
Abstract
The 3 biological domains delineated based on small subunit ribosomal RNAs (SSU rRNAs) are confronted by uncertainties regarding the relationship between Archaea and Bacteria, and the origin of Eukarya. The similarities between the paralogous valyl-tRNA and isoleucyl-tRNA synthetases in 5398 species estimated by BLASTP, which decreased from Archaea to Bacteria and further to Eukarya, were consistent with vertical gene transmission from an archaeal root of life close to Methanopyrus kandleri through a Primitive Archaea Cluster to an Ancestral Bacteria Cluster, and to Eukarya. The predominant similarities of the ribosomal proteins (rProts) of eukaryotes toward archaeal rProts relative to bacterial rProts established that an archaeal parent rather than a bacterial parent underwent genome merger with bacteria to generate eukaryotes with mitochondria. Eukaryogenesis benefited from the predominantly archaeal accelerated gene adoption (AGA) phenotype pertaining to horizontally transferred genes from other prokaryotes and expedited genome evolution via both gene-content mutations and nucleotidyl mutations. Archaeons endowed with substantial AGA activity were accordingly favored as candidate archaeal parents. Based on the top similarity bitscores displayed by their proteomes toward the eukaryotic proteomes of Giardia and Trichomonas, and high AGA activity, the Aciduliprofundum archaea were identified as leading candidates of the archaeal parent. The Asgard archaeons and a number of bacterial species were among the foremost potential contributors of eukaryotic-like proteins to Eukarya.
Keywords
Introduction
Molecular evolution analysis of small subunit ribosomal RNAs (SSU rRNAs) yielded a universal but unrooted tree of life (ToL) that comprises the 3 biological domains of Archaea, Bacteria, and Eukarya. 1 A ToL of transfer RNAs (tRNAs) based on the genetic distances between the 20 classes of tRNA acceptors for different amino acids located the Last Universal Common Ancestor (LUCA) near the hyperthermophilic archaeal methanogen Methanopyrus kandleri (Mka). 2 The rooting is supported by a wide range of evidence,3-14 and the finding of the Methanopyrus lineage as the oldest lineage among living organisms. 15 However, the phylogenies of the 3 biological domains are beset by 2 fundamental problems regarding the evolutionary relationship between Archaea and Bacteria, and the nature of the Archaea-Bacteria collaboration that gave rise to Eukarya. As long as these 2 problems remain unresolved, the root of life and the origin of Eukarya would both be open to diverse formulations.16-20 Accordingly, the objective of this study was to examine the pathways of descent of Bacteria and Eukarya from an archaeal LUCA and the identity of the plausible archaeal parent of Eukarya.
Materials and Methods
Source of data and materials
Protein and SSU rRNA sequences were retrieved from NCBI GenBank release 231 (ftp://ftp.ncbi.nlm.nih.gov/genomes/).21,22 For species without available SSU rRNA information in NCBI, quality checked SSU rRNA sequences were downloaded from the SILVA database release 132 (https://www.arb-silva.de/). 23 For species with multiple SSU rRNA sequences, the one yielding the highest total bitscore (using BLASTN 24 with “-word_size” flag set to 4) with SSU rRNAs of other species from the same domain was employed for analysis. The accession numbers of SSU rRNAs analyzed were available in File S1 in Supplementary Materials. Eukaryotic mitochondrial DNA-encoded protein sequences were retrieved from the RefSeq mitochondrial reference genomes in the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein).
Estimation of nuclear or mitochondrial proteome similarity bitscores
When comparing proteome similarities, the proteomes of all subject species were used to construct a local BLAST database using makeblastdb, 24 and every query proteome is searched against the local database using BLASTP with a BLOSUM62 matrix and thresholds setting to evalue <1 × 10−5, percent identity >25%, and query coverage >50%. Only the query and subject sequences that were the best match of each other, viz when query sequence n from species 1 exhibited the highest bitscore toward subject sequence m among all proteins of species 2 and vice versa, were included in the estimation of inter-proteome similarity, which was given by the sum of BLASTP bitscores of all such best-matched proteins between the 2 proteomes.
