
Abstract
Halenia elliptica is a popular Chinese medicinal herb that is used to treat jaundice disease and virus hepatitis, and its wild populations have been reduced significantly due to overharvesting recently. However, effective conservation could not be implemented because of the lack of genomic information and genetic markers. In this study, a de novo transcriptome of H elliptica was sequenced using the NGS Illumina, and 132 695 unigenes with the length >200 bp (base pairs) were obtained. Among them, a total of 32 109 unigenes were scanned to develop simple sequence repeats (SSRs). Based on NCBI (National Center for Biotechnology Information) nonredundant database (Nr), these SSR sequences were annotated and assigned into gene ontology categories. In addition, we designed 126 pairs of SSR primers for polymerase chain reaction amplification, of which 12 pairs were identified to be polymorphic among 40 individuals from 8 populations. We then used the 12 polymorphic SSRs to construct a UPGMA dendrogram of the 40 individuals. In addition, a significant correlation between the genetic relationship and the geographic distance was found, suggesting a phylogeographic structure in H elliptica. Moreover, 2 of these SSRs were also successfully amplified in a related species Veratrilla baillonii, suggesting their cross-species transferability. Generally, the SSR markers with high polymorphisms identified in this study provide valuable genetic resources and represent an initial step for exploring the genetic diversity and population histories of H elliptica and its related species.
Keywords
Introduction
Over the past decade, life sciences were greatly advanced based on the genome sequencing technologies, especially the next-generation sequencing (NGS) that provides a strategy of a low cost in sequencing and large quantities of genomic data. Based on NGS, a great number of sequenced genomes have been obtained in a short time, which enhanced our understanding on variations of genome sequence. 1 RNA-Seq of NGS is advantageous over chip technology on the digital region and can produce explicit transcriptome data for nonmodel species. The genome-scale transcriptome analysis is powerful in nonmodel species by revealing differential expressions of genes in time and spaces, determining the genetic basis of specific phenotypes, and outlining genomic diversity.2,3 In addition, a lot of simple sequence repeat (SSR) markers can be rapidly developed based on the genome-scale transcriptome analysis, which would be of great help in analyzing population genetic structure.4,5
The SSRs consist of short tandem repeats of 1 to 6 bp (base pair) nucleotides and are abundant in protein-coding and noncoding regions. The SSRs are highly diverse, codominant, and stable and thus were extensively used in many research subjects, such as evolutionary biology, population genetics, and conservation genetics. 6 In the past, it takes long time and high cost to obtain SSR markers, whereas RNA-Seq makes it easy to develop a great deal of SSR markers in the present time,3,7,8 which promoted research works in the genetic diversity and evolutionary biology. 9
Halenia elliptica D. Don, a biennial herb in the Gentianaceae family, is a popular Chinese medicinal herb that is widely used to treat jaundice disease and virus hepatitis. This species is mainly distributed at elevations ranging from 700 to 4000 m in Yunnan, Sichuan, Qinghai, and Tibet. 10 Due to its effective therapeutic effects, H elliptica was overexploited, leading to a decrease in the population size and genetic diversity in recent years. It is difficult to propose effective conservation methods without genome information and genetic markers.
In this study, we sequenced a de novo transcriptome for H elliptica on the Illumina platform and assembled the transcriptome sequences with software Trinity. As far as we know, this is the first exhibit of transcriptome results for H elliptica. In addition, we screened SSR markers in the transcriptome sequences and randomly selected markers to verify their amplification and polymorphism. The transcriptome sequence and polymorphic SSR markers developed in this work are believed to provide valuable genetic resources to study genetic diversity and population demographic history of H elliptica and its related species.
Materials and methods
Plant material
On July 2016, the fresh leaves of 10 H elliptica individuals were collected from Shangri-La in northwest Yunnan (28°31ʹ0ʹʹN, 99°57ʹ0ʹʹE, alt. 4514 m) and were kept immediately and separately in liquid nitrogen. In addition, a total of 40 individuals from 8 populations (Supplementary Table 1) were sampled and the leaves were stored in silica gel for polymorphic SSR markers validation.
RNA extraction and sequencing
We extracted the total RNA of each individual with a CTAB method 11 and measured the integrity of the RNA samples using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). To satisfy the criteria for RNA sequencing, equal amount of RNA from each replicate RNA was pooled together. We constructed the complementary DNA library using poly-A–enriched RNA method and fragmented the messenger RNA with fragmentation buffer based on Illumina protocols (San Diego, CA, USA). Random hexamer primers were used to synthesize the double strands. The short fragments were purified using the QIAquick PCR Purification Kit (Qiagen Inc., Courtaboeuf, France). Ultimately, the purified DNA libraries were first amplified by polymerase chain reaction (PCR) and then sequenced on Illumina HiSeq 2000 platform.
