
Abstract
Obligate intracellular chlamydiae diverged into pathogenic and environmental chlamydiae 0.7-1.4 billion years ago. While pathogenic chlamydiae have adapted to a wide range of vertebrates, environmental chlamydiae inhabit unicellular amoebae, the free-living Acanthamoeba. However, how and why this divergence occurred remains unclear. Meanwhile, giant viruses consisting of protozoa-related and protozoa-unrelated viruses have been discovered, with the former group being suggested to have more influenced environmental chlamydiae during their evolution while cohabiting host amoebae. Against this background, we attempted to visualize genes of giant viruses in chlamydial genomes by bioinformatic analysis mainly with comparative genome and phylogenic analysis, seeking genes present in chlamydiae that are specifically shared with protozoa-related giant viruses. As a result, in contrast to protozoa-unrelated giant viruses, the genes of protozoa-related giant viruses were significantly shared in both the chlamydia genomes depending on the giant virus type. In particular, the prevalence of Mimiviridae genes among the protozoa-related giant virus genes in chlamydial genomes was significantly high. Meanwhile, the prevalence of protozoa-related giant virus genes in pathogenic chlamydia genomes was consistently higher than those of environmental chlamydiae; the actual number of sequences similar to giant virus was also significantly predominant compared with those in the environmental chlamydial genomes. Among them, the most prevalent of giant virus was in the case of chlamydiae with Megavirus chiliensis; total of 1338 genes of the chlamydiae were found to be shared with the virus (444 genes specific to environmental chlamydiae, 892 genes shared between both chlamydiae, only two genes in the pathogenic chlamydiae). Phylogenic analysis with most prevalent sets (Megavirus chiliensis and Protochlamydia EI2 or Chlamydia trachomatis L2 434Bu) showed the presence of orthologs between these with several clustered. In addition, Pearson’s single regression analysis revealed that almost the prevalence of the genes from the giant viruses in chlamydial genomes was negatively and specifically correlated with the number of chlamydial open reading frames (ORFs). Thus, these results indicated the trace of lateral gene transfer between protozoa-related giant viruses of family Mimiviridae and chlamydiae. This is the first demonstration of a putative linkage between chlamydiae and the giant viruses, providing us with a hint to understand chlamydial evolution.
Introduction
Obligate intracellular chlamydiae separated into the environmental chlamydiae (eg, Parachlamydia, Protochlamydia, Neochlamydia) and the pathogenic chlamydiae (eg, Chlamydia trachomatis, C. pneumoniae) 0.7-1.4 billion years ago. 1 Pathogenic chlamydiae, which are the causative agents of human infectious diseases including sexually transmitted diseases and pneumonia, have adapted to a wide range of vertebrates.2–4 In contrast, environmental chlamydiae inhabit unicellular amoebae, the free-living Acanthamoeba, in a symbiotic relationship, being distributed across a huge range of environments, including soil, ponds, and places where people live and work.1–5 However, it remains unclear whether the amoebal symbiotic chlamydiae can also cause infectious diseases in humans.6,7 Meanwhile, ancestral amoebae are thought to have emerged 1 billion years ago, corresponding to the time at which the chlamydial ancestor diverged into two types, 8 which presumably occurred in a setting that facilitated chlamydial evolution.
A number of recent studies have revealed that the genomes of environmental chlamydiae (2.0-3.0 Mb) are more than double the size of those of pathogenic chlamydiae (1.0-1.2 Mb).1,9–12 It is thus clear that environmental chlamydiae still possess certain genes that pathogenic chlamydiae have lost. Meanwhile, similar to pathogenic chlamydiae, environmental chlamydiae undergo a unique developmental process, consisting of two distinct forms: the elementary body, its infectious form, and the reticulate body, its replicative form.12,13 We also found that some environmental chlamydiae could grow in immortalized human cells.12,13 It is thus clear that these two types of chlamydiae share similar backgrounds. However, the environmental factors that are responsible for promoting the divergence that occurred during chlamydial evolution and resulted in these two groups remain unknown.