Estimation of rProt similarity bitscores
To identify rProt sequences in Gla, Trv, Sce, and Hsa (see species name abbreviations in Table 1), eukaryotic proteomes were cleared of mitochondrial or mitochondrial DNA-encoded proteins, and then searched against the Pfam database 25 using RPSBLAST 24 at a threshold set by the “-evalue” flag at 0.01. For each of the 88 rProt families analyzed (Table S1), only the protein sequence from each species that yielded the highest bitscore toward the rProt family was analyzed further. On this basis, 79, 81, 84, and 86 out of the 88 rProt families were found in the Gla, Trv, Sce, and Hsa proteomes, respectively. These eukaryotic rProts were blasted against all the prokaryotic proteomes using BLASTP. Prokaryotic proteins passing the threshold of evalue <0.05 were searched against the Pfam database using RPSBLAST, and false-positive sequences that failed to map to the targeted rProt family were removed. The similarities between the rProt sequences identified from eukaryotes and prokaryotes were estimated based on the maximum BLASTP bitscores.
Partial list of species analyzed.
Note: C. in front of species name stands for Candidatus. Detailed species information is given in Table S2.
Estimation of non-rProt similarity bitscores
To identify Gla-like protein families in various prokaryotes, every sequence in the Gla proteome was blasted against the 82 prokaryotic proteomes in Table 1 (except for Psy from preprint form), and the best matches passing the threshold of evalue <0.05 were mapped to the Pfam database using the NCBI Batch CD-search Tool. 26 To remove false-positive pairs, only cases where both query and subject sequences belonged to the same targeted protein family were analyzed, and the Gla sequences that were relatively rare in prokaryotes, displaying similarity bitscores toward ⩽10 out of the 82 prokaryotic proteomes tested, were classified as Gla-like proteins.
Results and Discussion
Similarity between VARS-IARS paralogues
The relative antiquity of proteins could be approximated, except for proteins that have undergone extraordinarily extensive evolution, based on the increasing divergence of paralogous proteins in time. 27 Accordingly, BLASTP was performed between the intraspecies valyl-tRNA synthetase (VARS) and isoleucyl-tRNA synthetase (IARS) in the genomes of 5398 species in NCBI Genbank. When the bitscores obtained were arranged in descending order (Table S2), or in part on a distribution curve (Figure 1), Mka yielded a top bitscore of 473. BLASTP, which provided indication of similarity but not necessarily phylogenetic relationship, 28 was a fitting tool for evaluating the intracellular divergence of VARS-IARS which carried no phylogenetic implication: 2 neighboring species on the distribution curve could belong to 2 different biological domains. As the 119 highest scoring species were all archaeons, the top-scoring bacterium Mau gave only a bitscore of 378 and the top-scoring eukaryote Esi gave only a bitscore of 240, the smallest VARS-IARS divergences were clearly confined to Archaea, in keeping with the descent of Bacteria from Archaea, and descent of Eukarya from either Archaea or an Archaea-Bacteria collaboration. The foremost antiquity of Mka indicated by its bitscore was in accordance with the Mka-proximal LUCA identified by the genetic distances between alloacceptor tRNAs, 2 and the unchanging environment throughout the ages at the hydrothermal vents inhabited by Mka. It was also consistent with the datings of the sn1,2 chemistries of archaeal lipids, and the core of archaeal formylmethanofuran dehydrogenase, prior to the rise of LUCA. 29

Ranking of similarity bitscores of intraspecies VARS-IARS for various species in descending order (from left to right). The bitscores for 1185 archaeal, 3621 bacterial, and 592 eukaryotic species from NCBI are given in Table S2. IARS indicates isoleucyl-tRNA synthetase; NCBI, National Center for Biotechnology Information; VARS, valyl-tRNA synthetase.
The positions of some of the species analyzed in Figure 1 were indicated on the SSU rRNA tree, with their intraspecies VARS-IARS bitscores expressed in circles colored according to the thermal scale (Figure 2A).
