
Abstract
We assessed the quasispecies heterogeneity of a human astrovirus MLB2 (HAstV-MLB2-YJMGK) in immunocompromised patients following hematopoietic stem cell transplantation and performed genetic and evolutionary analyses of HAstV isolates circulating worldwide. The result showed that the virus had diversified variants and a strong positive selection in the patient, indicating that such patients may be a reservoir for astrovirus. The time to the most recent common ancestor of MLB2 and classic HAstVs was around 1800 years, and it has a decline in effective population size of HAstVs in the late 100 years.
Introduction
Human astroviruses (HAstVs) are divided into 3 major clades: classic human astroviruses (HAstV 1–8), MLB (HAstV-MLB 1–3), and VA (HAstV-VA 1–5). The prototype MLB strain was first identified in 2008 in the stool samples of children suffering from diarrhea in Melbourne, Australia, 1 and is genetically closer to astroviruses detected from other animal species than to the 8 classic HAstVs. HAstV-MLB2 was detected from the plasma and nasopharynx of 2 febrile children,2,3 from the cerebrospinal fluid of 2 adult patients (one of whom was immunocompromised), 4 and most recently from the sera of febrile children. 5 The tropism of HAstV-MLB2 strains may not be restricted to the gastrointestinal tract, and it has been hypothesized that HAstV-MLB may affect extraenteric tissues.
Recombination and mutations are important factors for RNA viruses, and several evolutionary processes have led to the existence of diverse astroviruses that infect different hosts. 6 Although HAstV infections were initially considered species specific, this paradigm is being challenged; eg, MLB2 was detected in a chimpanzee with diarrhea in China. 7 Because MLB2 strains are genetically similar to astroviruses that infect nonhuman primates, the origin of MLB2 strains warrants further investigation.
The enhanced immunity of patients undergoing hematopoietic stem cell transplantation (HSCT) exerts selective pressure on viruses during the recovery stage; thus, such patients may act as a reservoir for novel virus strains, such as norovirus. 8 We report here the identification of complete genome of HAstV-MLB2-YJMGK and its diversified variants in two patients following allo-HSCT and determine the evolutionary rates and patterns of the human astrovirus.
Materials and Methods
Samples and high-throughput sequencing
Fifty fecal samples were collected from 27 recipients of allo-HSCT at the Beijing University First Hospital from May to August 2016 and April to June 2018. The fecal specimens were transported on dry ice and stored at −80°C until further analyses. Thirty fecal samples collected from 10 patients were subject for high throughout sequencing (MiSeq platform, Illumina). Sample preparation for the MiSeq sequencing and informatics pipeline for analyzing the data were reported in our previous study. 9
Informed consent for analyses was obtained from the patients or their legal guardians. The study protocol was approved by the Ethics Committee of the National Institute for Viral Disease Control and Prevention, China Center for Disease Control, according to Chinese ethics laws and regulations.
Detection of HAstV-MLB2-YJMGK
The presence of HAstV-MLB2-YJMGK was confirmed by polymerase chain reaction (PCR) using primers designed based on the contig sequences obtained by MiSeq high-throughput sequencing (Table 1). All of the 50 collected samples from allo-HSCT recipients were screened for the virus using primers targeting a 702 bp fragment in the ORF1b region (forward, 5′-GACTGGACACGATTTGATGG-3′; reverse, 5′-CTCCAATTGTCTCCGAGTGA-3′). Positive and negative controls were included in each PCR run.
The primers designed based on the contig sequences obtained by MiSeq high-throughput sequencing.