During the last 10 years, giant viruses, which can be visualized under a light microscope, have been discovered and shown to have similar genes to those in other organisms, particularly those in several types of bacteria and in eukaryotes.14–16 The giant viruses consist of two distinct groups, protozoan-related and protozoan-unrelated types. The protozoan-related giant viruses include the families Mimiviridae (eg, Mimivirus), Marseilleviridae, Pandoraviridae, Pithovirus, and Mollivirus, all of which can infect amoebae; they are ubiquitous in the environment, including in soil and the water supply. 16 The protozoan-unrelated giant viruses include the families Ascovirus, Irdovirus, and Poxvirus, all of which were isolated from vertebrates and invertebrates. 16 Meanwhile, some giant viruses have also been isolated from patients suffering from pneumonia, indicating their potential pathogenicity to humans, although this remains to be confirmed.15,17,18 As mentioned above, similar to environmental chlamydiae, protozoa-related giant viruses also need to infect amoebae in order to replicate, indicating that the giant viruses could encounter environmental chlamydiae during the course of their life span; this would also have been the case for ancestral chlamydiae.
In the present study, we thus attempted to visualize genes of giant viruses in chlamydial genomes by bioinformatic analysis mainly with comparative genome and phylogenic analysis, seeking genes present in chlamydiae that are specifically shared with protozoa-related giant viruses. For the first time, we show a linkage between chlamydiae and protozoan-related giant viruses.
Materials and Methods
Data sets
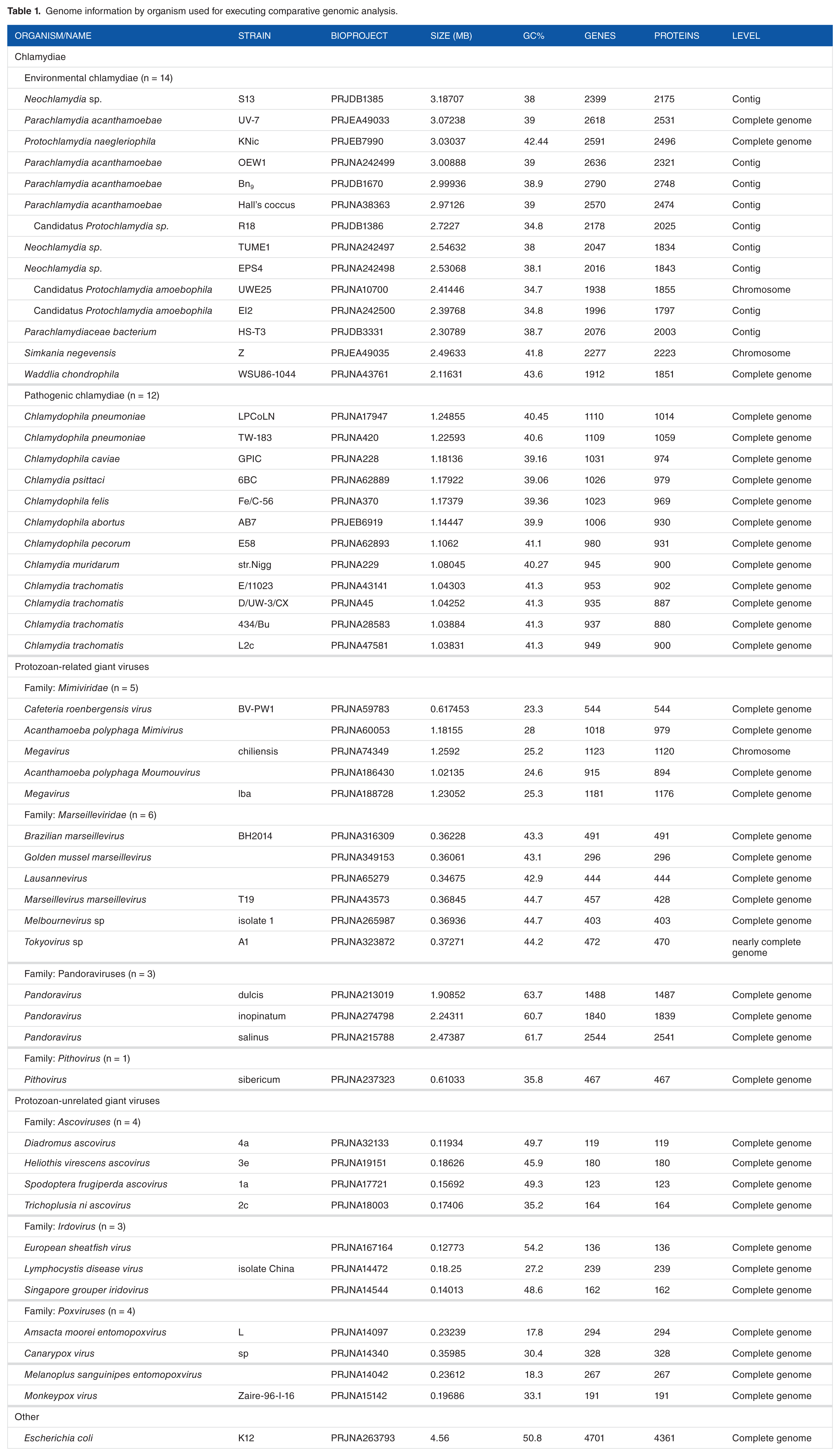
Chlamydiae and others (environmental chlamydiae n = 14, pathogenic chlamydiae n = 12, protozoan-related giant viruses n = 15, protozoan-unrelated giant viruses n = 11, others n = 1 (Escherichia coli K12)) were used for this study (Table 1).
Genome information by organism used for executing comparative genomic analysis.
Analysis flow
The genome (or contig) information was obtained from the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/genome/browse/), and the obtained sequence information was reconstructed as data sets with functional annotations into Rapid Annotation using Subsystem Technology (RAST) (http://rast.nmpdr.org/), which is an open-access genomic analysis tool that acts as a fully automated service for genomic annotation with Basic Local Alignment Search Tool (BLAST) analysis. 