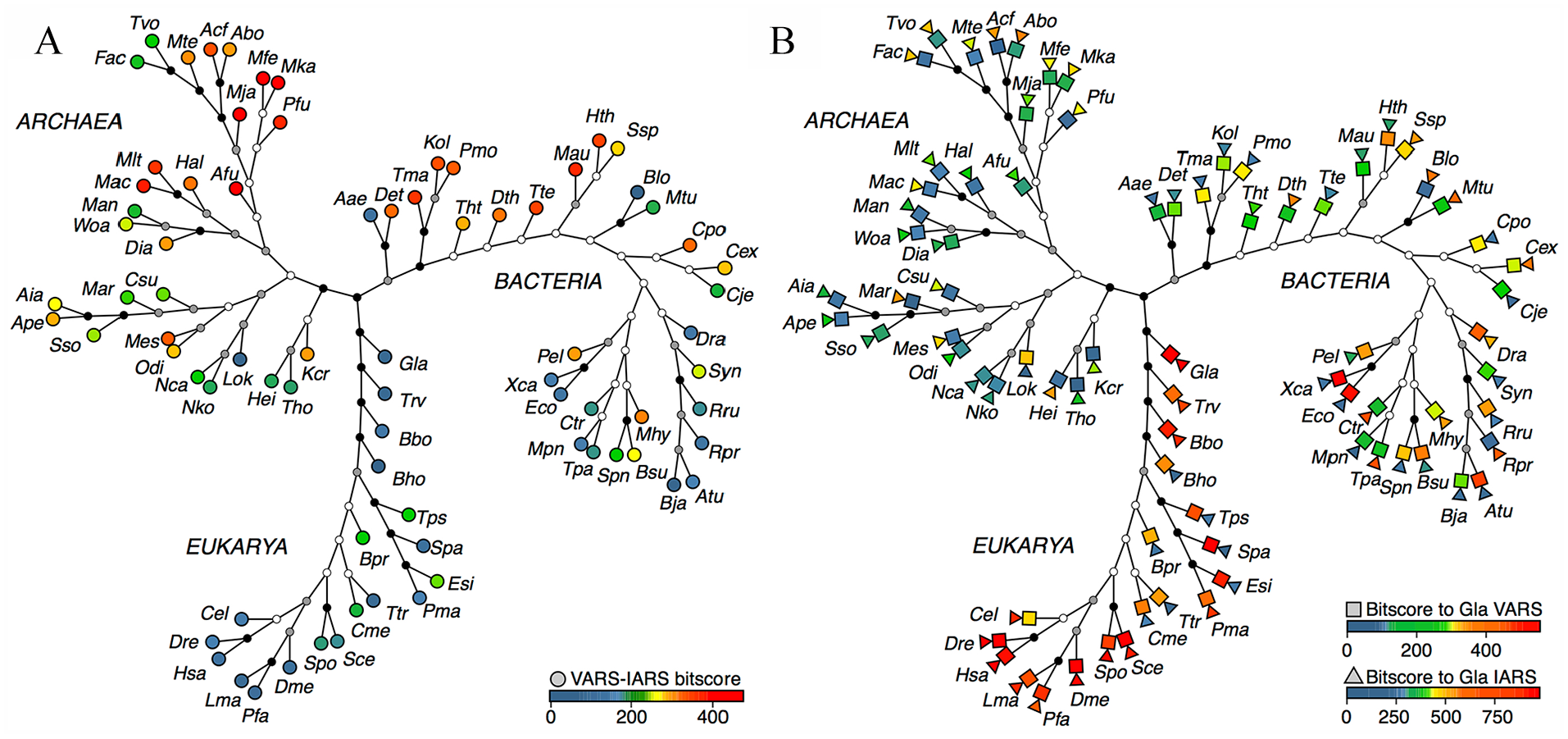
Distribution of similarity bitscores relating to VARS and IARS on SSU rRNA tree. (A) Bitscores for VARS-IARS pairs. (B) Bitscores for VARS (squares), or IARS (triangles), between Gla and other organisms. For building the consensus maximum parsimony tree of SSU rRNAs for 29 archaeal, 31 bacterial, and 19 eukaryotic species using PHYLIP version 3.698, 30 the sequences were aligned in Clustal Omega. 31 One thousand sets of bootstrap-resampled sequence alignments were generated using SEQBOOT and inputted into DNAPARS to construct maximum parsimony trees. The consensus tree was produced based on the 1000 sets of maximum parsimony trees using CONSENSE. The nodes indicate more than 85% bootstrap support (black), more than 50% (gray), or less than or equal to 50% (white). IARS indicates isoleucyl-tRNA synthetase; SSU rRNA, small subunit ribosomal RNA; VARS, valyl-tRNA synthetase.
