
Abstract
Background:
Increasing evidence has indicated that protein-protein interactions (PPIs) play important roles in various aspects of the structural and functional organization of a cell. Thus, continuing to uncover potential PPIs is an important topic in the biomedical domain. Although various feature extraction methods with machine learning approaches have enhanced the prediction of PPIs. There remains room for improvement by developing novel and effective feature extraction methods and classifier approaches to identify PPIs.
Method:
In this study, we proposed a sequence-based feature extraction method called LCPSSMMF, which combined local coding position-specific scoring matrix (PSSM) with multifeatures fusion. First, we used a novel local coding method based on PSSM to build a new PSSM (CPSSM); the advantage of this method is that it incorporated global and local feature extraction, which can account for the interactions between residues in both continuous and discontinuous regions of amino acid sequences. Second, we adopted 2 different feature extraction methods (Local Average Group [LAG] and Bigram Probability [BP]) to capture multiple key feature information by employing the evolutionary information embedded in the CPSSM matrix. Finally, feature vectors were acquired by using multifeatures fusion method.
Result:
To evaluate the performance of the proposed feature extraction approach, we employed support vector machine (SVM) as a prediction classifier and applied this method to yeast and human PPI datasets. The prediction accuracies of LCPSSMMF were 93.43% and 90.41% on the yeast and human datasets, respectively. Moreover, we also compared the proposed method with the previous sequence-based approaches on the yeast datasets by using the same SVM classifier. The experimental results indicated that the performance of LCPSSMMF significantly exceeded that of several other state-of-the-art methods. It is proven that the LCPSSMMF approach can capture more local and global discriminatory information than almost all previous methods and can function remarkably well in identifying PPIs. To facilitate extensive research in future proteomics studies, we developed a LCPSSMMFSVM server, which is freely available for academic use at http://219.219.62.123:8888/LCPSSMMFSVM.
Keywords
Introduction
Proteins are one of the most fundamental elements in living organisms and make important contributions in nearly all fundamental biological processes in the cell. However, proteins perform their functions by interacting with other proteins. Increasing studies have indicated that protein-protein interactions (PPIs) play a critical role in many important biological processes, including transcription regulation, signal transduction, foreign molecules recognition, translation, and so on. As indicated by increasing evidences, knowledge of PPIs can provide a deeper understanding of the molecular functions of biological processes, suggest novel methods for their practical use in medicine, and yield insight into disease mechanisms. Although many high-throughput methods, including the yeast 2-hybrid system,1-4 protein chips,5-7 and immunoprecipitation,8-10 are typically used to identify PPIs, experimental methods for identifying associations between proteins are expensive and time-consuming and suffer from high rates of false positives and false negatives.11-15 Thus, increasing numbers of studies have focused on computational approaches to identify PPIs.16-18 As a result, a large number of computational approaches using multiple data types, including protein domain, and genomic protein structure information have been developed to predict PPIs. However, most of these methods are limited due to difficulty in computing and dependence on a large number of homologous proteins.7,13,19-21 Therefore, it is very important to identify the close relationship between proteins by exploiting efficient computational approaches based on protein sequence information.
To date, many computational approaches based on protein information have been developed for predicting PPIs.19,22-24 An et al 25 proposed a novel feature extraction method based on protein sequence called Local Phase Quantization (LPQ), which was combined with RVM classifier to detect PPIs. Xia et al 26 developed a computational approach, which used rotation forest as a classifier and autocorrelation to represent protein sequences for identifying PPIs. Guo et al 27 exploited a new computational approach by combining autocovariance (AC) with SVM, where AC could take advantage of the interactions between residues a certain distance apart in the sequence. Yu et al 28 proposed a novel feature extraction method called local descriptors (LD), which accounts for the interactions between sequentially distant but spatially close amino acid residues and can fully capture multiple feature information in region in continuous and discontinuous regions within a protein sequence. Huang et al 29 proposed a novel computational model based on protein sequence by using weighted sparse representation as a prediction classifier (WSRC) and employed global encoding (GE) as feature extraction approach for predicting PPIs, which achieved better prediction results. However, there is still room to improve the prediction accuracy of the existing methods.