19 These reconstructed RAST data sets with annotated amino acid sequences are shown into Tables S1 to S4 (Table S1, protozoan-related giant viruses; Table S2, protozoan-unrelated giant viruses; Table S3, pathogenic chlamydiae; Table S4a and b, environmental chlamydiae). Then, BLAST analysis was performed using the RAST data sets with the default settings (cut-off 10−10 identity >10%), and these sequences were furthermore selected with bidirectional hits and length cut-off (>30 amino acid residues). Numbers of orthologs were normalized with genome sizes of both chlamydiae and viruses. Specifically, the normalized numbers were obtained from raw numbers divided with each of the chlamydia and virus genome sizes; it is shown as ortholog numbers of giant virus assumed with 1 Mbp of genome size per 1 Mbp of chlamydial genome. Also, the cut-off value (>1.48%) as a background was defined by the prevalence of genes from Mimiviridae (Cafeteria roenbergensis virus, Megavirus (lba and chiliensis), Moumouvirus, Mimivirus) in the genome of Escherichia coli K12, which has never adapted to protozoa (mean ± 2SD: 1.28 ± 0.2%) (Table S5). The identity of the extracted genes was finally determined by Simple Modular Architecture Research Tool (SMART; http://smart.embl-heidelberg.de/), which is a domain research tool. 20 Functional annotation was also performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) 21 or the Universal Protein Resource (UniProt) (http://www.uniprot.org/). 22 Annotated functions were classified into “Metabolic process” (associated with the metabolism of proteins, lipid, DNA, RNA, and so on), “Regulation/modification” (associated with DNA/RNA repair, homologous recombination, chaperones and folding catalysts, protein-protein interaction, and so on), “Structure” (flagella, outer membrane protein, and so on), and “Others” (associated with structure and including those with an unknown function). Phylogenic analysis was performed with a maximum parsimony method by using MAFFT version 7 (https://mafft.cbrc.jp/alignment/software/). 23
Statistical analysis
Comparison of the prevalence of giant virus genes between pathogenic and environmental chlamydiae was performed by Mann–Whitney’s U test. The presence of a correlation between the prevalence of genes from giant viruses within chlamydial genomes and annotated chlamydial ORF numbers was determined by Pearson’s single regression analysis. A correlation coefficient value of >0.5 or <−0.05 with a P-value of less than .05 was considered significant. Calculations were performed in Excel for Mac (2011) with Statcel3C.