There was a concentration of euryarchaeons with high VARS-IARS similarity in a “Primitive Archaea Cluster” centered between Pfu and Mac. In the Bacteria domain, there was likewise a concentration of species with high VARS-IARS similarity in an “Ancestral Bacteria Cluster” centered between Det and Hth. The deepest branching species in the Bacteria domain were 2 members of the Aquificae phylum, viz the anaerobic Det with high VARS-IARS similarity, and the microaerobic Aae with low similarity. As mutations could cause loss of similarity more easily than gain, this suggests that Aae has evolved far from the ancestral Aquificae species possibly as part of the wave of radical changes undergone by some former anaerobes in response to the appearance of atmospheric oxygen,32,33 thereby sustaining extensive evolutionary erosion of its VARS-IARS similarity. The enhanced resistance of paralogue similarity to perturbation by horizontal gene transfer (HGT), due to the difficulty of transfer of a pair of genes compared to the transfer of a single gene, was illustrated by the preservation of low VARS-IARS bitscores in the proteobacterial region of the tree against large shifts caused by HGT events.
Given the relative paucity of HGT effects on VARS-IARS similarity, the parallel prominences of high VARS-IARS similarity-bitscore species in the Primitive Archaea Cluster and the Ancestral Bacteria Cluster were explicable by vertical genetic transmission of the VARS and IARS genes from an Mka-proximal root of life to the archaeal cluster, and in turn to the bacterial cluster. As the top-ranked bacterial bitscore of Mau at 378 was between those of archaeons Mac at 382 and Pfu at 369, the results indicated that the Ancestral Bacteria Cluster branched off from the Primitive Archaea Cluster near the Mka-proximal root of life. The medium VARS-IARS bitscores of Esi, Tps, Bpr, and Cme among the Eukarya (Figure 2A) also pointed to the conservation of intraspecies VARS-IARS similarity in this domain. The much higher VARS (colored squares) and IARS (colored triangles) bitscores between Gla and various bacterial species compared to archaeal species, except for the high similarity exhibited by Gla IARS toward that of Abo, suggests that Eukarya received VARS from Bacteria and IARS from Abo or a bacterium (Figure 2B).
Sequence alignments
The aligned segments of VARS and IARS (Figure 3) from Mka, Mau, and Esi, viz the archaeon, bacterium, and eukaryote displaying the highest VARS-IARS similarity within their respective domains, included 42 of 207 columns where all 6 sequences carried the same amino acid, in support of sequence conservation of this pair of paralogous genes among all 3 living domains. Together with the higher rankings of VARS-IARS similarity attained by archaeons relative to both bacteria and eukaryotes (Figure 1), the sequence conservation observed represented strong evidence for the vertical transmission of the VARS and IARS genes from Archaea to both Bacteria and Eukarya.

Segments of the aligned VARS and IARS sequences of Mka, Mau, and Esi. Sequences were aligned using Clustal Omega, and the numbers indicate the positions of amino acid residues on the complete sequence alignment (Figure S1). Similar amino acids in the same column are colored in orange, and ⩾50% conserved ones in blue. Asterisks mark the 6 positions where a V or L residue is found in all 6 sequences. IARS indicates isoleucyl-tRNA synthetase; VARS, valyl-tRNA synthetase.
