
Abstract
Recent studies reveal that even the smallest genomes such as viruses evolve through complex and stochastic processes, and the assumption of independent alleles is not valid in most applications. Advances in sequencing technologies produce multiple time-point whole-genome data, which enable potential interactions between these alleles to be investigated empirically. To investigate these interactions, we represent alleles as distributed vectors that encode for relationships with other alleles in the course of evolution and apply artificial neural networks to time-sampled whole-genome datasets for feature learning. We build this platform using methods and algorithms derived from natural language processing (NLP), and we denote it as the nucleotide skip-gram neural network. We learn distributed vectors of alleles using the changes in allele frequency of echovirus 11 in the presence or absence of the disinfectant (ClO2) from the experimental evolution data. Results from the training using a new open-source software TensorFlow show that the learned distributed vectors can be clustered using principal component analysis and hierarchical clustering to reveal a list of non-synonymous mutations that arise on the structural protein VP1 in connection to the candidate mutation for ClO2 adaptation. Furthermore, this method can account for recombination rates by setting the extent of interactions as a biological hyper-parameter, and the results show that the most realistic scenario of mid-range interactions across the genome is most consistent with the previous studies.
Keywords
Introduction
In evolutionary biology, statistical models are used to investigate the evolutionary processes that generated the vast diversity of life. Our current understanding of evolution stems from the work of many biologists who derived theoretical models from the observations in nature, such as selection, genetic drift, migration, and speciation.1–5 However, these models are established under some assumptions that simplify the underlying principles, as most biological processes are dynamic and complex. For example, the Wright-Fisher model is one of the fundamental population genetic models, which represents the process of genetic drift as a binomial sampling of 2N gene copies between generations in an idealized population of N.2,3 This idealized population has no overlapping generations, and each gene copy is independently drawn to the next generation at random in a fixed population. Despite these simplifying assumptions, the Wright-Fisher model is still widely used in population genetic methods, for instance in a recent analysis of forward processes in time-sampled datasets.6–8 Some assumptions of the model are valid in most applications, as the model has been proved to give intuitive approximations of more complex real cases. 9 However, the assumption of independent alleles is problematic in many cases, particularly as advances in molecular genetics unveil how alleles interact in intricate networks to produce unexpected outcomes, even for viruses with comparatively small and simple genomes.10–13 Furthermore, advances in sequencing technologies generate datasets such as whole-genome sequences where the relations between alleles can potentially be investigated, particularly in time-serial cases.6–8,14 To investigate these complex interactions, we propose to represent alleles as distributed vectors that encode for relationships with other alleles in the course of evolution, rather than as discrete entities as in the case of conventional models like the Wright-Fisher model. This novel concept of representing alleles with distributed vectors is inspired from artificial neural networks (ANNs) and natural language processing (NLP), and we use a model-free approach to learn these distributed vectors of alleles directly from genetic sequence data.
Artificial neural networks are biologically inspired computing elements that can be interconnected to process and learn multiple levels of representation from external input information such as sound, image, or characters. Artificial neural networks are becoming increasingly popular in many fields, as they provide a flexible framework where learned features are easy to adapt and learn, without manual over-specification of features like other machine learning techniques. For instance, popular machine learning algorithms such as logistic regression or naive Bayes depend heavily on the representation of raw data, as the choice of representation has a critical effect on the performance of these algorithms. Since 2010, multilayer neural networks started outperforming other machine learning techniques in speech and vision with the availability of big data and faster machines. 15 There has been a recent interest to apply ANNs in medicine and biology due to the exponential growth in data production with technological advances, such as in structural biology, regulatory genomics, drug discovery, and cell imaging.16,17 However, in the evolutionary context, only a few attempts have been made to apply ANNs in classic population genetic problems such as inference of selection and demography from natural populations. 18 Here, we use ANNs in the evolutionary framework to exploit their ability to learn from high-dimensional biological data without explicit programming or modeling or prior knowledge through feature learning. The algorithms and training methods are derived from the skip-gram neural network model in NLP that aims to represent the meaning of a word in distributed vectors rather than treating words as atomic units.19,20 As opposed to discrete representations where there is no natural notion of similarity, distributed representations capture the context of a word over a large vector space. Distributional similarity-based representations define a word by means of its neighbors, where a word vector is trained using ANNs to predict neighboring words given a center word in the skip-gram neural network model. In a similar approach, we attempt to represent alleles by distributional similarity-based representations by predicting between a center allele and its neighboring alleles, with the aim of deciphering genetic interactions between these alleles along the course of a time-serial experimental evolution.
