
Abstract
Understanding the evolution of flower diversity is a central topic in plant evolutionary ecology, and natural selection on floral traits via male fitness could be estimated quantitatively using microsatellites. Here, based on RNA sequencing, we developed simple sequence repeat primers and verified polymorphisms in 2 wild populations of Herpetospermum pedunculosum (Cucurbitaceae), a dioecious annual plants native to the Himalaya Mountains. A total of 131 paired primers were designed; 15 paired primers were found to be polymorphic, with the expected heterozygosity varying between 0.280 and 0.767. We also identified 58 genotypes in 20 plants from the 2 populations. Conclusively, these primers could be effective in examining male fitness and population genetic structure of H pedunculosum in future studies.
Introduction
The incredible diversity of flowers is attractive to plant evolutionary biologists since Darwin 1 proposed the theory of natural selection in 1859, and, since then, floral evolution of plant species became one of the central topics in plant evolutionary ecology. Generally, floral diversity of flowering plants is strongly associated with their dominant pollinators, with aims of pollen dispersal from anthers and pollen receipt to stigmas.2,3 Thus, floral diversity could be attributed to the achievements of the maximum of male and female reproductive success of plant species. However, to the present, most studies measuring natural selection on floral traits are based on female reproductive success (seed set), 4 which could result from the easy estimations of seed set. In contrast, examinations of natural selection are still rare on floral traits based on male reproductive success, although achieving maximum of male reproductive success is considered the primary driver to the selection of floral traits.5,6 Estimations of male reproductive success could be performed by tracing pollen flows with fluorescent powder and dying pollen grains. 7 However, the number of pollen grains that deposited on the stigma might not be associated with seed production. 8 Accordingly, by tracing pollen grains, male reproductive success could be estimated only but not be quantified. The development of molecular markers could help us quantify male reproductive success.9,10 For example, a significant stabilizing selection was found for corona width and flower length through male function based on microsatellite markers in the tristylous daffodil Narcissus triandrus. 11 In Polemonium brandegeei, plants with more nectar sugar and narrow corolla tubes had high siring success via male fitness. 12 Therefore, microsatellites are the most powerful markers to determine male reproductive success quantitatively. 13
Herpetospermum pedunculosum is an annual liana native to Himalaya Mountains inhabiting altitudes ranging from 2300 to 3500 m. 14 This species is dioecious, and thus pollinators are necessary for seed production. Seeds of H pedunculosum are widely used in traditional Tibetan medicine. Infield populations, sex ratios are strongly male-biased (ca. 70%), 15 indicating that male-male competition might occur in pollinating female flowers. In our field observations, we found that flower size changed significantly among different plants, indicating that large flowers could attract more pollinators than small flowers. Therefore, to measure the effects of flower size on male reproductive success quantitatively in future researches, we developed microsatellite markers of H pedunculosum based on RNA sequencing in this research.
Materials and Methods
Materials and sample collections
Seeds of H pedunculosum were collected from Shangri-La Alpine Botanical Garden in 2017 and 50 of them were sowed in pots separately in the greenhouse of Yunnan Normal University in 2018. Seedlings with 2 euphylla were transplanted to the experimental lands in Yunnan Normal University, and frames were built separately for each seedling to meet their climbing habits. All seedlings were watered periodically to prevent drought-induced death. When plants were in the flowering time, we collected fresh leaves from 2 male plants and 2 female plants and kept them in liquid nitrogen separately. Besides, in each of the 2 wild populations in Shangri-La (Yunnan) and Nyingchi (Tibet), respectively, fresh leaves of 10 plants (5 male and 5 female plants) were collected and kept in silica gel separately.
RNA of each of the 4 individuals was extracted using a CTAB method, 16 and the integrity of RNA was measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA integrity number (RIN) of each of the 4 individuals ranged from 9.0 to 10.0, showing no sign of degradation. Total DNA of dried leaves from 20 plants was extracted separately using a modified CTAB method for validation of simple sequence repeat (SSR) markers.