Process of eukaryogenesis
Extensive evidence supports that an endosymbiotic event between an archaeal parent and an alphaproteobacterium played a key role in the development of Eukarya.34,35 Proposals regarding the identity of the archaeal parent have focused on a range of archaeons including Thermoplasmata where the lack of a rigid cell wall could facilitate engulfment of the alphaproteobacterium36,37; and the Asgard archaeons38,39 that were enriched in eukaryotic signature proteins (ESPs). 40 There is a phylogenomic impasse regarding these, as well as other, choices.41,42
Upon BLASTP comparisons of the 79 Gla, 81 Trv, 84 Sce, and 86 Hsa rProt families with prokaryotic rProts, 69/69 Gla, 71/72 Trv, 71/72 Sce, and 71/71 Hsa ones with prokaryotic resemblance showed higher similarity toward archaeons than bacteria; thus, only 1 of 72 of Trv (rProt L29) or Sce (rProt S4) ones showed higher similarity toward bacteria than archaeons (Figures 4A and S2), clearly indicating that eukaryogenesis was hosted by an archaeal parent instead of a bacterial parent.36,37 Those rProts in Table S1 without any prokaryotic resemblance might be derived from a prokaryote not analyzed in this study, invented by the eukaryogenic lineage, or diminished in their resemblances by evolutionary changes to beyond recognition by BLASTP.

Protein sequence similarities between Gla and prokaryotic species. (A) Maximum BLASTP bitscores between Gla rProts and prokaryotic rProts. (B) Bitscores of PEP-utilizing enzyme mobile domain (PF00391) between Gla and prokaryotes. (C) Bitscores between some of the Gla-like proteins from Table S3 and potentially homologous proteins in various prokaryotes. (D) Numbers of the 162 Gla-like proteins found in various prokaryotes. The color coding and order of different prokaryotic species on the x-axis in (B), (C), and (D) are the same as those in (A). PEP indicates phosphoenolpyruvate.
Among the 6502 proteins in the Gla proteome, 3203 of them showed finite similarity bitscores toward the sequences of one or more of the 82 prokaryotes tested, and the phosphoenolpyruvate (PEP)-utilizing enzyme mobile domain of Gla yielded the highest combined BLASTP bitscore of any Gla protein toward prokaryotic protein families, with Acf, Abo, and Mac (2nd, 1st, and 14th red columns from the right in Figure 4B) showing the top 3 archaeal bitscores. The bitscores were high for Tho and Hei but low for Odi and nil for Lok (3rd, 4th, 2nd, and 1st purple columns from the right) among the Asgard archaea, and high for Tvo and Tac but low for Mte, Min, and Fac among the Thermoplasmata (5th, 6th, 3rd, 4th, and 7th red columns from the right).
Figure 4C shows the distribution of potential archaeal and bacterial homologues of some of the 162 Gla-like proteins that were either ESPs or relatively rare proteins found in less than 10 of the 82 prokaryotes analyzed (Table S3). The Asgard archaeons (purple columns) and a number of bacterial species (green columns) were prominently endowed with the ESPs or rare proteins required for eukaryogenesis (Figure 4D and Table S4). However, the highest scoring Tho, Odi, Xca, and Lok in this regard harbored only 26, 19, 17, and 16 of the 162 Gla-like proteins, respectively, which underlined the difficulty for any archaeon or bacterium to accumulate a sufficient number of eukaryote-type proteins to launch the Eukarya domain by itself. On the other hand, it was impressive that one or more potential prokaryotic sources could be located for each of the 162 Gla-like proteins targeted despite the modest spectrum of prokaryotes analyzed in Figure 4C, demonstrating that the obstacle to eukaryogenesis posed by an ESP deficit could be overcome readily if some efficient mechanism was available for collecting the requisite protein genes from a broad spectrum of prokaryotes. With respect to the problem of inadequacy of ESPs occurring in any single archaeon,43,44 it was suggested that HGTs might provide a solution, 39 but the actual adoption of HGT-transferred genes by recipients might be a limiting factor, 45 as illustrated by the fact that few members of the alphaproteobacterial and Asgard groups had spread a large fraction of their Gla-like proteins to all other members of the same group through HGTs (Figure 4C).