Viruses are the most straightforward experimental models to study the interactions between alleles, and they have been investigated through theoretical modeling and empirical analysis using site-directed mutagenesis and mutation-accumulation experiments.21–25 These studies reveal that virus genomes evolve under complex patterns of genetic interactions such as epistasis and clonal interference to shape the landscape of fitness. Here, we learn these interactions between alleles as biological features through neural networks from mutation-accumulation experiments, where no model assumptions are needed as in the previous investigations. We use population-level whole-genome sequences from the time-sampled experimental evolution of echovirus 11 in the presence or absence of the disinfectant treatment to train this ANN platform, where features to learn are distributed vector representations of the alleles. After training, we cluster these alleles by similarity in the evolutionary trajectory using principal component analysis (PCA) and hierarchical clustering, and we compare these clusters of potential genetic interactions with the previous investigation and further discuss the advantages of representing alleles as distributed vectors rather than discrete entities. Through the nucleotide skip-gram neural network, we extract evolutionary features from the genetic data of a time-sampled evolution experiment and achieve the same level of knowledge as the virology expertise acquired through a series of previously published experiments.
Methods
The nucleotide skip-gram neural network
The skip-gram model uses distributed vector representations of words to predict for every center word its context words.19,20 The model uses neural networks for learning these word vectors from large datasets, and we apply the analogous neural network architecture as shown in Figure 1 to find distributed vector representations of alleles to predict between every center allele and its nearby alleles from time-sampled allele frequency datasets. Here, an allele is defined as a nucleotide that increases in frequency more than the sequencing error between 2 sampling time points. Given a nucleotide sequence of training data a1, a2, …, aT, the objective function is to maximize the average log probability:

where w is the window size of the nearby alleles that defines the extent of interaction. The nearby alleles can be defined as those that are within the physical range of biological interactions such as epistasis, clonal interference, and linkage from the center allele

where ao are the nearby alleles, M is the size of the set of alleles in the dataset, and

Architecture of the nucleotide skip-gram neural network used to train distributed vectors of alleles, with 1 linear hidden layer (Projection) of N neurons and 1 output layer (Softmax classifier) of M neurons. The weight matrix W containing rows of distributed vectors reveals genetic interactions between the alleles during the course of evolution. These evolutionary features to be learned using the training dataset are denoted as “allele embeddings.”
Noise-contrastive estimation
In practice, an approximate of the full softmax is computed by noise-contrastive estimation (NCE), as normalizing each probability at every training step is computationally expensive.