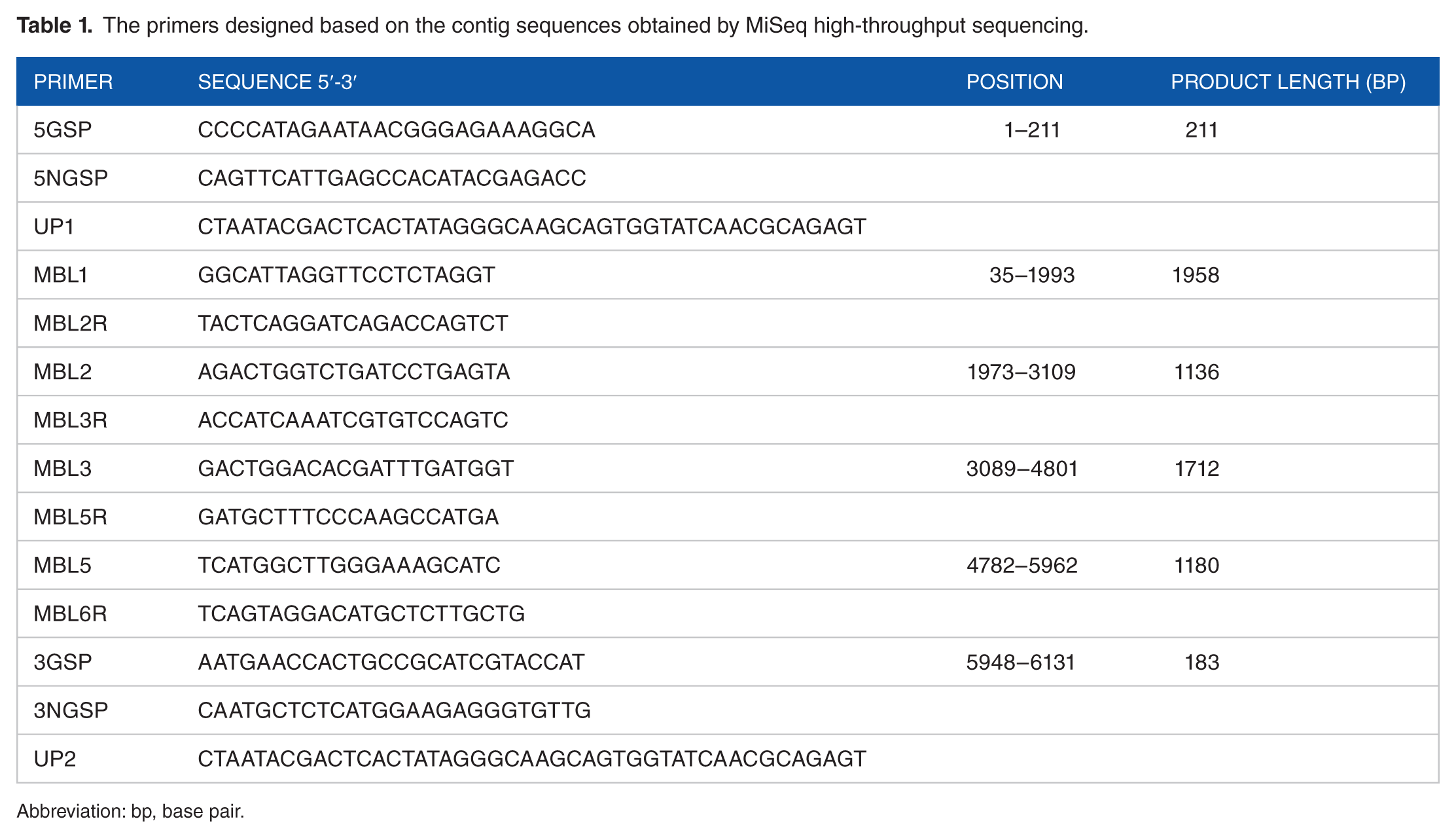
Abbreviation: bp, base pair.
Amplification of the full-length genome
To acquire the complete genome of HAstV-MLB2-YJMGK, primers were designed based first on the contigs obtained by high-throughput sequencing and second on the newly amplified sequences. The extreme 5′ and 3′ ends of the genome were determined using a SMART RACE cDNA Amplification Kit (Clontech, USA). After Sanger sequencing, sequences were assembled and edited manually to determine the sequence of the viral genome.
Recombination and phylogenetic analyses
The complete genomes of known HAstV-MLB2s were downloaded from GenBank. To detect recombination, aligned sequences were analyzed using the bootscanning method and a neighbor-joining algorithm was run with 100 pseudoreplicates in Simplot software. Phylogenetic trees were constructed using the nucleotide (nt) sequences of ORF2 by the neighbor-joining method and were subjected to bootstrap analyses with 1000 replicates to determine the relationship between HAstV-MLB2 and other astroviruses. Tree figures were produced using MEGA software (v. 6).