Nature of archaeal parent
Eukaryogenesis could follow a mitochondria-early scenario or a mitochondria-late scenario, 46 and there is no consensus on these 2 scenarios.47-49 Previously, the proteome of the eukaryote Sce was found to contain a rich variety of bacterial proteins, and also some archaeal ones, and it was suggested that the influx of bacterial genes into Sce was not explicable by a merger between archaeal parent and another bacterium besides an alphaproteobacterium, or by uptake of bacterial genes through ingestion of bacteria as food. 35 When the eukaryotic Gla and Trv proteomes were employed as probes for BLASTP query against various prokaryotic proteomes, they gave rise to so many hits with a range of archaea and bacteria (Table S5) that the influx of bacterial and archaeal genes into the eukaryogenic lineage would need to be mediated by some specially efficient form of HGT. Comparable yet nonidentical spectra of inter-proteome similarities were exhibited by Gla and Trv toward the prokaryotes, with archaeal bitscores surpassing bacterial ones in the case of Gla but vice versa in the case of Trv (Figure 5A). It was suggested that actin-associated proteins and regulators were introduced into archaea from diverse bacteria 50 ; and the influx of a large number of bacterial genes into a methanogen was found to precede its evolution into the haloarchaeans. 51 Accordingly, an influx of prokaryotic genes into the eukaryogenic lineage, likely beginning prior to the emergence of the archaeal parent and continuing through to the Last Eukaryotic Common Ancestor (LECA) and the early eukaryotes, could play a crucial role in eukaryogenesis.

Inter-proteome similarity bitscores. (A) Total similarity bitscores of Gla and Trv proteomes toward individual prokaryotic proteomes. Relationships of average bitscore per best-match hit (y-axis) with the number of best-match hits (x-axis): (B) between prokaryotic and Gla proteomes and (C) between prokaryotic and Trv proteomes.
Based on the premise that the free-living archaeal parent might still retain recognizable similarity toward eukaryotes, 46 archaeal proteomes were compared regarding their relationships with the proteomes of Gla and Trv. Figure 5B and C showed that the proteome of the Aciduliprofundum archaeon Abo displayed the highest average similarity bitscores among archaeons toward the proteomes of both Gla and Trv, which identified Abo and its companion species Acf as candidate archaeal parents. The Asgard archaeons Hei, Odi, Tho, Lok, and the cultivatable Psy52,53 constituted an unusually inventive group with both some high average similarity bitscores and a rich store of ESPs, even though their average similarity bitscores were lower than those of Abo. Among all the prokaryotic species, Psy also yielded the highest number of similarity hits toward both Gla and Trv, indicating that the archaeal parent contained more genes derived from Psy than any other archaeon. For the bacterial species, once any bacterial protein entered into the eukaryogenic lineage, its eukaryotic version and free-living bacterial version became segregated irreversibly and evolved independently; the divergence between the 2 versions would increase with time as in the case of paralogues such as VARS and IARS. Accordingly, the higher inter-proteome bitscores of Tpa toward Gla and Trv compared to Mpn could be at least in part the result of later entry of Tpa genes than Mpn genes into the eukaryotes. These findings thus suggest that the entries of various bacterial proteins at different times into the eukaryogenic lineage would furnish useful landmarks for deciphering the chronicle of eukaryogenesis. The determinants of the bitscores of archaeons outside of the archaeal parent were more complex, for they would depend not only on the time of entry of their proteins into the eukaryotes but also on the extent of their kinship with the archaeal parent.