In this study, we proposed a sequence-based feature extraction method called LCPSSMMF, which combined local coding position-specific scoring matrix (PSSM) with multifeatures fusion. First, we used a novel local coding method based on PSSM to build a new PSSM (CPSSM); the advantage of this method is that it incorporated global and local feature extraction, which can account for the interactions between residues in both continuous and discontinuous regions of amino acid sequences. Second, we adopted 2 different feature extraction methods (Local Average Group [LAG] and Bigram Probability [BP]) to capture multiple key feature information by employing the evolutionary information embedded in the CPSSM matrix. Finally, feature vectors were acquired by using multifeatures fusion method.
Result
To evaluate the performance of the proposed feature extraction approach, we employed support vector machine (SVM) as a prediction classifier and applied this method to yeast and human PPI datasets. The prediction accuracies of LCPSSMMF were 93.43% and 90.41% on the yeast and human datasets, respectively. Moreover, we also compared the proposed method with the previous sequence-based approaches on the yeast datasets by using the same SVM classifier. The experimental results indicated that the performance of LCPSSMMF significantly exceeded that of several other state-of-the-art methods. It is proven that the LCPSSMMF approach can capture more local and global discriminatory information than almost all previous methods and can function remarkably well in identifying PPIs. To facilitate extensive research in future proteomics studies, we developed an LCPSSMMFSVM server, which is freely available for academic use at http://219.219.62.123:8888/LCPSSMMFSVM.
Materials
Dataset
In this study, yeast and human datasets that can be obtained from the publicly available Database of Interacting Proteins (DIP) were used to evaluate the proposed method. 30 To better carry out our method, some protein sequence pairs were removed, if they were fragments with less than 50 residues in length. For eliminating bias of homologous protein sequence pairs, sequence pairs with ⩾40% sequence identity were considered to be homologous. Thus, these protein sequence pairs were also removed. As a result, we constructed the yeast dataset, which contained 5594 positive protein pairs and 5594 negative protein pairs. In the same way, the human dataset was constructed, which contained 3899 positive protein pairs and 4262 negative protein pairs. Consequently, the yeast dataset contains 11 188 protein pairs and the human dataset contains 8161 protein pairs.
Feature Extraction Method
From a computational perspective, the key to identify PPIs is to develop an effective computational approach based on protein sequences, which is usually divided into the following 2 steps:
Step 1. Analyzing and designing a feature extraction method that not only captures protein-protein interaction information but also extracts more key feature information contained in the protein sequence.
Step 2. Designing and selecting an appropriate prediction classifier.
The above-mentioned 2 steps are closely related and complement each other. A drawback of the design process of either step is that bias will be introduced that may influence the performance of the prediction model. The pattern of feature extraction is generally divided into 2 classes: (1) the original protein sequence is directly represented as a feature vector by mathematical description and (2) a given protein sequence is first transformed a matrix; and second, the feature vector is created by mathematical description based on the sequence matrix.
Increasing studies have demonstrated that prediction accuracy of the second class is better than that of the first class. As a result, we presented a new feature extraction method based in this article on the second class.
Position-specific scoring matrix
Position-specific scoring matrix (PSSM which was originally used to detect distantly related proteins) is a very helpful tool for representing protein sequences as a matrix. Thus, we also transformed each protein sequence into a PSSM by employing position-specific iterated BLAST (PSI-BLAST) 31 :