Evolutionary analyses
To precisely estimate the substitution rate, a Bayesian Markov Chain Monte Carlo (MCMC) approach was implemented using BEAST software (v. 1.8.2). jModelTest software (v. 2.1.7) was used to identify the optimal evolutionary model. The Akaike information criterion and hierarchical likelihood ratio test suggested that the general time reversible (GTR) + Γ (gamma distributed rate variation) model best fitted the sequences. The MCMC analyses were performed with 50 million generations and sampling every 1000 generations with 10% burn-in. The results were computed and analyzed using Tracer v. 1.6. The effective sample size values for the estimated parameters in the MCMC analyses were greater than 200. Statistical uncertainty in the data was reflected by 95% highest probability density (HPD) values.
Bayesian skyline plot
The GTR + Γ + UCLD (the uncorrelated log-normal distribution relaxed clock) with constant growth demographic population dynamic models, as implemented in BEAST software, were used as the fittest model to the dataset of HAstVs. The MCMC analyses were performed over 40 million generations and sampling every 2000 generations with 10% burn-in. The Bayesian skyline plot was analyzed using Tracer.
Calculation of genetic diversity
All of the reads from sample LHC2 sequenced by high-throughput sequencing of HAstV-MLB2 were mapped to the reference (HAstV-MLB2-YJMGK) genome using Geneious (v. 6.1.4). Diversity was quantified as the mean genetic distance calculated for all pairs of nt sequences using MEGA software (v. 5). The rates of synonymous substitutions per synonymous site (dS) and nonsynonymous substitutions per nonsynonymous site (dN) were calculated using the method of Nei and Gojobori with the Jukes-Cantor correction for multiple substitutions using MEGA software (v. 4.1). The dN/dS ratio is an indicator of the strength of the positive (>1) or negative (<1) selection pressure on a quasispecies.
Results
Complete genome of the virus
After removing the reads with quality mean less than 25, a total of 724 757 000 clean reads was obtained from the 30 fecal samples, among which 852 matched HAstV-MLB2. Two unique sequences with lengths of 1694 and 4286 bp assembled from the initial sequencing data matched HAstV-MLB2, with 98% nt identity and 100% coverage. Six fragments with overlaps based on the initial sequences were amplified to generate the genome sequences, and the nearly full-length genome sequence (6131 bp) was acquired and deposited in GenBank under accession number MK327365. The whole sequence exhibited 98% nt identity to that of MLB2-GUP187 (AB829252.1), with 100% query coverage and comprises 3 open reading frames (ORFs)—ORF1a, ORF1b, ORF2—and 3′ untranslated region (UTR), similar to previously reported HAstVs. However, compared with MLB2-GUP187, a 17 bp in the 5′ end of the genome of our MLB2-YJMGK, of which 14 bp in the 5’ UTR region and 3 bp in the ORF1a region, was not acquired.
Detection of HAstV-MLB2-YJMGK
The presence of HAstV-MLB2-YJMGK was confirmed by PCR. Results showed that 5 of the 50 samples from 2 patients (patients 1 and 2) were positive for the virus, the amplified 702 bp sequences targeting ORF1b region showed 99% nt similarities. The other samples were negative. Patient 1 is 53 years old, suffering from multiple myeloma, while patient 2 is 62 years old, suffering from plasmacytoma; they both underwent a bone marrow transplant. Immunosuppressive agents cyclosporine A, mycophenolate mofetil, and methylprednisolone were administered to control the graft-versus-host disease. The first sample of patient 1 (LHC1) was collected on the day of stem cell transfusion, and the second sample (LHC2) was collected 24 days later, while the first sample of patient 2 (XZJ1) was collected on the first day of admission, the interval between his first and third samples was 26 days, and his second sample was collected on day 18.
Phylogenetic and evolutionary analyses
Recombination analyses showed no evidence of recombination for HAstV-MLB2-YJMGK strain. Phylogenetic analyses of ORF2 region showed that the HAstV-MLB2-YJMGK strain in this study was genetically similar to previously reported MLB2 astroviruses, and to primate astroviruses, and they formed an independent branch from other HAstVs. The MLB2 strain analyzed in this work was closer to MLB3 than to MLB1 (Figure 1).

Phylogenetic analyses of the nucleotide sequence of the ORF2 region of astroviruses. The tree was constructed using the neighbor-joining method with MEGA v. 5 using 1000 bootstrap replicates. • indicates virus analyzed in this study. Bootstrap values are shown on the branches. The results showed that MLB2 represents a distinct lineage in the astrovirus family.