When the bacterial-gene contents of different archaeons were compared regarding their abilities to acquire bacterial genes, Hla, Hgi, and Mac with their large proteomes (3704 to 4469 protein-coding genes) displayed high similarity bitscores toward a wide range of bacteria (Figure 6, left panel). However, when the bitscore of each archaeon was normalized with respect to the number of protein-coding genes in its genome, the normalized bitscores of the smaller Abo, Acf, Mte, Tvo, and Tac (each with <1600 protein-coding genes), Mfe (1283 protein-coding genes), and Mlt (1291 protein-coding genes) became more prominent (Figure 6, right panel). The medium-sized Pfu (2065 protein-coding genes) gave much the same result with or without normalization. Notably, the high similarity bitscores exhibited by these archaeal proteomes toward multiple bacterial proteomes suggest that they had efficiently adopted exogenous genes received by them from HGT into their own genomes. In contrast, the bacterial proteomes of Bja, Tht, Pel, Dth, Tte, and the DNA transformation-active Bsu exhibited only modest bitscores toward smaller number of archaeons. This enhanced ability of some archaeons to adopt exogenous genes may be referred to as an accelerated gene adoption (AGA) phenotype. The prominence of AGA in some archaeons was consistent with the finding that 44% of Mja gene products were derived from bacteria. 54 A possible determinant of the AGA phenotype could be the “Darwinian Threshold,” viz organisms below a given threshold level of organizational connectedness adopt genes received from HGTs more readily than organisms above the threshold. 55 Other determinants might include a full-fledged or partial scavenger lifestyle, 56 tetraethers in their membranes,56,57 or the presence of rudimentary phagocytosis.58,59 Previously, it was suggested that eukaryotes could ingest bacteria as proto-organelles, and upon lysis transfer their genes to the eukaryotic nuclear genome through a recycling rachet.60,61 The plausible deployment of the dissimilar AGA and recycling rachet mechanisms for gene transfer in eukaryogenesis underlines the significance of prokaryotic genes in eukaryogenesis. Importantly, the bacterial species Rpr, Bap, Ctr, Mpn, and Tpa furnished few genes to the AGA-active archaeons (Figure 6), and their proteins were also depleted in the proteomes of both Gla and Trv (Figure 5A), clearly indicating that AGA played a major role in governing the entry of bacterial genes into Eukarya.

Similarity bitscores between archaeal proteomes (y-axis) and bacterial proteomes (x-axis) without (left) or with (right) normalization based on the number of protein-coding genes in each archaeon. Data for the heat maps are given in Table S6.
On account of the large variety and numbers of prokaryotic genes to be included in eukaryotic genomes (Figure 5A), it would be essential for the archaeal parent to be highly active in AGA, so that it could assemble beneficial genes from wide ranging prokaryotic sources and incorporate them into its own genome in the course of eukaryogenesis. Besides AGA activity, Abo the first cultivatable archaeon from the “Deep-sea hydrothermal vent euryarchaeotic 2” (DHVE2) group, and its facultatively anaerobic companion species Acf,57,62,63 possess an exceptionally flexible cell surface which can form small blebbing vesicles that bud off and anneal with other cells. While all prokaryotic cells evolve on the basis of nucleotidyl mutations through the replacement, addition, and subtraction of nucleotides, AGA would enable the archaeal parent to evolve on the basis of gene-content mutations as well through the replacement, addition, and subtraction of genes, or gene clusters, expediting eukaryogenesis by orders of magnitude. The AGA-active Tac for example succeeded in acquiring gene clusters from other organisms for rProts, NADH dehydrogenase, precorrin biosynthesis, flagellar proteins, and a protein degradation pathway amounting to 32% of its total open reading frames via its AGA which was considerably less active than that of Abo and Acf (Figure 6, right panel). 56 The blebbing vesicles of Abo and Acf could further mediate gene exchanges between individual cells engaged in eukaryogenesis to advance the process. Overall, therefore, based on their highest archaeal BLASTP bitscores toward the PEP-utilizing enzyme mobile domain of Gla (Figure 4B), highest average archaeal bitscores toward the Gla and Trv proteomes (Figure 5B and C), front-rank AGA activity, blebbing membrane vesicles, and almost complete Embden-Meyerhof-Parnas pathway 62 that could evolve readily into a glycolytic pathway to link up with mitochondrial respiration, Abo and Acf were endowed with a range of advantageous attributes as candidates for the archaeal-parent role. 64 Acf and Abo are highly similar, although the facultatively anaerobic nature of Acf could enable it to explore more ecological niches than anaerobic Abo to collect and adopt useful genes from HGT donors.