Results and Discussion
Several genes of protozoa-related giant viruses of the family Mimiviridae are significantly conserved in the genomes of both chlamydiae
To explore the traces of an encounter with giant viruses in chlamydiae, the prevalence of genes from giant viruses in chlamydiae was assessed by BLAST analysis using RAST with genomic information from multiple species from each group (environmental chlamydiae n = 14, pathogenic chlamydiae n = 12, protozoan-related giant viruses n = 15, protozoan-unrelated giant viruses n = 11)(see Table 1). To ensure a uniform annotation of all the genes, pre-existing annotations in the database were re-annotated by RAST (http://rast.nmpdr.org/), which is an open-access genomic analysis tool that acts as a fully automated service for genomic annotation with BLAST analysis. Also, the cut-off value (>1.48%) as a background was defined by the prevalence of genes from Mimiviridae in the genome of Escherichia coli K12, which has never adapted to protozoa (see Table S5). As a result, in contrast to protozoa-unrelated giant viruses, the genes of protozoa-related giant viruses were significantly shared in both the chlamydia genomes depending on the giant virus type (Figure 1). In particular, the prevalence of Mimiviridae genes among the protozoa-related giant virus genes in chlamydial genomes was significantly high, exceeded the cut-off value (Figure 1A, green circles into Mimiviridae). Meanwhile, the prevalence of the genes of giant viruses within chlamydial genomes varied from 0.2% to 3.5% depending on not only the giant virus type but also the chlamydial strain (Figures S1 and S2). Furthermore, the prevalence of protozoa-related giant virus genes in pathogenic chlamydia genomes was consistently higher than those of environmental chlamydiae (Figure 1A and B), corresponding to number of sequences normalized with genome sizes of both chlamydiae and viruses similar to giant virus that was significantly predominant as compared with those in the environmental chlamydial genomes (Figure S3). In addition, phylogenic analysis with most prevalent sets (Megavirus chiliensis and Protochlamydia EI2 or Chlamydia trachomatis L2 434Bu) clearly showed that there was several clusters, indicating the presence of orthologs between the chlamydiae and the giant viruses (Figure 2).

Comparisons of the prevalence rates of giant virus genes in chlamydial genomes and of the trend of dispersion on the prevalence of giant virus genes between pathogenic and environmental chlamydiae. Panels (A) and (B) show protozoa-related giant viruses and protozoa-unrelated giant viruses, respectively. Blue and red bars show the prevalence of giant virus genes in environmental and pathogenic chlamydial genomes, respectively. Comparisons of the prevalence rate were conducted using Mann–Whitney’s U test. Stars show a significant difference (P < .05) between the prevalence values of environmental and pathogenic chlamydiae. Green circles show a significant difference in the prevalence rate of giant virus genes with values more than cut-off. Cut-off (1.48%) as a background value (dashed line) was defined by the prevalence of genes from Mimiviridae (Cafeteria roenbergensis virus, Megavirus chilensis, Megavirus lba, Mimivirus, Moumouvirus) in the Escherichia coli K12 genome (1.28 ± 0.19%) (see Table S5).