De novo assembly
The reads with many ambiguous bases (>8) and with more than 50% low-quality bases (quality score ⩽5) in raw reads were filtered out using Perl scripts. The transcriptome sequences of H elliptica were assembled using Trinity software with default parameters. 12
SSR locus search, primer design, and validation
To detect the potential SSR loci, all the contigs were scanned by MicroSAtellite software (MISA, http://pgrc.ipk-gatersleben.de/misa). 13 In general, the SSR locus search minimum requirements were 5 repeats for the simple motifs and 3 repeats for the complex motifs. In this study, we set the minimum repeat unit as 10 for mononucleotides, 6 for dinucleotides, and 5 for trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides in MISA. The SSR primer pairs were designed with Primer 3.0. 14 The designed primer pairs in the study were excellent, and each target SSR was required to contain at least 5 repeats, with the length of PCR products ranging from 80 to 500 bp. Based on the above criteria, 126 primer pairs were randomly synthesized for validating SSR locus (Supplementary Table 2), of which 62 primer pairs were successful in the PCR products. Then, the successful primer pairs in PCR amplification were used to detect the polymorphism among 40 individuals from 8 populations (Supplementary Table 1). Polymerase chain reaction was performed in a 25-μL volume, and the PCR reaction program was set as the following conditions: (1) DNA initial denaturation was 4 minutes at 94°C, 35 cycles of 1 minute 30 seconds at 94°C; (2) the annealing temperature ranged from 50°C to 60°C for 50 seconds, following 72°C for 45 seconds; and (3) an extension was 7 minutes at 72°C. The PCR products were sequenced on the ABI 3730 genetic analyzer (Applied Biosystems, Foster City, CA). The statistics of polymorphic SSR loci were calculated using POPGEN v1.32. 15
Functional annotation for contigs containing SSRs
All the SSR-containing contigs were used to search objective sequences in the NCBI’s NR protein database using BLASTx with the E-value threshold setting as 1e−6. The contigs were assigned with gene names according to best BLASTx hits. Functional annotation of contigs was conducted by the program Blast2GO. 16 Functional categories were classified with the program WEGO. 17
Genetic analyses in populations
We conducted phylogenetic analysis of the 40 individuals from 8 populations with the 12 primer pairs and used software MEGA6 18 to construct the dendrogram tree using the UPGMA method.
Results and Discussion
De novo assembly of H elliptica
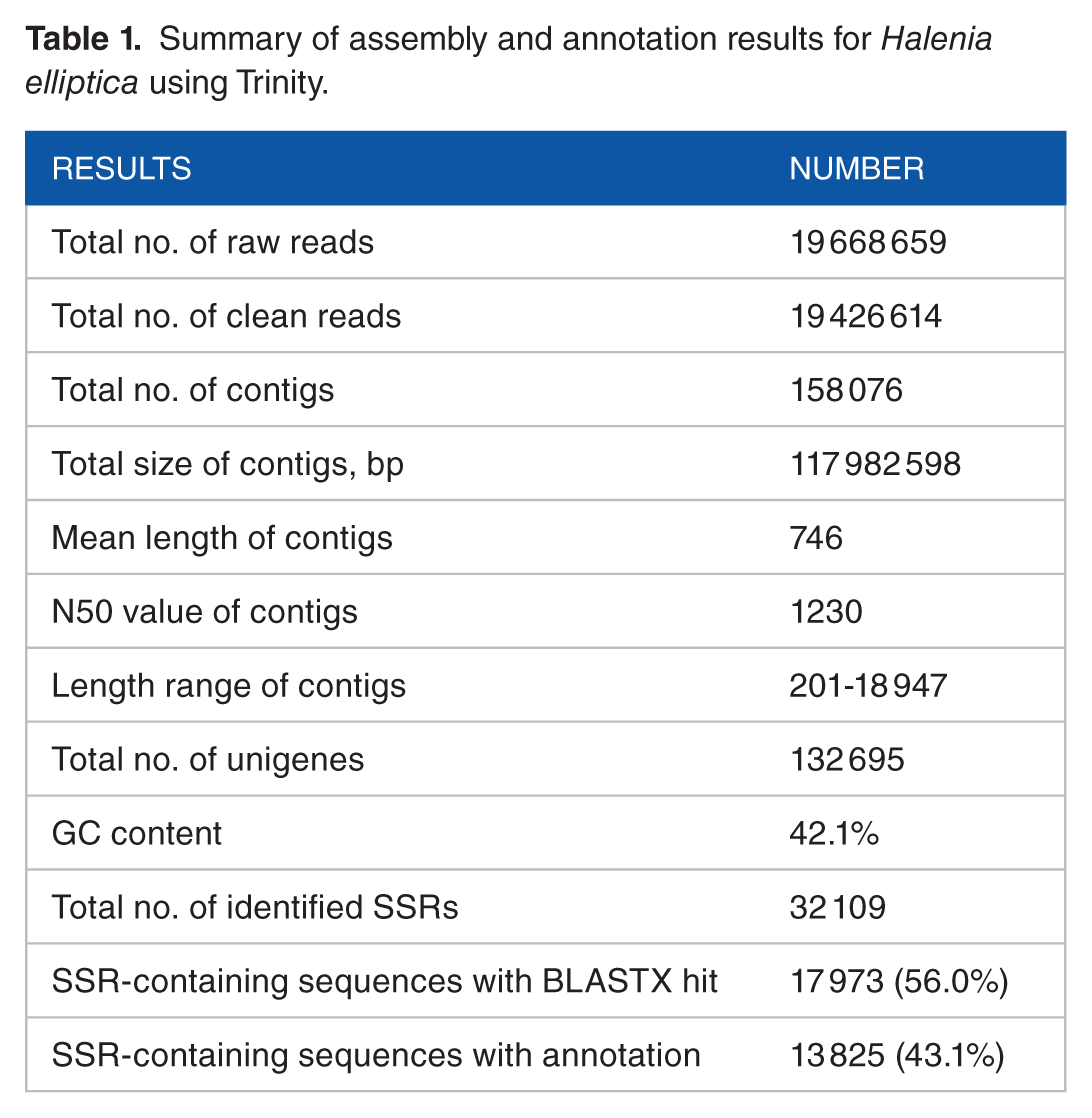
A total of 19 668 659 raw reads data were generated by the Illumina HiSeq sequencer. After all adaptor sequences were removed, the ambiguous and low-quality sequences were filtered, a total of 19 426 614 RNA-Seq clean reads remained for further analysis, which had been deposited into the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/) with accession number SRP126366. These clean reads were used to assemble the contigs with Trinity software, which generated 158 076 contigs with the length ranging from 201 to 18 947 bp. The median length and the N50 value of the contigs are 450 and 1230 bp, respectively. The high N50 value suggested the high quality of the assembly. The GC content of our contigs is 42.1% (Table 1). With the increase in contig length, the frequency of the contigs decreased, suggesting a power law–like distribution. The contigs with lengths ranging from 200 to 500 bp were dominant, making up 58.14% of the total contigs (Figure 1), as detected in Veratrilla baillonii 4 and Gentiana straminea 19 in the same family of Gentianaceae.