RNA sequencing and assembly
The cDNA library of each individual was built following Illumina’s recommendations (San Diego, CA, USA). Random hexamer primers were used to synthesize the double strands, and the QIAquick PCR Purification kit (Qiagen Inc) was employed to purify the short fragments. Then paired-end reads were generated on the Illumina HiSeq2500 platform at the Biomarker Technologies Co, China.
After filtering the adaptors and reads with >8 ambiguous bases and >50% of the bases (quality score ⩽ 5) in raw data of each individual, all clean reads of 4 individuals were pooled and then de novo assembled to transcripts using Trinity software. 17 To evaluate the quality of transcriptome assembly, we used BUSCO v4 18 software with plant ortholog data sets from OrthoDB v10 19 to assess completeness. The clean sequence data reported in this paper have been deposited in the Genome Sequence Archive 20 in BIG Data Center, 21 Beijing Institute of Genomics (BIG), Chinese Academy of Sciences.
SSR locus search, primer design, and validation
Before detecting SSR loci in transcripts, we followed several criteria in light of SSR’s traits. (1) There should be 5 and 3 repeats at least for simple and complex repeats, respectively. (2) The length of motif should be 2 to 10 bp, and the interruption distance between different SSRs should be a maximum of 100 bp for complex SSRs. (3) Only 2 to 6 nucleotide motifs were considered, and the minimum repeat unit was defined as 6 for di-, and 5 for tri-, tetra-, penta-, and hexanucleotides. Then, potential SSR loci were scanned in the transcripts using Micro-SAtellite (MISA). 22
By using Primer 3.0, 23 we designed paired primers for each unique SSR containing at least 5 repeats. We then performed polymerase chain reaction (PCR) in a 25-μL volume containing 20 to 30 ng DNA. The PCR reactions were carried out with the following conditions: DNA initial denaturation at 94°C for 4 minutes, 35 cycles of 94°C for 1 minute 30 seconds, annealing temperature ranging from 45°C to 60°C for 50 seconds, 72°C for 50 seconds, and a final extension at 72°C for 7 minutes. By using a TIAN quick Midi Purification Kit (Tiangen Biotech (Beijing) Co, Ltd, China), excess primers and deoxynucleotide triphosphates were removed to purify the PCR products. We employed those paired primers that were successful in the PCR amplification to detect the polymorphism among 18 to 20 individuals from 2 populations with POPGEN v1.32. 24 Sequencing reactions were then performed under the instruction of ABI Prism Sequencing Ready Reaction Kit with the same primers as PCRs and analyzed on the ABI 3730 genetic analyzer (Applied Biosystems).
Annotation for transcripts containing SSRs
We determined the objective sequences and gene names by the transcripts that included SSR in the National Center for Biotechnology Information’s (NCBI) NR protein database using BLASTx by setting the E-value threshold as 1e−6. We then conducted functional annotation of transcripts using the programs Blast2GO 25 and KEGG, 26 and classified the functional categories using the program WEGO. 27
Results and Discussion
RNA sequencing and assembly
After filtering, 30 598 513 and 26 227 138 clean reads were acquired for 2 male plants, and 30 620 799 and 29 180 298 clean reads were obtained for 2 female plants (Table 1). Assembly resulted in 254 706 transcripts, and the length of assembled transcripts ranged from 200 to above 6000 bp, with an average of 848 bp (Figure 1, Table 1). The N50 value of our assembly is 1860 bp and is similar to those from other species by transcriptome sequencing (Table 1), including Veratrilla baillonii Franch 28 and Halenia elliptica D. Don. 29 The GC content is more than 45% for both male and female plants (Table 1). With the increase in transcripts size, the number of transcripts decreased, indicating a power-law-like distribution (Figure 1), which is a common trait of transcripts in many plant species. Furthermore, the transcriptome of H pedunculosum included 350 of the 425 (82.35%) complete BUSCO genes, indicating the high quality of our assembly.
Characteristics of de novo samples and clustered transcripts.

Abbreviation: SSR, simple sequence repeat.