26
The score of the similarity measure between the center allele at and the nearby alleles ao is given as a dot product. The dot product,

where a′ is the set of alleles increasing above the sequencing error. However, computing the score for all other a′s in the current center allele at at every training step is expensive, and a full probabilistic model is not necessary for feature learning. The nucleotide skip-gram model uses a binary classification object to discriminate the nearby alleles ao from k noise alleles

where

where
Implementation
Biological model and training data
We used the nucleotide skip-gram neural network in an unsupervised setting to train the distributed vector representations of the alleles using the data from the experimental evolution of echovirus 11 under the presence or absence of the disinfectant, ClO2. 27 Echovirus 11 (enteric cytopathic human orphan) is a single-stranded RNA virus belonging to the species Enterovirus B with a small genome of 7400 bases. The virus has a high mutation rate of 9 × 10−5 per site per cell infection and an average recombination rate of 3 × 10−6 to 7 × 10−6 per site per generation. It is an infectious human pathogen, residing in the gastrointestinal tract where it causes opportunistic infections. In this experiment, a wild-type (WT) population of echovirus 11 was repeatedly exposed to chlorine dioxide (ClO2), which is a highly effective disinfectant that inactivates a broad range of waterborne viruses. Following inactivation, the surviving viruses were passaged onto a monolayer of BGMK cells for regrowth. For each disinfection stage, the virus population was subjected under ClO2 concentration so as to reduce the inactivation rate constant by approximately 50%. After 20 cycles of disinfection-regrowth in 2 replicates (EA and EB), the whole genomes of the WT and evolved virus populations were sequenced with next-generation sequencing (Illumina HiSeq 2500). As a control experiment, the same WT viruses were subjected to 10 cycles of bottleneck events by dilution without being exposed to ClO2 (NEA and NEB) and regrowth in cell culture, followed by the whole-genome sequencing of the evolved virus population.
The training datasets were generated from the whole-genome datasets using the increase in the minor allele frequencies between the 2 sampling time points (WT and evolved states). Here, a nucleotide sequence of minor alleles a1, a2, …, aT is defined as the SNP or standing variation with the highest frequency in the evolved populations. As the datasets were pool-sequenced from the short reads produced using the Illumina HISeq 2500, we reconstructed the difference in the raw reads of each allele between 2 sampling time points by simulating each site in the virus genome as the binomial distribution with the probability of success as the increase in the allele frequency, f:

where n is the number of raw reads and k is the number of minor alleles at each site. The genome-wide average of the coverage depth is approximated by taking the mode of the raw read depths across the virus genome at each sampling time point as shown in Table 1 and Figure S1. We only retained the minor alleles whose increase in frequency was above the sequencing error of the Illumina sequencer, which was previously validated to be 1% from the experimental study. 27 The combined number of the retained minor alleles in the exposure replicates (EA and EB) and control replicates (NEA and NEB) is 86 and 72, respectively.
Mode of coverage depth in the whole-genome sequencing of echovirus 11 with Illumina HiSeq 2500.

Training neural networks with TensorFlow
Skip-gram models are a generalization of n-grams that model sequences, in which components may be skipped over rather than being in consecutive order. Here, we have a simple neural network with a single hidden layer to train, where the weights of the hidden layer are allele embeddings that are the features to be learned as shown in Figure 1. The size of the allele embeddings to be learned, denoted as M in Figure 1, is 86 and 72 for the exposed experiment and control experiment, respectively. These weights are initialized randomly from a uniform distribution of [–1, 1]. For this feature learning, we trained the neural network in TensorFlow (Version 1.2.1) to optimize the probability for every allele in our datasets of being the nearby allele given the center allele, as illustrated with a computational graph of TensorBoard in Figure S2. From our datasets of the allele sequences, center and nearby allele pairs are randomly chosen as [input, output] within the given window size w as described in the “Methods” section. The NCE training objective defined above is optimized with stochastic gradient descent (SGD) using mini batches, where the batch size of 128 was used in our optimization step. The output vectors are the probabilities of all alleles in our datasets being the nearby allele for the chosen center allele, optimized using the mini batches of the generated [input, output] pairs.
Hyper-parameters and bio-parameter optimization
In our nucleotide skip-gram neural network, we have 3 hyper-parameters to consider: the dimension N of the distributed vectors in the hidden layer, the number of negative samples k per positive sample, and the learning rate r. After several optimization runs, we chose the simplest hyper-parameters among the test values as 128 neurons in the hidden layer, 16 negative samples, and the learning rate of 1 × 10−2, respectively. The workflow of training neural networks in TensorFlow is illustrated in Figure S3, including the steps of data processing, weight initialization, and hyper-parameter optimization.