where L is a given protein sequence length, 20 represent the 20 amino acids, and
In this article, to obtain highly and widely homologous protein sequences, the PSI_BLAST e-value parameter was set to 0.001, and 3 iterations were selected. Consequently, each protein sequence can be represented as 20-dimensional PSSM, which contain L × 20 elements, where L is the length of a given protein sequence and the columns are 20 amino acids.
Local coding based on PSSM matrix
As is well known, the key characteristics of PPIs are that “PPI usually occur in discontinuous segments in the protein sequence, where distant amino acid residues are brought into spatial proximity by protein folding.” As a result, we developed a novel local coding method based on PSSM; the advantage of the method is that it incorporates both global and local feature extraction, which can account for the interactions between residues in both continuous and discontinuous regions in amino acid sequences.
The steps for creating the CPSSM matrix are as follows: first, using PSI-BLAST, we constructed each protein sequence PSSM matrix; second, each PSSM was divided into 4 parts: A, B, C, and D. We intercepted the first 75%, the last 75%, and the middle 75% of the PSSM and named these as parts A, B, and C. The whole PSSM matrix is represented in part D. Finally, a new PSSM (CPSSM) is created by merging the A, B, C, and D sub-PSSM in parallel, where the length of the CPSSM is longer than the length of the PSSM. The advantage of the CPSSM matrix is that it can fully account for global and local feature information and the interactions between sequentially distant but spatially close amino acid residues. As a result, the CPSSM matrix can adequately capture multiple overlapping continuous and discontinuous binding patterns within a protein sequence and improve prediction accuracy. Figure 1 shows the procedure of local coding based on PSSM.
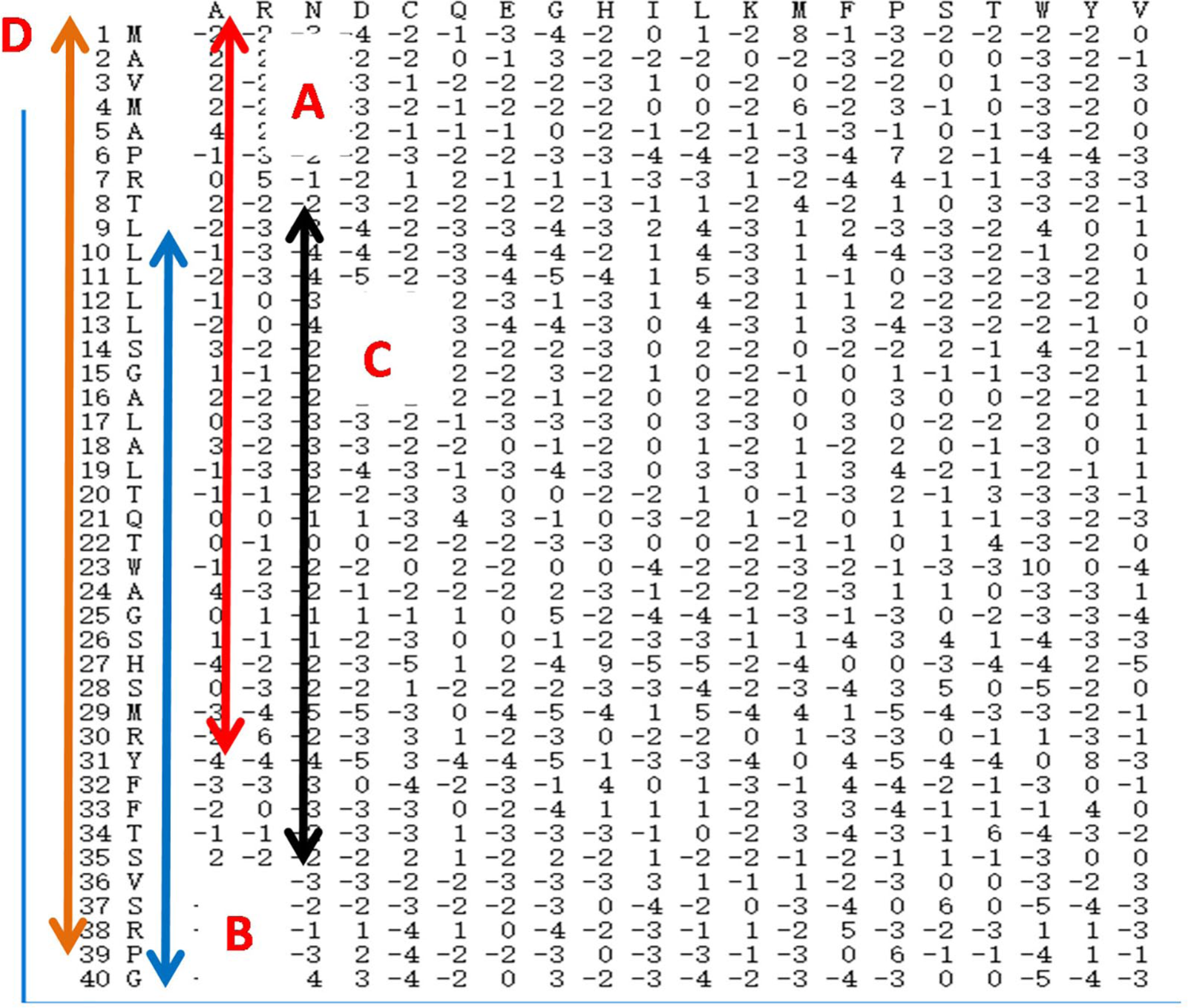
The flow diagram of local coding based on PSSM matrix. PSSM indicates position-specific scoring matrix.
Multi-features fusion
In previous studies, many sequence-based feature extraction methods used a single feature approach, whose drawbacks are that they cannot integrate multiple key feature information–contained protein sequence, and they cannot comprehensively consider the correlation of the various elements of a protein sequence. Therefore, there is considerable interest in developing a novel feature extraction approach by using multifeatures fusion, which is capable of increasing the quantity of feature vectors information and improving the prediction accuracy. Thus, LAG and BP were used to carry out multifeatures fusion.
Local Average Group
The LAG method divided each CPSSM into 20 groups. Each group contained 5% of the length of a CPSSM. Thus, each CPSSM was divided into 20 groups regardless of the length of the protein sequence, where each group consists of 20 features derived from the 20 columns of the CPSSM. The mathematical description is as follows:


where
Bigram Probabilities
N-gram models are usually employed to estimate the probability of a random sequence,32,33 such as the random sequence in the following:


where

where L represents the number of the CPSSM row. A CPSSM element
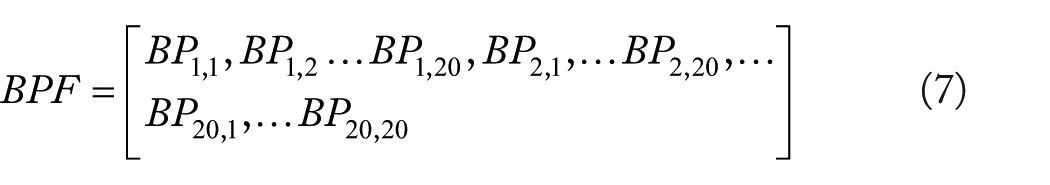

where
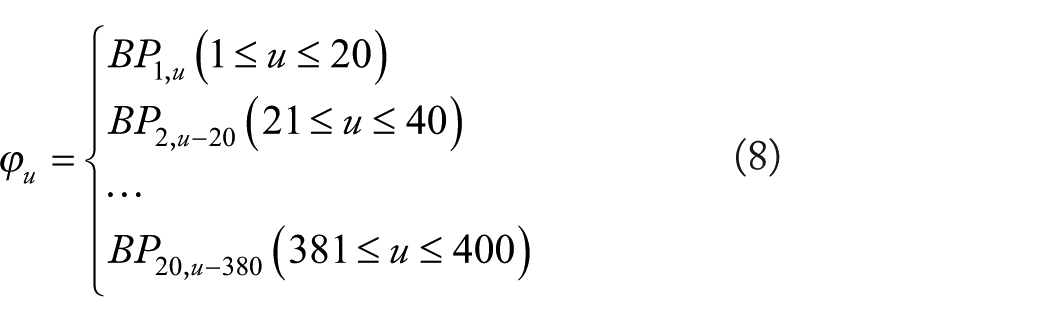
Finally, each CPSSM of a given protein sequence was converted into a 400-dimensional vector using the BP feature extraction method.
As a result, based on the above analysis, we proposed a new sequence-based feature extraction method by combining local coding based on PSSM and multifeatures fusion in this study. The proposed method fully considers the following key points:
Capturing protein interaction information by using local coding based on PSSM;
Increasing the amount of feature information through employing LAG and BP to carry out multifeatures fusion.
In this article, we proposed local coding method based on PSSM; the highlight of the method incorporates global and local feature extraction, which could account for the interactions between residues in both continuous and discontinuous regions of amino acid sequences. Using multifeatures fusion integrates multiple key pieces of feature information contained in protein sequences and comprehensively considers the relevant information of each feature element. The flow diagram of our feature extraction algorithm is shown in Figure 2