To determine the evolutionary relationship between the HAstV-MLB2 strains and related HAstVs, a Bayesian MCMC estimation of the time of the most recent common ancestor was performed using BEAST software. Under the best-fit model, the mean substitution rate for the HAstVs was 1.97 × 10–3 substitutions per site per year, with a 95% HPD of 2.76 × 10–4 to 5.77 × 10–4. The time of the most recent common ancestor of HAstV-MLB2 and MLB3 isolates was estimated to be around 180 years ago, while that of the MLB isolates (MLB1, MLB2, and MLB3) was estimated to be 410 years ago. The most recent common ancestor of HAstV-MLB and classic HAstVs was estimated to be more than 1800 years ago (Figure 2).

Bayesian Markov Chain Monte Carlo tree of the open reading frame 2 region of MLB2 and known human astrovirus (HAstVs). The estimated time of the most recent common ancestors of the major nodes of the lineages are shown. The results showed that MLB2 and the classic HAstVs diverged from their common ancestor 1800 years ago.
Bayesian skyline plot
The Bayesian skyline plot of the demographic population history for HAstVs suggested that the HAstV population was relatively stable over time, with the exception of a slight expansion 1500 years ago. The genetic diversity of HAstVs has declined in the last 100 years (Fig. 3).
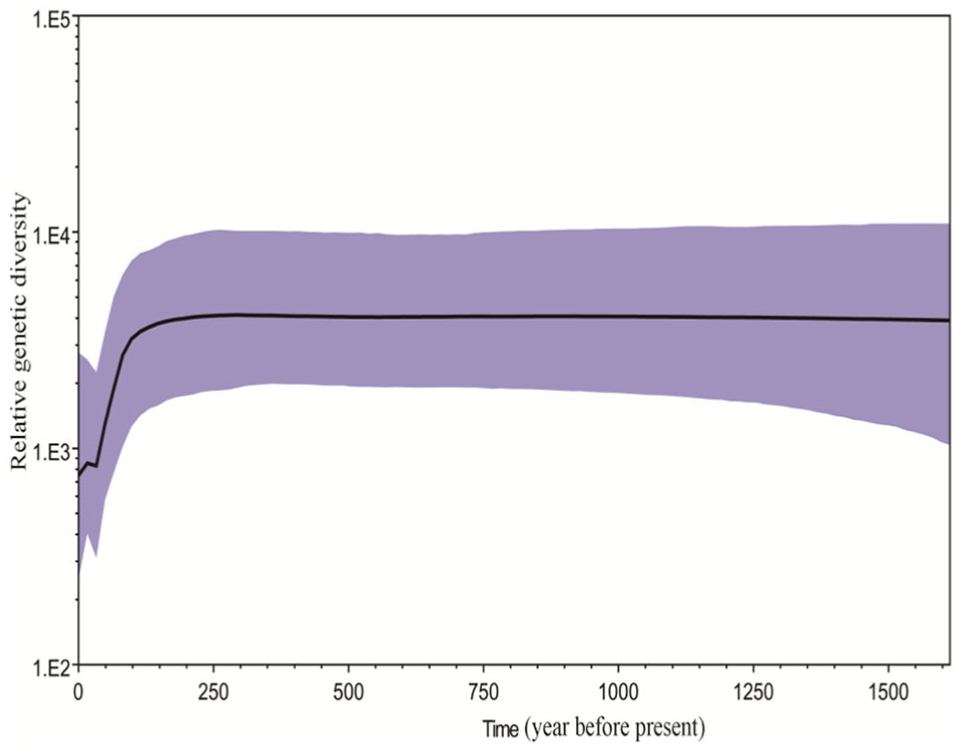
Bayesian skyline plot showing the historical dynamics of the total HAstV population. X-axis, years before present; y-axis, relative genetic diversity; thick solid line, mean estimates; purple shading, 95% highest probability density. The decline in relative genetic diversity in the last 100 years is evident.
Genetic diversity
Reads from different samples were separated by different barcodes, and the reads number in different samples varied greatly, with sample LHC2 from patient 1 accounting for 95.8% (816 reads) of all the MLB sequences, so the reads from sample LHC2 were used for the genetic diversity analysis. The distribution of HAstV-MLB2-YJMGK diversified variants was analyzed using the partial sequences of the capsid protein. We only called a variant if at least 2 differences were observed in a read to reduce false positives. In total, 67 of the 311 reads obtained by high-throughput sequencing exhibited 100% nt identity to the partial sequences of the capsid region of HAstV-MLB2-YJMGK. The reads shared 94.3% to 100% nt identities to the HAstV-MLB2-YJMGK. The others have different mutants in the sequences. The positions and frequency of substitutions in the sequences were shown in Figure 4, a total of 81 variants were found, indicating that a high genetic diversity of the virus existed in the patient. The dN/dS ratio of both the S and P domains in the capsid region was determined to be 1.667.