Here, we define the window size w as a biological hyper-parameter because its value depends on the architecture of a virus genome. The window size designates how far a center allele is assumed to interact with its nearby alleles in the genome, potentially representing the extent of biological factors such as epistatic interaction or clonal interference. Thus, the window size must depend on the recombination rate of the virus model under investigation. The recombination of echovirus 11 has not yet been measured directly, but its closely related species, Poliovirus, is known to have a recombination rate of 3 × 10−6 to 7 × 10−6 per nucleotide per generation. 28 For the genome size of 7 × 103 in echovirus 11, a similar recombination rate amounts approximately to 2 × 10−2 to 5 × 10−2 recombination events per generation. Thus, we considered following 3 cases where the center allele amid the set of M alleles is linked to:
Only the immediate neighbors (w = 1),
Quarter of the neighbors on either side (w = M/4 if M is even, or w = (M – 1)/4 if M is odd),
Half of the neighbors on either side (w = M/2 if M is even, or w = (M – 1)/2 if M is odd).
These 3 cases represent, respectively, (1) a high recombination rate that the center allele is linked to only neighbors located nearby, (2) a moderate recombination rate that the extent of interaction reaches approximately half of the genome, and (3) a low recombination rate that the extent of interaction reaches almost the entire genome.
Results
Visualization with TensorBoard
We applied the genetic data from the experimental evolution of echovirus 11 to the nucleotide skip-gram neural network using TensorFlow. The training data consist of approximately 3 × 104 genome samples from the 2 replicates of repeated bottleneck-regrowth cycles with or without the exposure to the disinfectant, ClO2. 27 The loss function over time for the training data is visualized as the average loss over every 2000 epochs in Figure 2. The loss curve steadily decreases until 20 000 epochs after which it stabilizes, indicating the chosen hyper-parameter as an optimal learning rate for this training set. After 105 training steps, the distributed vectors of the alleles were visualized using TensorBoard for the exposed and non-exposed experiments (Figures S4 to S6). The graphs from TensorBoard show the cosine distances between the allele embeddings in the original space learned from the nucleotide skip-gram neural network. Each point is indexed to the nucleotide position in the genome, and the allele of interest in the exposed population (P129Q denoted as Position 2844) and the non-exposed population (H215N denoted as Position 3101) is highlighted, to indicate candidate mutations of adaptation. According to the previous literature, these 2 non-synonymous mutations are potential drivers that give rise to the new functional adaptation to the echovirus under the given challenging conditions.27,29 Echovirus 11 has a small genome encoding for 4 structural proteins (VP1-4) and 7 nonstructural proteins (2A-C, 3A-D). Zhong et al 30 previously demonstrated that ClO2 impairs host binding, thus the evidence of resistance to ClO2 in these experiments indicates the echovirus is able to evolve an enhanced binding mechanism that can counteract the disinfectant. Further studies show that these adaptive mutations are likely to be located on the structural proteins of VP1 and VP2, as these specific mutations allow echovirus to use an alternative co-receptor that strengthens virus binding.27,29 As the candidate allele on VP2 was already present as a major allele in the WT at position 139 in our experimental datasets, the candidate mutation for enhanced host binding was deduced to have arisen on VP1—as K259E in the exposed population and as H215N in the non-exposed population. These mutations may be one of the factors that render the echovirus with replicative advantages compared to the WT under the challenges of repeated bottlenecks. Furthermore, another mutation at P129Q is important under the presence of ClO2 in the exposed population, as it causes the substitution of a ClO2-reactive amino acid (proline) to a ClO2-stable amino acid (glutamine), increasing the ClO2 endurance of the protein capsid. 27

Average loss over every 2000 epochs during learning in TensorFlow (shown for Exposed with w = M/2). The loss curve decreases steadily until 20 000 after which it stabilizes, indicating the chosen hyper-parameter as an optimal learning rate.