Summary of assembly and annotation results for Halenia elliptica using Trinity.

Number of sequences for all 158 076 transcriptome contigs for Halenia elliptica.
Validation and distribution of SSRs
Using MISA software, we analyzed the contigs, and 32 109 SSRs of total 158 076 contigs were validated. The number of SSRs identified in H elliptica was higher than that in G straminea (14 561), but lower than that in V baillonii (40 885). The density of SSRs for H elliptica was 244.3 per MB, similar to V baillonii (243.3).
Based on different sizes, SSR loci were classified into 2 categories of genetic markers. Class I was hypervariable markers with the length of SSRs more than 20 bp, and Class II was potentially variable markers with the length of SSRs ranging from 12 to 20 bp. Because of the large sizes and long repeats, SSRs of Class I generally have much information and high polymorphism, which are beneficial in developing SSR markers. Owing to the small sizes, SSRs of Class II are less variable and thus are difficult to find polymorphism with SSR markers. In H elliptica, 23.9% of the SSRs were categorized as Class I and 76.1% as Class II. The proportion of Class I in H elliptica was higher than that in V baillonii (6.8%), but the fraction of SSRs was still deficient for further development of efficient and polymorphic SSR markers.
The detailed information of SSRs with different repeat styles is showed in Table 2. The result suggested the dominance of the SSRs with mononucleotide motifs, which accounted for 65.4% of the total. Without regard to the mononucleotide, trinucleotide, and dinucleotide motifs were predominant in quantity, accounting for 50.2% and 42.5% among the rest of these motifs, respectively. The total number of tetranucleotide, pentanucleotide, and hexanucleotide motifs was 2.8% of the total SSRs, close to V baillonii (2.7%). Moreover, the mode of SSRs distribution in H elliptica is similar to that of V baillonii. In mononucleotide motifs, the frequency of (A/T)n type was 96.8%, a situation found in many plant species.4,20 In dinucleotide repeat motifs, the frequencies of AT/AT, AG/CT, and AC/GT were 65.5%, 19.1%, and 14.4%, respectively. In contrast, the content of CG/CG was lower, contributing to 1% only in H elliptica. These results are similar to that of Gentianaceae family and also in accordance with other dicot genomes in which A/T-rich repeats in trinucleotide motifs were frequent. 21 In general, AAC/GTT, AAG/CTT, and AAT/ATT were extensive in dicot genomes,3,8 and our results are consistent with this conclusion. In H elliptica, the repeats AAC/GTT, AAG/CTT, and AAT/AAT were dominant with the frequencies of 6.6%, 18.0%, and 16.9%, respectively, and the total frequency of the trinucleotide motifs amounted to 41.5%.