The flow diagram of our feature extraction algorithm. BP indicates Bigram Probability; LAG, Local Average Group.
Performance evaluation
For evaluating the effectiveness of LCPSSMMF, 4 parameters, including sensitivity, accuracy, Matthews correlation coefficient (MCC), and precision, were calculated. The mathematical descriptions are as follows:




where FP represents false positives, TP represents true positives, FN represents false negatives, and TN represents true negatives. True positives represent the number of true interacting pairs that were correctly predicted. True negatives represent the count of true noninteracting pairs that were predicted correctly. False positives represented the number of noninteracting pairs that were falsely predicted, and false negatives represented true interacting pairs that were falsely predicted to be noninteracting pairs. Moreover, we drew a receiver operating characteristic (ROC) curve to further evaluate the effectiveness of our method.
Results and Discussion
Performance of the proposed method
For evaluating the efficiency of our feature extraction approach, we compared it with other feature extraction approaches, as shown in Table 1.
The abbreviations of different feature extraction methods.

Abbreviations: LCPSSMMF, local coding position-specific scoring matrix with multifeatures fusion; PSSM, position-specific scoring matrix; LAG, Local Average Group; BP, Bigram Probabilities.
In Table 1, PSSM, LC, and MF represent PSSM, local coding based on PSSM, and multifeatures fusion, respectively. LAG and BP represent LAG and BP feature extraction method, respectively.
The SVM classifier has been widely used to predict PPIs. As a result, for ensuring fairness, different feature extraction methods used the same SVM classifier on human and yeast datasets in the experiment. In addition, to avoid over fitting, we divided the datasets into training sets and independent test sets. Specifically, 4out of 5 of the datasets were selected as training sets and the remaining datasets were selected as independent test sets. Simultaneously, 5-fold cross validation was employed to benchmark the effectiveness of our feature extraction method. The LIBSVM tool 34 was used to carry out classification in the experiment. The radial basis function (RBF) kernel parameters of the SVM were optimized by using the grid search method, where c is 0.001 and g is 0.3, and other parameters were set to the default value.
It can be seen from Table 2 that the LCPSSMMF obtained an average prediction accuracy, sensitivity, precision, and MCC of 93.14%, 92.50%, 93.90%, and 87.41%, respectively, using the yeast dataset. As shown in Tables 3 to 5, the PSSMMF, LCPSSMLAG, and LCPSSMBP achieved an average prediction accuracy, sensitivity, precision, and MCC of 90.48%, 90.26%, 90.58%, and 82.84%; 87.09%, 86.57%, 87.13%, and 77.33%; and 88.83%, 86.22%, 87.67%, and 81.34% using the yeast dataset, respectively. Similarly, Table 6 shows that the LCPSSMMF obtained an average accuracy, sensitivity, precision, and MCC of 90.41%, 93.54%, 88.02%, and 82.62% using the human dataset. It can be seen from Tables 7 to 9 that the PSSMMF, LCPSSMAB, and LCPSSMBG obtained an average accuracy of 87.58%, 85.86%, and 86.42%; average sensitivity of 87.69%, 84.89%, and 86.03%; average precision of 87.24%, 86.58%, and 87.10%; and average MCC of 77.84%, 75.72%, and 76.85%, respectively, on the human dataset. Similarly, as shown in Figures 3 and 4, the ROC curves of LCPSSMMF are also significantly better than those of PSSMMF, LCPSSMLAG, and LCPSSMBP. As a result, these experimental results demonstrated that the predictive capability of LCPSSMMF is superior to other methods. This result clearly demonstrated that LCPSSMMF is an effective feature extraction method for predicting PPIs. The success of LCPSSMMF can be attributed to the following several factories: (1) we exploited local coding based on PSSM matrix, which incorporates global and local feature extraction, thus accounting for the interactions between residues in both continuous and discontinuous regions of amino acid sequences; (2) serial multifeatures fusion can integrate multiple key feature information contained in the sequence and comprehensively consider the relevant information of each element in the sequence; and (3) the LAG method based on the residue conservation tendencies in the same domain family is similar, and the locations of the domains in the same family are closely related to the length of the sequence. The BP approach represented each protein sequence by its PSSM and calculated the Bigram feature using the probability information contained in PSSM. This approach can significantly reduce the sparsity level and helps to improve recognition performance. Thus, information can be effectively captured from the new PSSMs by using the LAG and BP feature extraction methods. Therefore, the efficiency of the LCPSSMMF approach is clearly superior to other feature extraction methods.
The experimental results of the LCPSSMMF method on the yeast dataset.