Correlation analysis with PCA
Principal component analysis is a statistical technique that converts data of correlated variables to linearly uncorrelated variables called principal components. PCA was carried out on the learned allele embeddings for the 3 window sizes (w = 1, w = M/4, w = M/2) using TensorBoard (Figures S4 to S6). The total variance described with the first 3 PCA components is summarized in Table S1. The pair-wise correlation matrices were generated by calculating the cosine distances between the first 3 PCA components of the allele embeddings, and they were visualized in the genomic position order as correlation maps in A, B of Figures S7 to S9. These correlation maps display the correlation coefficients between 2 allele embeddings in color according to the similarity, with positive correlations in blue color and negative correlations in red color. As shown in Figures S7 to S9, color intensity is proportional to the correlation coefficients ranging from 1 to −1. The correlation maps reveal the pair-wise similarity of the alleles in terms of genetic interactions with the neighboring alleles during the course of evolution. To mine for hidden patterns, we applied hierarchical clustering to each matrix to obtain agglomerative correlation maps of the allele embeddings as shown in C, D of Figures S7 to S9. Each correlation map contains one empty box that is designated for all zeros in the data. As shown in the figures of hierarchical clustering (C, D of Figures S7 to S9), it is notable that the correlation matrices display clear clustering of the alleles by similarity (blue) as well as by dissimilarity (red). The patterns of similarity and dissimilarity are more distinct in the exposed experiments, indicating the evolution of echovirus 11 was more directional in the presence of the disinfectant ClO2 as compared to that of the control experiments.
The hierarchical clusters of the allele embeddings were compared to the previous results 27 whose results are summarized in Table S2, where the mutation clusters were identified by calculating Pearson correlation coefficients between 2 mutations using their allele frequencies and setting a cluster threshold of 0.95. The Pearson correlation method in this study was limited to pair-wise correlations in the allele frequencies between 2 mutations from a manually specified set, and it was unable to take into account of the complete datasets (eg, differences in replicates). We chose the cluster size of 7 as used in the previous analysis, and the results from the 2 approaches were compared as shown in Table 2 andTable S3. In Table 2, the clusters containing the alleles of interest (P129Q and H215N) in the exposed and non-exposed populations are shown in detail. The comparison reveals that the allele clusters from the nucleotide skip-gram neural network with the window size as M/4 are the most consistent with the Pearson correlation clusters by Zhong et al, 27 for both the exposed and non-exposed populations. These clusters with the window size as M/4 have the highest number of the overlapping alleles to the previous list and they all lie within the protein-coding genome. It is intriguing that this window size of M/4 represents a moderate recombination rate, as the alleles are assumed to interact between the quarter of the neighboring alleles on either side that are evenly distributed along the genome (Figure S10). Given the recombination event of 2 × 10−2 to 5 × 10−2 per generation, 28 the case with a moderate recombination rate where the extent of interaction reaches approximately half of the genome during the evolution is the closest representation of the echovirus biology.
Allele clusters identified by 2 approaches: (A) Pearson correlation coefficient 27 and (B) skip-gram neural network.

For the nucleotide skip-gram neural network, the order of the alleles represents the order of similarity (see Table S3 for a complete list of clusters). For the nucleotide skip-gram neural network, the cluster results using 3 different window sizes (w = 1, w = M/4, and w = M/2) are shown. Each cell shows Protein: Nucleotide position with the major and minor alleles: Nucleotide position within the protein with the original amino acid (and the substituted amino acid for non-synonymous mutations). The red highlight indicates the candidate mutation of adaptation in the presence of the disinfectant ClO2, and the blue highlight indicates the candidate mutation of adaptation for alternative receptor binding.
indicates STOP codon.