Frequency of mono- to hexanucleotide repeat motifs in Halenia elliptica.

In general, the feature of SSRs distribution in H elliptica was similar to that of V baillonii, indicating the close phylogenetic relationship and high similarity in transcriptome level between the 2 species. However, the similarity between the 2 species may also suggest their similar evolutionary histories because H elliptica originated from the East Asia 22 and V baillonii are restricted in the Hengduan Mountains. 10
Functional annotation based on SSR-containing coding sequences of H elliptica
Using BLASTx, a lot of the SSR-containing contigs (17 973) were detected to have no less than one hit in the NCBI’s NR protein database, but the percentage (56.0%) was lower than that of V baillonii (70.7%). A further analysis of the contigs was implemented by the program Blast2GO. The contigs were assigned with gene names based on best BLASTx hits, and 13 825 SSR sequences were annotated. We conducted WEGO 17 to achieve functional categories. The contigs were classified into 3 categories of gene ontology terms, respectively, as cellular component, molecular function, and biological processes (Figure 2). Within the cellular component category, the cell and cell part were the most abundant types. Within the molecular function category, catalytic activity was the most dominant group, followed by binding. For the biological processes category, cellular process and metabolic process were the most common.

GO classification of SSRs in coding regions. GO indicates gene ontology; SSRs, simple sequence repeats.
Polymorphism of SSR markers and phylogenetic analysis
To obtain polymorphic SSR markers, we designed 126 pairs of SSR primers for PCR amplification in 8 populations, and the PCR amplification of the 62 SSRs pairs was successful. The validated primers were used to screen genetic polymorphism of H elliptica with 40 individuals (Supplementary Table 1). And 12 pairs of these SSRs with polymorphism were found. The number of alleles per locus varied from 3 to 10, and the expected heterozygosity ranged from 0.023 to 0.530, whereas the observed heterozygosity varied between 0.000 and 0.400 (Table 3). Polymorphism information content values of the SSR markers varied between 0.0714 and 0.7852. Then, we used UPGMA to construct a phylogenetic tree of the 40 individuals. There was a significant correlation between the genetic relationship and the geographic distance in the phylogenetic tree of H elliptica (Figure 3), suggesting a phylogeographic structure in H elliptica. Further study with more populations is required to reveal the dynamic evolutionary history of H elliptica.

UPGMA dendrogram constructed among 40 individuals from 8 populations based on 12 SSR markers developed in this study. SSR indicates simple sequence repeats.
Results of primer screening through 40 diversified accessions in Halenia elliptica.

Abbreviations: He, expected heterozygosity; Ho, observed heterozygosity; Na, number of alleles; Ne, effective number of alleles; PIC, polymorphism information content; size, size of cloned allele; Ta, annealing temperature.
The 12 SSRs validated in this study exhibited high-quality and high genetic polymorphism, which allows us to analyze the genetic diversity and dynamic evolutionary history of H elliptica. The further analysis will provide us conservation strategies for this traditional medicinal plant. In addition, 2 pairs of these SSRs primers are also available in V baillonii, which suggests their transferability in the related species of H elliptica.
Conclusions
In this study, the de novo transcriptome for H elliptica was determined with RNA-Seq. A number of microsatellite markers were identified and 126 pairs of SSR primers were designed for PCR amplification. We found 12 SSR markers to be polymorphic, which can be used for future studies in H elliptica. The SSR markers with polymorphism identified in this study provide valuable genetic resources and represent an initial step for exploring the genetic diversity and population history of H elliptica and its related species.
Supplemental Material
Supplementary_Table_of_SSR_marker_for_Halenia_elliptica – Supplemental material for Transcriptome Analysis and Microsatellite Markers Development of a Traditional Chinese Medicinal Herb Halenia elliptica D. Don (Gentianaceae)
Supplemental material, Supplementary_Table_of_SSR_marker_for_Halenia_elliptica for Transcriptome Analysis and Microsatellite Markers Development of a Traditional Chinese Medicinal Herb Halenia elliptica D. Don (Gentianaceae) by Mingliu Yang, Nanyu Han, Heng Li and Lihua Meng in Evolutionary Bioinformatics
Footnotes
Acknowledgements
The authors are grateful to Dr Yuanwen Duan for the field sampling and Dr Dongrui Jia for English editing.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (31460096).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MY conducted the sample collections, the laboratory experiments, and statistical analyses. NH and HL assisted with bioinformatics tools. LM designed the study, conducted statistical analyses, and drafted the manuscript. All authors read and reviewed the final manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.