Abbreviations: LCPSSMMF, local coding position-specific scoring matrix with multifeatures fusion; MCC, Matthews correlation coefficient.
The experimental results of the PSSMMF method on the yeast dataset.
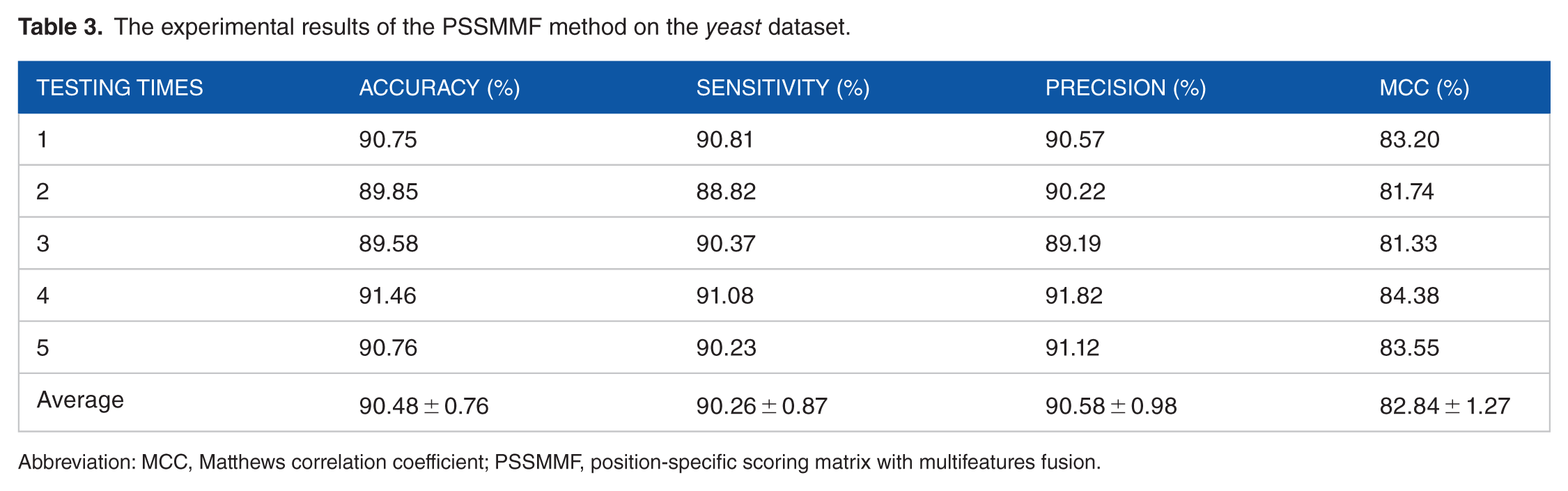
Abbreviation: MCC, Matthews correlation coefficient; PSSMMF, position-specific scoring matrix with multifeatures fusion.
The experimental results of the LCPSSMAB method on the yeast dataset.

Abbreviation: MCC, Matthews correlation coefficient.
The experimental results of the LCPSSMBG method on the yeast dataset.

Abbreviation: MCC, Matthews correlation coefficient.
The experimental results of the LCPSSMMF method on the human dataset.

