divergence sites were predicted. The number of identified critical amino acid sites responsible for Type II functional divergence was 234, indicating that rapid change in amino acid physiochemical properties played a leading role in plant TPS functional divergence during the process of evolution (Figure 2). The large number of functionally divergent sites indicated that the TPS family underwent a strong divergence during the evolution process. Also, 87 critical amino acid sites existed both in Type I and Type II functional divergence (Figure 2), suggesting that these sites might play a key role in the subgroup-specific functional evolution of the TPS genes.

Venn diagram of Type I and Type II functional divergence as well as positively selected amino acid sites.
Positively selected residues in the TPS gene family
To identify the positive selection of specific amino acid sites in the TPS family, the site models in the CODEML program of PAML v4.4 was used to detect positive selection.38,39 Two pairs of models (M0/M3 and M7/M8) were selected and compared. Also, to test for variable omega ratios among lineages, we applied the LRT to compare the 2 extreme models. 40 The log-likelihood values under the M0 (one-ratio) and the M3 (discrete) model were determined to be −91 850.046 and −89 430.183, respectively. Twice the log-likelihood rate difference value, 2ΔlnL = 4839.726, markedly exceeded the critical value of 13.28 (P < .01), indicating that the discrete model M3 was significantly better than the one-ratio M0 model. Moreover, the log likelihood values under the M7 and M8 models were −89 241.465 and −106 676.322, respectively. Twice the log-likelihood rate difference value, 2ΔlnL = 34 869.714, also markedly exceeded the critical value of 9.21 (P < .01), indicating that some sites were under strong positive selection. As a result, 46 sites were identified to constitute positively selected amino acid sites based on the Bayes Empirical Bayes analysis in the M8 model (P < .05) (Table 2).
Positive selection among codons of TPS genes using site-specific models.
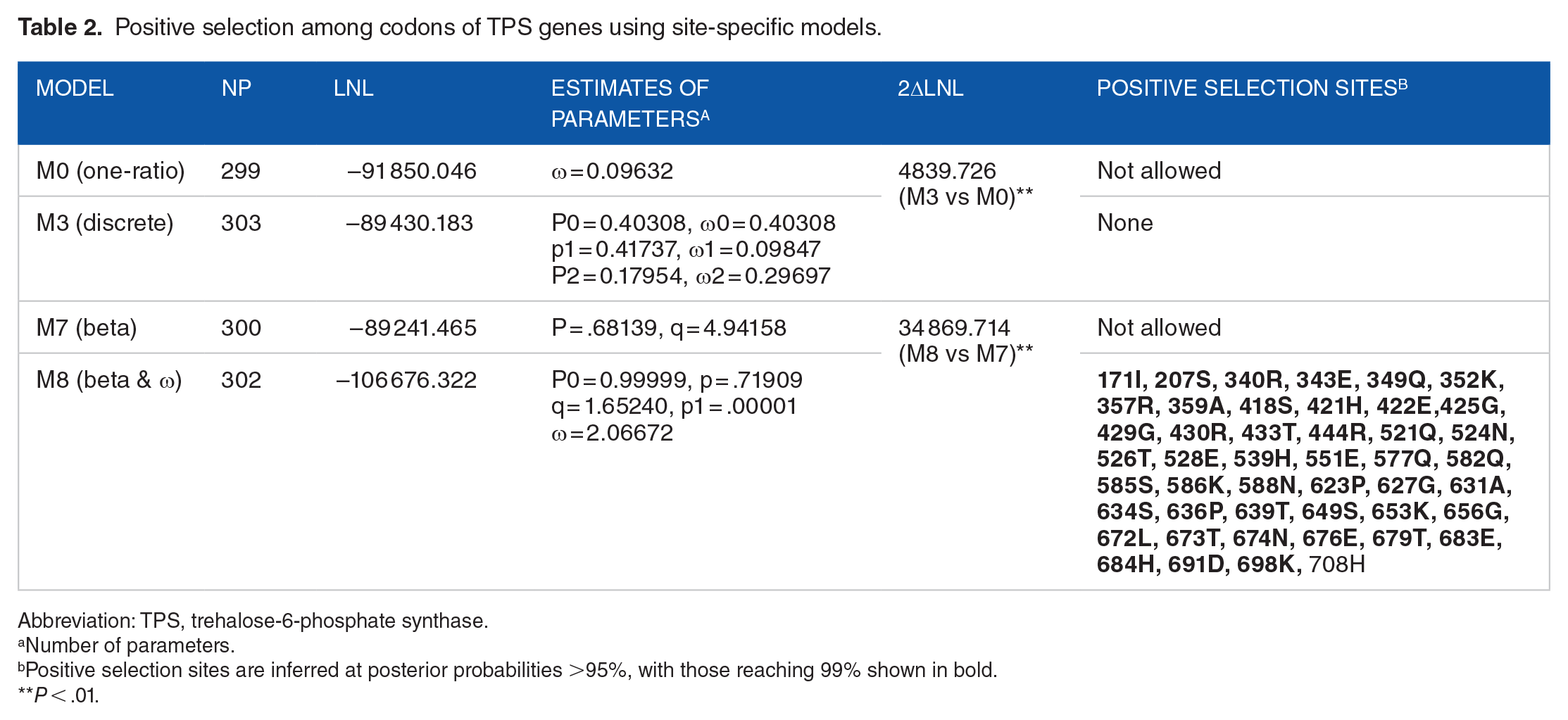
Abbreviation: TPS, trehalose-6-phosphate synthase.