Phylogenic analysis with most prevalent sets (Megavirus chiliensis and Protochlamydia EI2 or Chlamydia trachomatis L2 434Bu) showing several clusters. Trees (A) and (B) show Megavirus chiliensis (MegaVirus) with Protochlamydia EI2 (Proto_EI2) and with Chlamydia trachomatis L2 434Bu (Chlt_L2), respectively. Additional numbers (peg) show gene ID numbers assigned by RAST (see Table S1 to S4b). Black circles show these chlamydial genes. Phylogenic trees were constructed with a maximum parsimony method by using MAFFT version 7 (https://mafft.cbrc.jp/alignment/software/). 23
As expected, we found that, in contrast to the protozoa-unrelated viruses, several genes of protozoa-related giant viruses, the family Mimiviridae (Megavirus chiliensis was most prevalent) were significantly conserved in the genomes of both the chlamydiae. Meanwhile, as compared with those of pathogenic chlamydiae, the prevalence of Mimiviridae genes was found to more differ among the various genera of environmental chlamydiae, being particularly high in Neochlamydia (S13, TUM1, EPS4) and Protochlamydia (UWE25, R18) and contrastingly low in Parachlamydia (UV-7, KNic, OEW1, Bn9, Hall’s coccus) (see Figure S1). It is possible that the selection and maintenance of the giant virus genes occurred preferentially in some environmental chlamydiae through the ongoing interaction, presumably into cohabiting amoebae. Thus, these findings indicated that, in contrast to the protozoa-unrelated viruses, several orthologs of protozoa-related giant viruses, in particular Mimiviridae, were more conserved in the genomes of either environmental or pathogenic chlamydiae, suggesting that chlamydiae and Mimiviridae did interact in the host cells that both cohabited.
The prevalence of genes from protozoa-related giant viruses in chlamydiae is negatively and specifically correlated with chlamydial ORF numbers
If the prevalence of genes from giant viruses in chlamydial genomes specifically revealed that chlamydiae had encountered protozoa-related giant viruses presumably in ancestral amoebae, this would also suggest that this encounter resulted in specific modifications of the chlamydial genome, such as changes of the ORF numbers. To assess this hypothesis, the correlation between the prevalence of giant virus genes in chlamydial genomes and the chlamydial ORF numbers was assessed by Pearson’s single regression analysis. The results showed significant correlation coefficients of <−0.5 with a P-value <.05 for almost combinations of chlamydiae with giant viruses, indicating the prevalence of the genes from giant virus in chlamydial genomes could be a factor predicting the number of chlamydial open reading frames (ORFs) (Table S6).
The prevalence of the genes from almost giant viruses in each of the chlamydiae was negatively and specifically correlated with the number of chlamydial ORFs. These results suggest that these giant viruses changed the chlamydial genome and influenced chlamydial evolution. Interestingly, studies have shown that Protochlamydia (UWE25 or R18) can induce cell death such as apoptosis in insect cells or human immortal HEp-2 cells,24–26 while Neochlamydia (S13) was found to exhibit complete loss of its ability to perform secondary infection of amoebae. 27 These findings also suggest the presence of a sympatric lifestyle between the viruses and chlamydiae and that such selection and maintenance of the giant virus genes were required for the successful specific adaptation of chlamydiae to the host niche.
In contrast to the pathogenic chlamydiae, the environmental chlamydiae specifically possess genes conserved among the Mimiviridae (Megavirus chiliensis)
Since information on the specific genetic material that was shared would be critical for understanding the forces driving the evolution and divergence of chlamydiae, we explored the specific genes of chlamydiae commonly shared with protozoa-related giant viruses, the Mimiviridae. Meanwhile, because of most prevalent, Megavirus chiliensis as a representative virus was used for this analysis. As shown in Figure 3 and Table S7, a total of 1,338 genes of the chlamydiae were found to be shared with the virus (444 genes specific to environmental chlamydiae, 892 genes shared between both chlamydiae, only two genes in the pathogenic chlamydiae). Although these genes were classified into the categories of “Metabolic process,” “Regulation/modification,” “Structure”, and “Others”, almost genes (approximately 60%) were assigned to “Metabolic process” regardless of pathogenic or environmental chlamyidae (Figure 3, pie charts in the center). Meanwhile, as well as some genes of “Metabolic process”, the genes assigned to “Structure” (surface protein Sur1, phage tail fiber protein, outer membrane lipoprotein Blc, flagellar hook-length control protein FliK) was specifically seen into environmental chlamydiae (Figure 3 and Table S3). Furthermore, few genes were multiply conserved in almost all of the environmental chlamydiae used in this study (Figure 3 and Table S3).