Virus protein evolution
We further analyze the most realistic scenario of the allelic interactions within the window size of M/4. Figure 3 shows that the correlation map of the allele embeddings can be arranged in positively correlated clusters using hierarchical clustering, and these clusters also show strong patterns of negative correlation as well. This result is consistent with the fact that when alleles are similar by distributed vector representations, they should also display a similar pattern of dissimilarity. This indicates that a cluster of alleles that increases in frequency in the similar context may behave antagonistically in a collective manner to a cluster of alleles of another context; however, biological interpretations to this result should be investigated further with experimental validations.

Pairwise correlation map of the first 3 PCA components of the allele embeddings from TensorBoard with w = M/4. The alleles are arranged in the genomic order: (A) exposed and (B) non-exposed, and in the hierarchical clustering of 7 clusters: (C) exposed and (D) non-exposed. The green boxes indicate the cluster containing the mutation of interest, in the exposed population (P129Q denoted as Position 2844) and in the non-exposed population (H215N denoted as Position 3101). Positive correlations are in blue and negative correlations are in red, with the color intensity proportional to the correlation coefficients ranging from 1 to −1.
For the exposed population, the cluster that contains the candidate mutation P129Q that enhances host binding in the presence of ClO227,30 is highlighted on the structural protein of VP1 in Figure 4. Most of the mutations on the virus structural proteins in this cluster are overlapping with the list from Zhong et al, 27 except for the 2 mutations (T2849A, C2850A) on the VP1 protein. Interestingly, all but one of the mutations on VP1 in this cluster are non-synonymous, supporting the hypothesis of the important role of VP1 in adapting to the disinfectant. Furthermore, this cluster contains another candidate mutation (K259E) that was previously identified as contributing to adaptation, and this finding suggests a potential genetic interaction between these 2 candidate mutations, P129Q and K259E, during the course of experimental evolution. Contrary to the list from Zhong et al, 27 this cluster also contains additional mutations from the nonstructural proteins, such as the proteins related to the viral polymerase (3C and 3D) and NTPase (2C). However, all mutations but one are synonymous, which indicates that this interaction may be an artifact of genetic drift or linkage rather than these mutations on the nonstructural proteins playing an adaptive role in the challenging environments.