Number of parameters.
Positive selection sites are inferred at posterior probabilities >95%, with those reaching 99% shown in bold.
P < .01.
Relationships between amino acid sites under positive selection and functional divergence were also compared; the results are shown in Figure 2. As indicated, sites 171I, 207S, 623P, 672L, and 698K were under both positive selection and Type I functional divergence; 20 sites were under both positive selection and Type II functional divergence. Sites 340R, 352K, 421H, 425G, 429G, 430R, 444R, 521Q, 528E, 539H, 586K, 627G, 636P, 639T, 649S, 674N, 683E, and 691D were under positive selection in addition to Type I and Type II functional divergence, suggesting that these 18 sites may play important roles in TPS family evolution. We visualized these 18 sites in the 3D structure of the reference sequence At1G78580 to investigate their structural characteristics (Figure 3). Among these sites, 11 were located in the Glyco_transf_20 domain (PF00982), and the remaining 7 sites were located in the Trehalose_PPase domain (PF02358) (Figure 3). Notably, all sites located in the PF00982 domain were involved in helix secondary structure except for 586K, whereas all sites located in the PF02358 domain were involved in loop secondary structure except for 683E. The distribution of these sites further suggested their critical roles in the evolution process of this protein family, which provides insight helpful for future research on TPS family proteins.

Critical evolutionary amino acid sites mapping in the 3D structure.
Co-evolution of TPS amino acid sites
To analyze sites of co-evolution in TPS proteins, CAPS analysis was conducted using protein multiple sequence alignment, which tends to be significantly more sensitive than other methods and robust at a wide range of amino acid distances and alignment length. 41 Three groups of co-evolved amino acid sites were identified with each group containing 2 amino acids: 11S and 12H, 33S and 34N, and 109G and 110E, respectively. Notably, these 3 group sites were adjacent concerning their primary structures, which are located in the N-terminal region of the AT1G78580 protein (Figure 4). Furthermore, no amino acid sites overlapped with those identified from the functional divergence and positive selection results.

Co-evolutionary amino acid sites mapping in the 3D structure.
Critical structural and functional sites in the TPS family
To predict the sites representing pivotal structural and functional amino acids in the TPS protein, the CLIPS-4D online tool was used to identify the catalysis, ligand-binding, or protein stability function for each residue-position of a protein. 42 We identified 77 amino acid sites using CLIPS-4D (Online Additional File 2), which were regarded as structurally and functionally conserved sites in TPS proteins. Comparison of these sites with the critical evolutionarily conserved sites detected by positive selection, functional divergence, and co-evolution revealed that none of the 77 amino acid sites overlapped with the latter except for site 713D, which was identified by both CLIPS-4D and Type I functional divergence.
Discussion
Evolution of the TPS family in higher plants
In this study, we identified 150 TPS genes from 13 species representing 2 main plant lineages by genomic analysis. A Bayesian tree including 150 protein sequences demonstrated that these genes could be divided into 2 subfamilies: clade A and clade B (Figure 1). The number of TPS genes in clade A was substantively lower than that in clade B, which was consistent with the previous research and might be due to the loss of TPS genes during the long period of evolution. 13 The classification was further supported by the exon-intron analyses. The 4 branches in clade B indicated that genes in the subclades had undergone expansion during evolution. Notably, clades A and B contained both monocotyledonous and dicotyledonous members in all 13 species, indicating that the TPS subclades might have existed as distinct entities before the divergence of monocotyledon and dicotyledon 200 million years ago. Also, all sequences from Arabidopsis tightly clustered with sequences from B rapa, indicating that they might have a similar function. For example, AT1G78580 (AtTPS1) plays a role in postembryonic development and the regulation of sugar metabolism;49-51 therefore, the 2 TPS genes in Brassica may play similar functions.