Similarity bitscores displayed by the proteomes of 225 different archaeons, alphaproteobacterial genera, and other bacteria toward the total mitochondrial DNA-encoded proteins of different eukaryotes indicated that the prokaryotic proteomes displaying top similarity toward each of 19 mitochondrial proteomes were all alphaproteobacterial ones (Table S7). The distributions of the bitscores of the prokaryotic proteomes toward the mitochondrial DNA-encoded proteins of R americana, M paleacea, and P falciparum, viz mitochondria with the highest total score, mitochondria with the second highest total score, and the mitochondria with a small number of mitochondrial DNA-encoded proteins, respectively, are illustrated in Figure 7; the 3 top-scoring alphaproteobacteria in each instance are indicated with their bitscores in parentheses. These findings demonstrated the dominance of alphaproteobacterial precursors in mitochondrial evolution among extant eukaryotes.

Similarity bitscores between mitochondrial DNA-encoded proteins and prokaryotic proteins. Total bitscores displayed by 46 archaeons, 150 alphaproteobacterial genera, and 29 other kinds of bacteria toward 3 species of mitochondrial DNA-encoded proteins are shown in the 3 panels. In each case, the 3 top-scoring prokaryotes are indicated with their individual total bitscores inside parentheses.
Conclusions
In this study, Methanopyrus kandleri was found to be the top-ranked organism with respect to the similarity between intraspecies VARS-IARS among 5398 species from the 3 biological domains and therefore closest to LUCA. Moreover, the parallel clusters of archaeal and bacterial species with high VARS-IARS similarity delineated a pathway of descent of these genes from the Primitive Archaea Cluster to the Ancestral Bacteria Cluster, branching early from the Archaea domain. The asterisked columns in Figure 3, where all 6 aligned protein sequences uniformly showed a Val or Leu residue despite the ease with which Val, Leu, and Ile can be interchanged in evolution, conveyed a surprising level of protein sequence conservation across 2 different proteins, 3 biological domains, and a time span of more than 2 billion years in support of the descent of Bacteria and Eukarya from an archaeal root of life. With respect to eukaryogenesis, the preeminent eukaryotic-archaeal similarities pertaining to rProts compared to eukaryotic-bacterial similarities showed that the prokaryotic parent which hosted the process of eukaryogenesis was an archaeal parent rather than a bacterial parent. Evidence suggests that the archaeal parent was an archaeon enriched with eukaryote-homologous proteins and expert in the acquisition of exogenous genes through AGA, as exemplified by the Aciduliprofundum archaeons.
Supplemental Material
FigureS1-S2_xyz322784b82bfeb – Supplemental material for Descent of Bacteria and Eukarya From an Archaeal Root of Life
Supplemental material, FigureS1-S2_xyz322784b82bfeb for Descent of Bacteria and Eukarya From an Archaeal Root of Life by Xi Long, Hong Xue and J Tze-Fei Wong in Evolutionary Bioinformatics
Supplemental Material
TableS1-S7_xyz3227876cd61d9 – Supplemental material for Descent of Bacteria and Eukarya From an Archaeal Root of Life
Supplemental material, TableS1-S7_xyz3227876cd61d9 for Descent of Bacteria and Eukarya From an Archaeal Root of Life by Xi Long, Hong Xue and J Tze-Fei Wong in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Innovation and Technology Commission of Hong Kong SAR (grant number ITS/113/15FP).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
JT-FW and HX conceived the study; XL collected the data and performed computational analysis; and JT-FW, HX and XL wrote the paper. All authors read and approved the final manuscript.
Data Availability
Supporting data for the present study are provided in online Supplementary Materials.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.