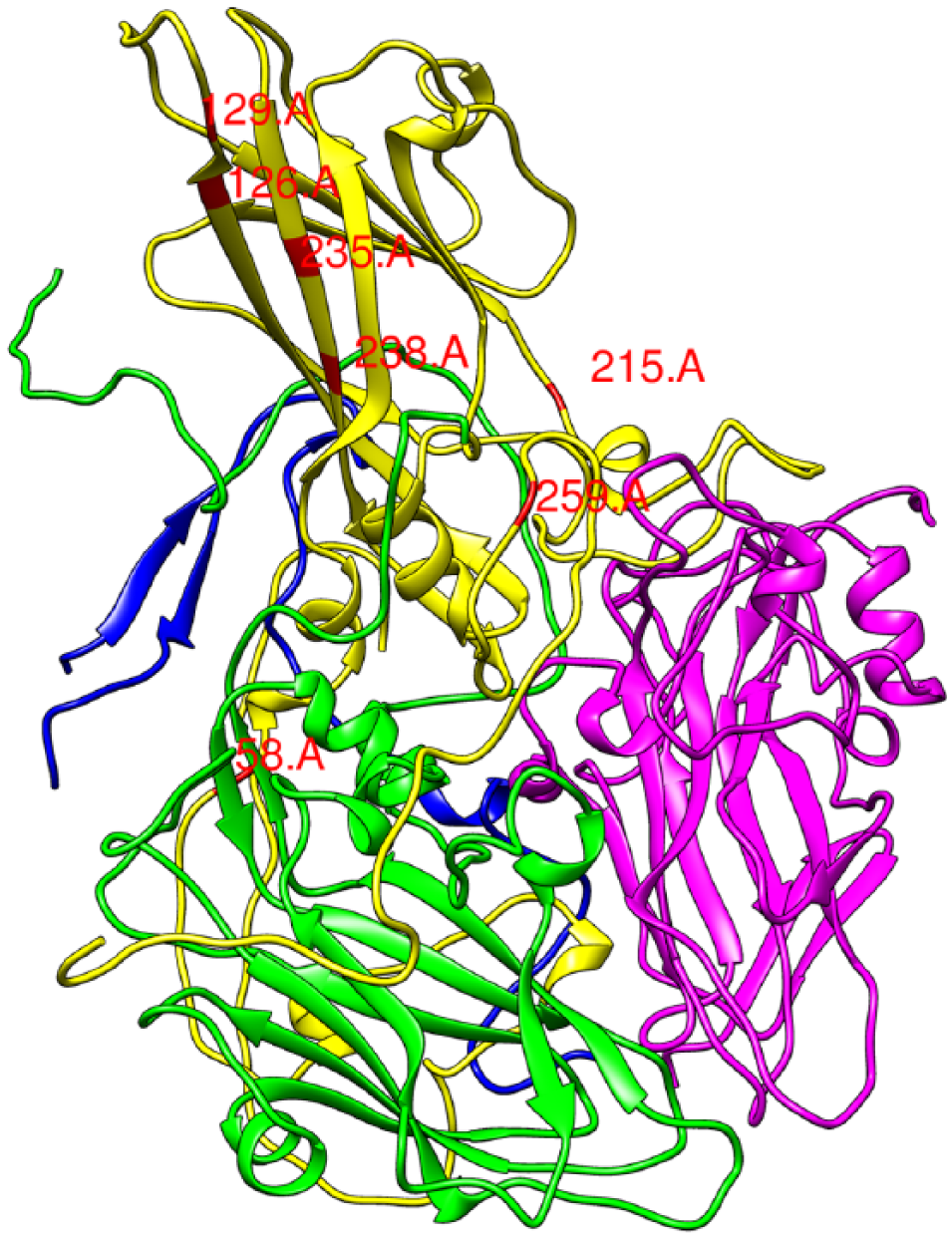
Non-synonymous mutations on the echovirus structural proteins (VP1: yellow, VP2: green, VP3: magenta, VP4: blue) identified as the cluster of potential adaptation to the disinfectant ClO2 by the nucleotide skip-gram neural network. The cluster of mutations on the VP1 is associated with the 2 candidate mutations (K259E and P129Q shown as 259.A and 129.A, respectively) that are previously identified as contributing to disinfectant adaptation. The images were generated by Chimera (Version 1.11.2) based on PDB entry 1H8T.
For the non-exposed population, the candidate mutation that gives a fitness advantage to the wild type is H215N on the VP1 protein. We also compare the cluster containing this mutation, which may help usage of an alternative receptor binding, to the previous result. 27 Contrary to the list of Zhong et al 27 with only 2 mutations on the VP1 protein, the cluster of the nucleotide skip-gram neural network contains several mutations from both the structural and nonstructural proteins. Besides the 2 mutations on VP1 previously identified, there are a few non-synonymous mutations from the viral polymerase proteins (3A and 3D) 31 which are to be investigated further empirically whether their clustering with the candidate mutation can potentially result from genetic interactions.
Discussion
We present an application of ANNs that learns biological features from time-sampled datasets of virus genome evolution. This application uses methods and algorithms derived from NLP, such as the skip-gram model and NCE, to learn distributed vector representations of the alleles that increase in frequency above the sequencing error during the time-serial experimental evolution. To the best of our knowledge, this is the first attempt to represent alleles as distributed vectors instead of discrete entities as in conventional evolutionary models, enabling the relationships between these alleles to be encoded in a continuous vector space of low dimension. We learn these features through the neural networks by predicting for every center allele its neighboring alleles from the changes in allele frequency, and the data application using the time-sampled whole-genome sequences of echovirus 11 was carried out. The results show that the alleles rising above the sequencing error can be represented effectively as distributed vectors from genetic sequence data, as compared to being represented as discrete and independent entities in most classic population genetic models. Distributed vector representations of alleles learned from the nucleotide skip-gram neural network have the advantage of incorporating the comprehensive interactions from a given window of neighboring alleles entirely from input data without model assumptions.