Alternatively, a large number of dicot and monocot TPS genes were clustered with AtTPS1 as a subclade in clade A, whereas only B rapa genes clustered with AtTPS2 (At1G16980), AtTPS3 (At1G17000), and AtTPS4 (At4G27550) in the same subgroup without monocot members (Figure 1), indicating that this subgroup may have a specific function in Brassicaceae different from that in other species. In addition to the induction of expression of AtTPS1 by exogenous sources of sucrose, 52 heterologous expression of AtTPS2 and AtTPS4 in the tps1 and tps2 yeast mutants restores the ability of the yeast to synthesize T6P and trehalose. 53 However, as the AtTPS2 and AtTPS4 genes are not widespread in species outside of the Brassicaceae, functions of these 2 genes in T6P synthesis are as-yet unexplored.
In contrast, clade B contained a larger number of TPS members that could be further divided into 4 subclades, B1-B4. The AtTPS5 (At4G17770) gene from clade B1 is strongly induced by sucrose, whereas other TPS genes (AtTPS8-AtTPS11) from clades B2 and B3 are strongly repressed by sucrose, yet their roles in regulating T6P levels remain unknown. 52 Nevertheless, 2 maize genes Zm00001d043468 (GRMZM2G304274) and Zm00001d043469 (GRMZM2G123277) in clade B4 and 1 gene Zm00001d032311 (GRMZM2G008226) in clade B3 are associated with domestication improvement. 30 Thus, although genes clustered in the same subclade may have similar functions, these genes from maize and Arabidopsis may have diverged independently following the monocot-eudicot separation. Moreover, in clade B4, a maize gene GRMZM2G118462, which is tightly clustered with GRMZM2G304274 and GRMZM2G123277 in a minor clade, is not associated with domestication, indicating the existence of complex functional mechanisms in this family.
Functional divergence and positive selection in the TPS family
Type I and Type II functional divergence between gene clusters of TPS subfamilies was estimated using DIVERGE analysis. Our results showed that 138 sites were predicted by Type I functional divergence and 234 sites by Type II functional divergence. A total of 86 sites were identified as co-occurring sites for both Type I and Type II functional divergence. Among these, 53 were in the conserved domain Gly_transf_20 of the N-terminal region of TPS and 33 were in the conserved domain Trehalose_PPase at the C-terminal region. The analysis showed that the Gly_transf_20 domain exhibited significant divergence, whereas the Trehalose_PPase domain was comparatively much more highly conserved. A larger number of sites exhibited Type II divergence, which indicated that the TPS family had undergone site-specific property shifts. Following gene duplication or species differentiation, the constraints on genes lead to the preservation of beneficial sequence. Moreover, multiple sites underwent both Type I and Type II divergence, especially Type II, suggesting that when selection was relaxed, more sites would be subject to evolutionary change.
Analysis of the selective pressure at the amino acid level serves as an indirect method to assess functionality. 54 Using sequences from 81 Arabidopsis accession numbers, the ratio of nonsynonymous to synonymous SNPs (single nucleotide polymorphisms) was calculated for each gene to assess selective pressure. 14 The analysis revealed that TPS5, TPS6, and TPS7 are extremely conserved with a much lower ratio of nonsynonymous to synonymous SNPs. TPS2 and TPS3 were found to contain a premature stop signal, leading to protein truncation, suggesting they are likely to be dispensable pseudogenes as suggested previously. 46 To explore whether positive selection drove the evolution of the TPS gene family, more comprehensive codon models implemented using the PAML program were used as a selective pressure test. 55 The results detected 46 sites, thus providing evidence for adaptive evolution. Among these sites, 21 were located in the conserved domain (Glyco_transf_20) of the N-terminal region and 19 were in the conserved domain of the C-terminal region (Trehalose_PPase). Furthermore, 18 sites overlapped with those flagged as exhibiting functional divergence. As these amino acid sites appear to be involved in both functional divergence and positive selection, they may, therefore, play important roles in the evolutionary process of TPS protein, providing competitive advantages. Moreover, as this evolvability property is valuable for protein engineering,56,57 targeting these sites may allow the improvement of protein stability through protein engineering.