Total number of functional genes in each of the chlamydial genomes shared among protozoa-related giant viruses in the Mimiviridae (Megavirus chilensis). The genes shared between protozoa-related giant viruses in the Mimiviridae (Megavirus chilensis) and each of the chlamydiae were extracted, from a comparative genome analysis with RAST (see filter conditions into Material and Methods). Functional annotation was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) 23 or the Universal Protein Resource (UniProt). 24 Upper panel: specific to environmental chlamydiae; Middle panel: shared between both chlamydiae; Lower panel: specific to pathogenic chlamydiae. Colors show distinct gene functions annotated by KEGG or UniProt. Pie charts in the center show the prevalence of genes classified into the categories of “Metabolic process,” “Regulation/modification,” “Structure,” and “Others.”
Thus, these findings indicated that, in contrast to the pathogenic chlamydiae, the environmental chlamydiae specifically possessed functional genes conserved among the Mimiviridae responsible for “Metabolic process” or “structure” presumably as a platform for survival into harsh natural environments and as a trace of ongoing interaction of the chlamydiae with giant viruses. It is possible that because of the presence of genes from Mimiviridae presumably with adverse effects, the loss of such genes in chlamydiae may have been a critical event required for adaptation to mammalian cells. Furthermore, a large number of protozoa-related giant virus genes shared between both chlamydiae. It appeared that the some giant virus genes were passed down through the generations and became fixed evenly in both environmental and pathogenic chlamydiae, implying before dividing two chlamydial lineages, ancestral chlamydiae had encountered giant viruses. Interestingly, only two genes specific to the pathogenic chlamydiae were detected into Megavirus chiliensis. The results revealed that in contrast to environmental chlamydiae, ongoing interaction of pathogenic chlamydiae with giant viruses may be minimal, presumably prompting pathogenic chlamydial genome reduction. 28
Conclusions
Altogether, our study showed a putative linkage between chlamydiae and protozoa-related giant viruses, in particular Mimiviridae. These results indicated the trace of lateral gene transfer between protozoa-related giant viruses of family Mimiviridae and chlamydiae. This is the first demonstration of the linkage, providing us with a hint to understand chlamydial evolution via encounters with giant viruses in host niche.
Supplemental Material
Supplemental_Material – Supplemental material for Lateral Gene Transfer Between Protozoa-Related Giant Viruses of Family Mimiviridae and Chlamydiae
Supplemental material, Supplemental_Material for Lateral Gene Transfer Between Protozoa-Related Giant Viruses of Family Mimiviridae and Chlamydiae by Takanori Watanabe, Sumire Yamazaki, Chinatsu Maita, Mizue Matushita, Junji Matsuo, Torahiko Okubo and Hiroyuki Yamaguchi in Evolutionary Bioinformatics
Footnotes
Acknowledgements
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants-in-aid for Japan Society for scientific research KAKENHI (grant number: 16K15270). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
HY conceived and designed the project; TW, SY, CM, MM, JM, TO, and HY contributed toward the analysis and confirmation; YH, SY, JM, TO, and HY contributed toward critical editing; HY wrote the manuscript. All authors read and approved the final manuscript.
Ethical approval
The study reported in this manuscript did not involve any human participants, human data, human tissue, data on specific individuals, or animal experiments.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.