Length distribution of all transcripts in Herpetospermum pedunculosum.
Distribution and annotation of SSRs
In total, 18 510 potential SSRs were identified, and 95% of them were trinucleotide and dinucleotide as we did not consider mononucleotide in SSR searching (Tables 1 and 2). Generally, all potential SSR loci could be classified into 2 categories in light of the size of genetic markers. Those with length more than 20 bp were hypervariable markers and could be considered as class I. Those with a length between 12 and 20 bp might be variable markers potentially and could be considered as class II. SSRs of class I could be more variable than those of class II and thus might be with high polymorphism. In the potential SSRs of H pedunculosum, 32.6% belonged to class I.
Characteristics of SSR.

Abbreviation: SSR, simple sequence repeat.
Results of BLAST2GO suggested that 9183 transcripts containing SSRs of H pedunculosum could be annotated against the GO database (Table 1, Figure 2) and classified them into 3 categories: cellular components, molecular functions, and biological process. Specifically, of the cellular components, cell, cell part and organelle part were 3 highly represented classes. Catalytic activity and binding were the most matched among 6 molecular functions. In contrast, cell and metabolic processes were the most matched 2 classes of biological processes (Figure 3). However, results of KEGG annotation suggested that only 924 transcripts including SSRs of H pedunculosum could be annotated. In the top 20 KEGG pathways, genes including SSRs associated with metabolic pathways (10.63%) were most representative, followed by biosynthesis of secondary metabolites (5.26%) and biosynthesis of antibiotics (2.30%) (Figure 4A). Besides, several genes containing SSRs participated in ethylene and jasmonic acid signal transduction pathways, which may involve in flower sex determination of H pedunculosum30-32(Figure 4B).

Matching results by BLASTx for Herpetospermum pedunculosum.

GO classification of SSRs for Herpetospermum pedunculosum. SSRs indicate simple sequence repeat.
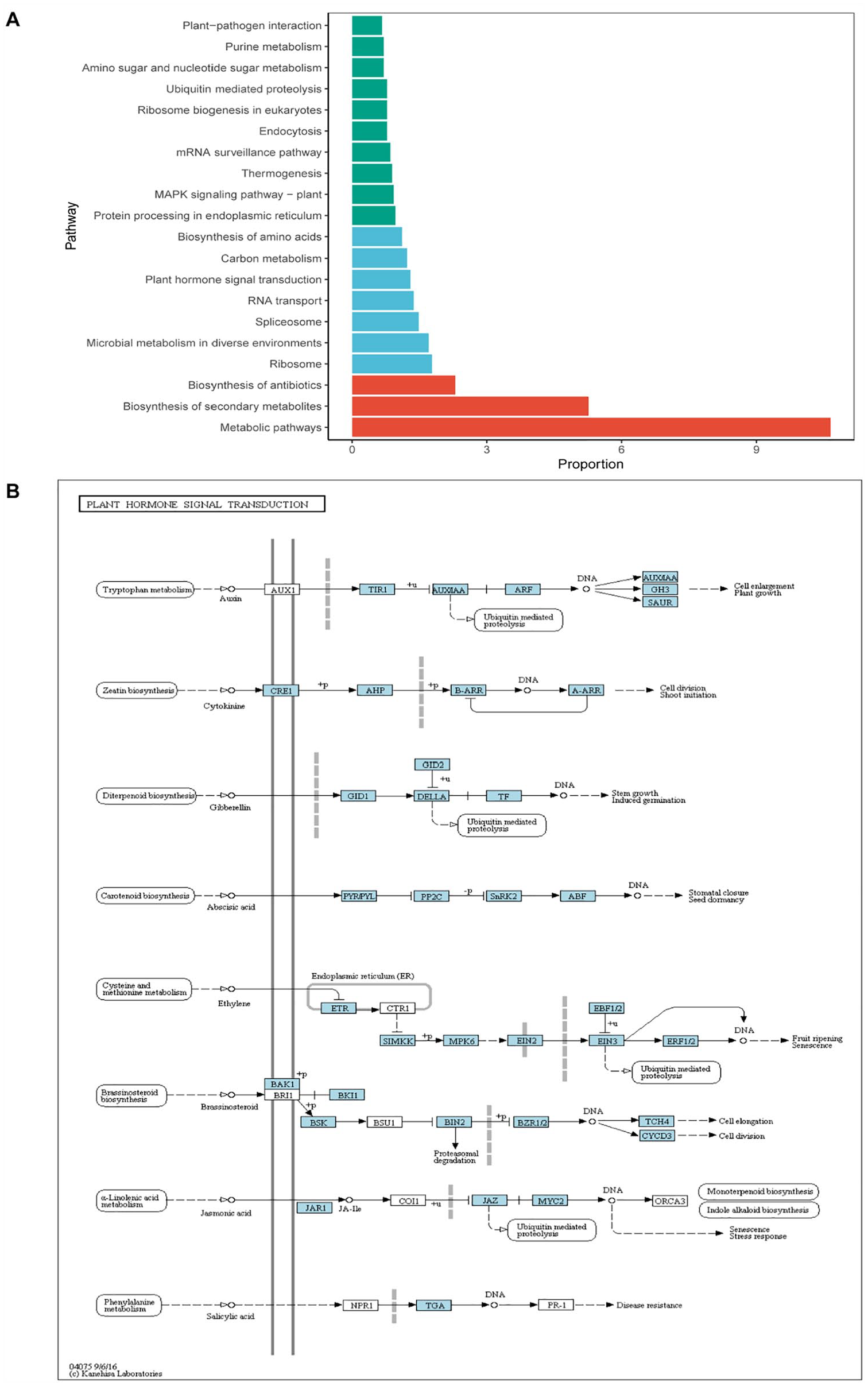