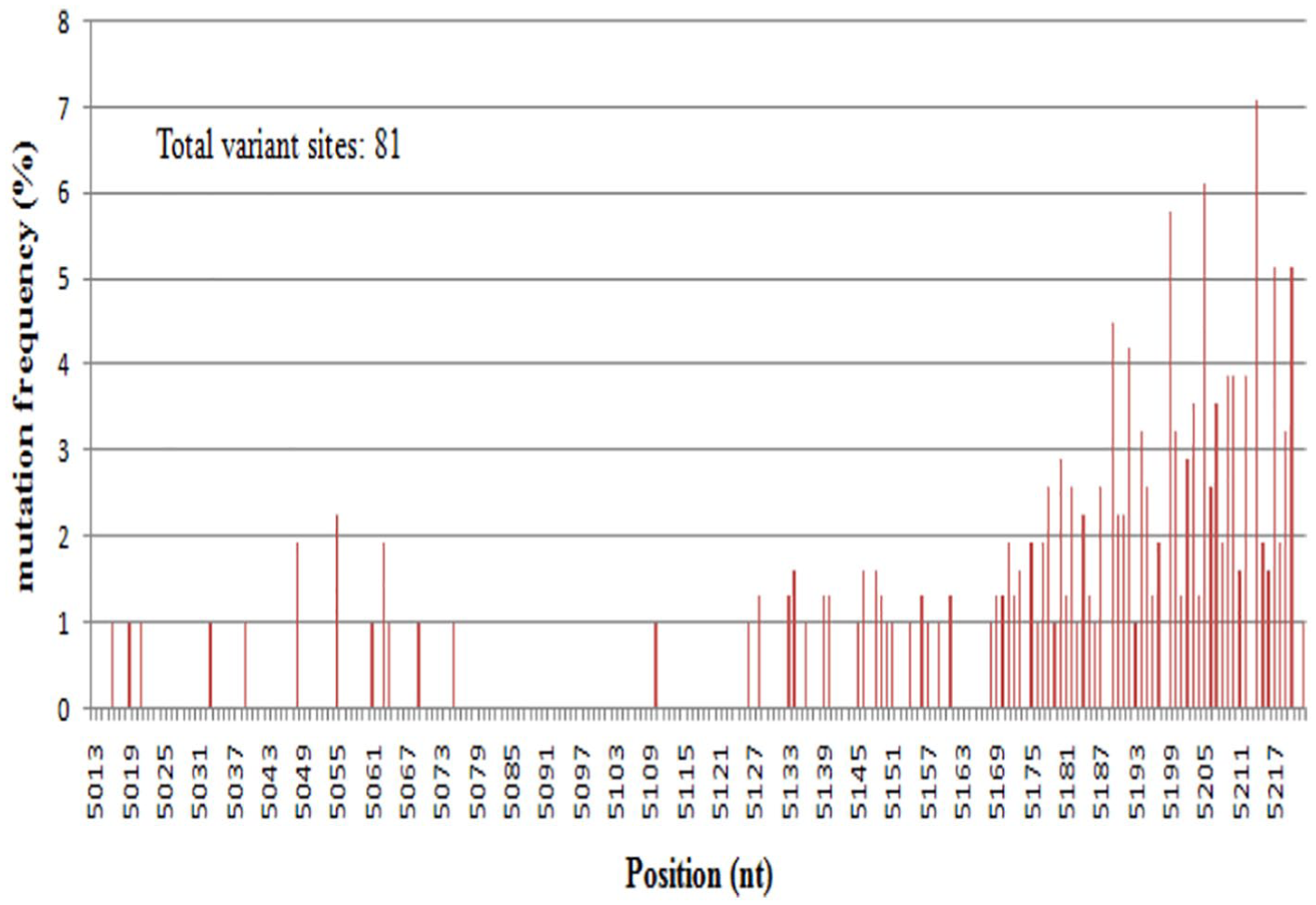
Distribution of HAstV-MLB2-YJMGK mutation frequencies across the partial sequences of open reading frame 2 region (nt 5013-5217). A total of 81 variant sites were found in the 210 bp length sequence. We only called a variant if at least 2 differences were observed in a read to reduce false positives.