Previously, several TPS proteins in eubacteria, archaea, plants, fungi, and animals were chosen for a selection study, 54 which indicated that TPS proteins are under strong purifying selection. However, in the present study, we found that numerous sites are under both functional divergence and positive selection. Therefore, the TPS family must maintain some functionality, perhaps related to their original enzymatic activity, and is not either in the process of becoming pseudogenes or under strong adaptive selection. 54
Also, we found that 486D in Type I coincided with the UDP binding site in bacteria and that 262T and 476E were comparable with the G6P and UDP-G binding sites in bacteria, respectively. These sites may thus play a similar role in the TPS plant family as in bacteria. 12
Co-evolution and CLIPS-4D analysis of TPS family
Unveiling the mechanisms of natural selection whereby proteins evolve constitutes a fundamental aim of evolutionary genetics studies. The identification of genes showing particular amino acid residues that have undergone adaptive evolution is the key to determining functionally or structurally important protein regions. 58 Testing for co-evolution between sites is an essential step to complement the molecular analysis and provide more biologically realistic results. Toward this end, in the present study, we detected 3 pairs of co-evolved amino acid sites: 11S and 12H, 33S and 34N, and 109G and 110E. Among these sites, 11S and 12H are located in the N-terminal region of AT1G78580 (Figure 4). The plant-specific N-terminal region may act as an inhibitory domain allowing modulation of TPS activity. 59 This pair of sites may, therefore, be constructive in maintaining the normal function of the N-terminal region. Similarly, the sites 109G and 110E are in the Glyco_transf_20 domain (Figure 4). These results demonstrated that complementary mutations existing in the co-evolved residues of TPS families might play a vital role in maintaining the structural and functional stability of TPS proteins. Moreover, the co-evolutionary relationship between each of the 2 sites in each pair represents an evolutionary limitation. There was no overlap between these pairs and the previously identified evolutionary amino acid sites, revealing that co-evolutionary amino acid sites were not involved in functional divergence and positive selection. This may reflect in part the observation that the co-evolved sites of TPS proteins play more important roles in structural and functional stability rather than divergence.
Also, CLIPS-4D analysis of the TPS proteins detected 77 sites that were related to the catalysis, ligand-binding, or stability of TPS proteins. As these sites are responsible for maintaining protein structural stability, they have therefore been subjected to selection constraints and could be considered as conservative amino acid sites.
Overall, the lack of overlapping sites among functional divergence, positive selection, and co-evolution, CLIPS-4D analyses demonstrated that 2 types of sites existed in the TPS gene family: one type exhibits both functional divergence and positive selection and is evolvable and the other type has only minimal chance to evolve, as reflected by the sites in the co-evolution and CLIPS-4D analyses. These results indicated that the evolutionary amino acid sites were rarely involved in the main structure and function of the protein. Thus, these evolutionarily conserved amino acid sites had more flexibility to alternate with other amino acids while concurrently preserving the basic structure and function of the protein. This attractive feature may provide target amino acid sites for the improvement of protein properties via gene engineering.
Conclusion
In conclusion, our study identified 150 genes in 13 higher plant species and constructed the associated phylogenetic tree, which divided the genes into 5 branches in 2 clades. We applied the DIVERGE program and identified 286 nonredundant functional divergence sites. With the use of the PAML program, 46 sites undergoing positive selection were detected. Finally, we identified 18 important sites that were subjected to both functional divergence and positive selection and were crucial evolvable sites. Conversely, 3 groups of sites noted by co-evolution and 77 sites from CLIPS-4D analyses appeared to have minimal opportunity to evolve. These results provide an improved understanding of the complexity of the TPS gene family and its function and evolution in higher plants.
Supplemental Material
Additional_File_1_Basic_information_of_TPS_members_xyz3109977a2a7da_1 – Supplemental material for Delineation of the Crucial Evolutionary Amino Acid Sites in Trehalose-6-Phosphate Synthase From Higher Plants
Supplemental material, Additional_File_1_Basic_information_of_TPS_members_xyz3109977a2a7da_1 for Delineation of the Crucial Evolutionary Amino Acid Sites in Trehalose-6-Phosphate Synthase From Higher Plants by Rong Wang, Congfen He, Kun Dong, Xin Zhao, Yaxuan Li and Yingkao Hu in Evolutionary Bioinformatics
Supplemental Material
Additional_file_2_Amino_acid_sites_predicted_by_CLIPS_xyz310992b98cb09 – Supplemental material for Delineation of the Crucial Evolutionary Amino Acid Sites in Trehalose-6-Phosphate Synthase From Higher Plants
Supplemental material, Additional_file_2_Amino_acid_sites_predicted_by_CLIPS_xyz310992b98cb09 for Delineation of the Crucial Evolutionary Amino Acid Sites in Trehalose-6-Phosphate Synthase From Higher Plants by Rong Wang, Congfen He, Kun Dong, Xin Zhao, Yaxuan Li and Yingkao Hu in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Beijing, China (6192002) and the Science and Technology Development Project of the Beijing Education Commission (KM201710028010).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RW, CH, and KD collected and verified all sequences and draw figures. RW, XZ, and YL performed the main bioinformatics analysis. YH, XZ, and YL conceived the study and planned experiments. YH, CH, and KD drafted the manuscript. All authors read and approved the final manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.