Abbreviation: LCPSSMMF, local coding position-specific scoring matrix with multifeatures fusion; MCC, Matthews correlation coefficient.
The experimental results of the PSSMMF method on the human dataset.

Abbreviations: PSSMMF, position-specific scoring matrix with multifeatures fusion; MCC, Matthews correlation coefficient.
The experimental results of the LCPSSMAB method on the human dataset.

Abbreviation: MCC, Matthews correlation coefficient.
The experimental results of the LCPSSMBG method on the human dataset.

Abbreviation: MCC, Matthews correlation coefficient.

Comparison of ROC curves based on different feature extraction methods and SVM classifier on the yeast dataset. ROC indicates receiver operating characteristic; SVM, support vector machine.
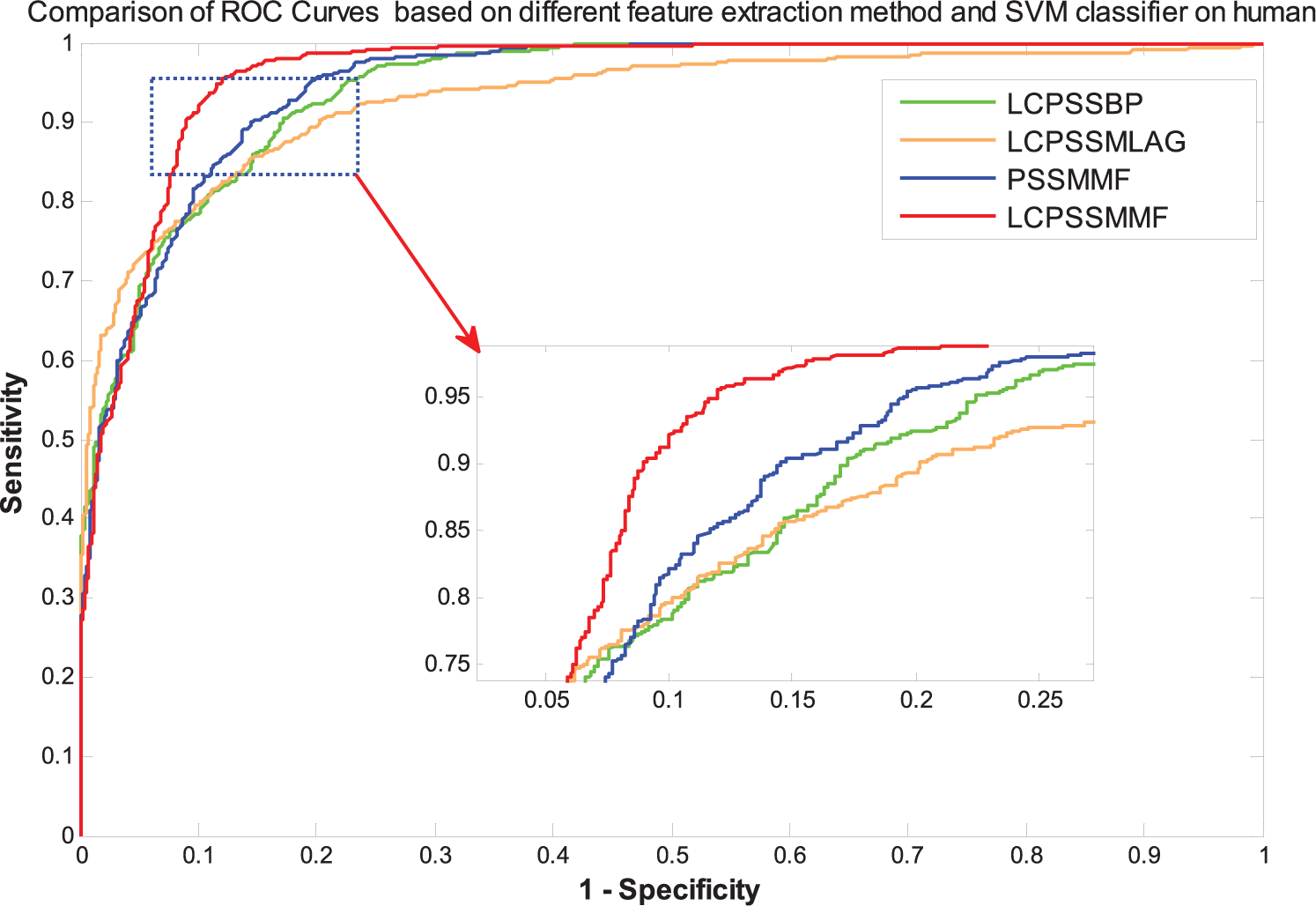
Comparison of ROC curves based on different feature extraction method and SVM classifier on the human dataset. LCPSSMMF indicates local coding position-specific scoring matrix (PSSM) with multifeatures fusion; ROC, receiver operating characteristic; SVM, support vector machine.
Comparison of SVM based on other feature extraction methods
Meanwhile, to further evaluate the effectiveness of LCPSSMMF, we compared the prediction capability of LCPSSMMF with that of existing methods by using the same SVM classifier on the yeast dataset. It is shown in Table 10 that 4 different approaches obtained average prediction accuracy between 87.36% and 91.73%, which is lower than that of the proposed LCPSSMMF method. Similarly, the sensitivity and precision of LCPSSMMF are also superior to other approaches. It is obvious from these experimental results that the proposed LCPSSMMF feature extraction method yielded significantly better prediction results than other existing approaches. All these results indicated that the LCPSSMMF can improve the prediction accuracy relative to current state-of-the-art methods.
The prediction results of different feature extraction methods on the yeast dataset.