Discussion
The prevalence and etiological role of HAstV-MLB strains of any of the 3 genotypes (MLB1–3) in humans remain unclear, and the origin of the virus is unknown. In this study, we analyzed HAstV-MLB2-YJMGK strains detected in recipients of HSCT. The ORF1a, ORF1b, and ORF2 regions of the virus shared high nt-sequence similarities to previously reported MLB2 strains. Five samples from 2 patients were positive for HAstV-MLB2-YJMGK. Neither patient exhibited symptoms like diarrhea and fever, suggesting that MLB2 may not cause disease or cause extra-intestinal tissues and other signs in the patients. However, in patients 1 and 2, the intervals between the first and last stool samples were 24 and 26 days, respectively, suggesting that MLB2 persisted in the patients. Although prolonged HAstV infections have been reported, particularly in immunocompromised patients, with virus shedding for 17 to 183 days,10,11 HAstV infections typically last for 1 to 4 days. These findings remind us that compromised innate and adaptive immunity before and after allo-HSCT may facilitate the evolution of MLB2, as for norovirus. 12
Numerous mammalian and avian astroviruses have been described, and the list of astrovirus continues to expand. The identification of the mutation may play a role in the host tropism and evolution of astroviruses. The HAstV mutation accumulation rate is 1.01 × 10–3 to 3.7 × 10–3 substitutions per site per year.13,14 In this study, the average mutation accumulation rate of HAstV based on the ORF2 fragment was 1.97 × 10–3 substitutions per site per year, which fall into the range of previous estimates. Use of an extended sequence enables reliable estimation of the rate of viral genome variation; however, because of the numerous recombination events in the evolutionary history of HAstV, 15 estimation of its evolutionary rate based on the complete genome sequence may yield nonsignificant results. To conclude, genomic recombination and the high mutation rate of HAstV make this virus a rapidly evolving infectious agent.
Phylogenetic analyses confirmed the existence of 3 independent clades of HAstVs: the classic HAstVs (HAstV genotypes 1–8), MLB (MLB genotypes 1–3), and VA (both VA and HMO genotypes). The MLB strains are closer to the classic HAstVs than to the VA strains. The MLB2 strains detected in this study shared high nt-sequence similarities with previously reported MLB2 strains and formed a single cluster in the MLB clade. Analyses of the chronogram for the ORF2 region demonstrated that the 3 clades diverged about 3800 years ago, while the classic HAstVs diverged from their common ancestor about 670 years ago, similar to the 690 years reported by Babkin et al. 14 The MLB isolates in this study diverged from their common ancestor around 410 years ago, and the VA isolates approximately 820 years ago. The MLB2 isolates diverged from their common ancestor around 30 years ago. Genetic diversity is equal to the product of the effective population size and the generation length in years. 16 The Bayesian skyline plot suggested that the HAstV population has remained relatively stable over time, but the effective population size has decreased in the last 10 to 20 years, possibly due to improved economic conditions and widely use of antivirals in the clinical.
Immunocompromised patients are susceptible to infection by pathogens that can persist for a long period of time. 12 Information on HAstV genome variation is important for predicting emergence of new variants of the virus. We detected a large number of mutant variants in this study, and the dN/dS ratio of the sequences was >1, indicating strong positive selection. Because immunocompromised patients act as a reservoir for novel virus strains and strongly antigenic positive selection sites are present in the capsid protein of an enterovirus, 17 the presence of positive selection sites in the capsid protein of MLB2 may explain its divergence in allo-HSCT patients. However, no study of the relationship between positive selection and the antigenicity of HAstV has been reported.
In conclusion, we detected diversified variants of HAstV-MLB2-YJMGK in hospitalized immunocompromised patients, suggesting that such patients may act as a reservoir for astrovirus variants; therefore, their excreta should be monitored after discharge from the hospital. Moreover, the high mutation rate of HAstV-MLB2 in this study warrants further investigation.
Footnotes
Acknowledgements
We thank Yu-jun Dong from Department of Hematology, Beijing University First Hospital, for providing the fecal samples.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant no. 81702007).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KG, JMY, and YY designed the study. QZ and LLL collected the samples and analyzed the data. JMY, QZ, and KG do the experiment. KG and JMY wrote the manuscript. JMY and YY reviewed the manuscript. All authors have read and approved the manuscript.