Using PCA, the pair-wise correlation map between these allele embeddings is generated and arranged by agglomerative hierarchical clustering, which unveils the similarity in the evolutionary trajectory between these alleles. In comparing with the previous result by Zhong et al, 27 the clusters with the window size of M/4 are the most plausible, representing a moderate recombination rate which is also consistent with the known echovirus biology. Furthermore, a few non-synonymous mutations are identified to have had evolved similarly to the candidate mutations of adaptation. For the exposed population, this new approach using the nucleotide skip-gram neural network identified 2 candidate mutations (P129Q and K259E) in the same cluster, which supports the presence of potential genetic interactions in the structural protein of VP1 in adapting to the challenging environment. For the non-exposed population, our analysis reveals a list of non-synonymous mutations in the same cluster as the candidate mutation (H215N) in contrary to the previous analysis. However, this feature learning with the nucleotide skip-gram neural network has the caveat that interacting alleles can only be identified when the candidate mutations are known a priori. Thus, it remains a future challenge to expand this approach to identify candidate mutations responsible for patterns of selection in genetic datasets without prior knowledge.
Through the application of neural networks in genetic data, we aim to develop a computational method that can be a tool of prediction for future experiments, as compared to a tool of inference from experimental data like the other likelihood-free methods.6–8 In this study, we were able to extract evolutionary features from the genetic data of a time-sampled evolution experiment and predict clusters of mutations that may be informative for future experiments such as functional validation or network analysis. The advantages of the nucleotide skip-gram neural network model include the absence of manual over-specification and model assumptions as needed in the previous studies of genetic interactions. Thus, the nucleotide skip-gram neural network is a flexible platform that applies to a wide range of genetic data to define alleles with distributed vectors, which encode information about their interactions with neighboring alleles during the course of evolution. The automated workflow of the platform can easily be adapted to each investigation, for instance, to consider new mutations arising in a replicate experiment. Furthermore, this neural network platform has the potential to be applied to larger and more complex datasets, such as for organisms with bigger genomes or for data from natural populations. The nucleotide skip-gram neural network has a simple architecture of 1 hidden layer that minimizes computational complexity, which makes the platform ideal for much bigger datasets such as human genomes with approximately 3 billion base pairs. The caveat of this current data application is that the time-sampled whole-genome virus datasets from this experimental evolution are actually “too small” for the capacity of neural networks which only takes a few minutes to train in TensorFlow, potentially leaving the results under-trained and sub-optimal. For future investigations, this method can be applied to time-sampled datasets from human cancer cells to decipher the interactions between the mutations that arise during the course of cancer evolution, or to spatial datasets from natural populations of humans or drosophila to decipher spatial rather than temporal interactions between the alleles of interest. Furthermore, biological factors such as recombination rate can be represented in more accurate ways to investigate whether the patterns of interaction are produced by deterministic or stochastic evolutionary forces.
Supplemental Material
Supplementary_Material – Supplemental material for Feature Learning of Virus Genome Evolution With the Nucleotide Skip-Gram Neural Network
Supplemental material, Supplementary_Material for Feature Learning of Virus Genome Evolution With the Nucleotide Skip-Gram Neural Network by Hyunjin Shim in Evolutionary Bioinformatics
Footnotes
Acknowledgements
We thank Christopher Bernido, Maria Victoria Carpio-Bernido, Valeria Montano, Chip Huyen, Minwoo Sun, and Diego Marcos Gonzalez for helpful discussions. We also thank Qingxia Zhong and Anna Carratalà for providing the experimental dataset of echovirus.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Swiss National Science Foundation (grant number P1ELP3_168490 to H.S.) and Firmenich EPFL-Stanford research exchange program.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
HS conceived of the presented idea, designed the analysis tool, performed the analysis and wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.