Abbreviations: AC, auto covariance; LCPSSMMF, local coding position-specific scoring matrix with multifeatures fusion; LD, local descriptors; ACC, auto cross covariance; GE, global encoding.
Conclusions
In this study, we proposed a sequence-based feature extraction method called LCPSSMMF, which combined local coding based on PSSM with multifeatures fusion. First, we used a novel local coding method based on PSSM to build a new PSSM (CPSSM), which incorporates global and local feature extraction to account for the interactions between residues in both continuous and discontinuous regions of amino acid sequences. Second, we adopted 2 different feature extraction methods (LAG and BP) to capture multiple key feature information by using the evolutionary information embedded in CPSSM. Finally, feature vectors were acquired by using the multifeatures fusion method.
The experimental results proved that the predictive capability of the LCPSSMMF is superior to that of other methods. The success of LCPSSMMF can be attributed to the following reasons: (1) we developed local coding based on PSSM, which can fully account for global and local feature information and the interactions between sequentially distant but spatially close amino acid residues. As a result, this method can adequately capture multiple overlapping continuous and discontinuous binding patterns within a protein sequence and improve prediction accuracy. (2) Serial multifeatures fusion can integrate multiple key feature information contained in the sequence and comprehensively consider the relevant information of each element in the sequence. (3) The LAG method based on the residue conservation tendencies in the same domain family is similar and the locations of domains in the same family are closely related to the length of the sequence. The BP approach represented each protein sequence by its PSSM and calculated the Bigram feature using the probability information contained in PSSM, which can significantly reduce the sparsity level and improve the recognition performance. Thus, information can be effectively captured from the new PSSMs by using the LAG and BP feature extraction methods. Therefore, the efficiency of LCPSSMMF approach is obviously superior to other feature extraction methods. This study clearly demonstrated that the LCPSSMMF is an effective feature extraction method for predicting PPIs.
Footnotes
Acknowledgements
The authors would like to thank all the guest editors and anonymous reviewers for their constructive advice.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by “The Fundamental Research Funds for the Central Universities (2019XKQYMS88).”
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Ji-Yong An and Zhou You conceived the algorithm, carried out analyses, prepared the data sets, carried out experiments, and wrote the manuscript; Yu-Jun Zhao and Zi-Ji Yan designed, performed, and analyzed experiments and wrote the manuscript; and all authors read and approved the final manuscript.
Availability of data and material
In this study, our experimental datasets contain yeast and human data, which can be obtained from the publicly available DIP. 30