The KEGG pathway of Herpetospermum pedunculosum: (A) frequency distribution of the KEGG pathway functions functions and (B) the plant hormone signal transduction pathway.
Polymorphism of microsatellites
To validate the polymorphism of screened SSRs using transcriptome sequencing, we designed 131 paired primers of H pedunculosum and performed PCR in 20 plants from 2 distinct populations (Supplementary Table 1). We then sequenced the PCR products and found that 15 paired primers were polymorphic. The expected heterozygosity per locus varied between 0.280 and 0.767 in total, with a range between 0 and 0.621 for the Yunnan population and a range between 0 and 0.574 for the Tibet population, respectively. We also identified 58 genotypes in 20 plants from the 2 populations (Table 3).
Polymorphism of microsatellites screening between 2 populations in Herpetospermum pedunculosum.

Abbreviations: He, expected heterozygosity; Ho, observed heterozygosity; No., number of samples; Na, number of alleles; Ne, effective number of alleles; PIC, polymorphism information content.
Conclusions
Microsatellites have increasingly been used in population genetics analysis and mating system estimation for their abundance in genomes, genetic codominance, high reproducibility and polymorphism, 22 and rapid development of next-generation sequencing advance the wide uses of SSRs. Here, we developed 15 paired SSR primers using transcriptome sequencing that showed polymorphic in 2 populations of H pedunculosum. There SSR primers could be useful in future quantification of male reproductive success and paternity analysis of this dioecious plant.
Supplemental Material
Supplementary_Table_of_microsatellite_markers_for_a_dioecious_Herpetospermum_pedunculosum_Cucurbitaceae_xyz307800943cb39 – Supplemental material for Development of Microsatellite Markers for a Dioecious Herpetospermum pedunculosum (Cucurbitaceae)
Supplemental material, Supplementary_Table_of_microsatellite_markers_for_a_dioecious_Herpetospermum_pedunculosum_Cucurbitaceae_xyz307800943cb39 for Development of Microsatellite Markers for a Dioecious Herpetospermum pedunculosum (Cucurbitaceae) by Zhu-Qing Chen, Zhi-Li Zhou, Lin-Lin Wang, Li-Hua Meng and Yuan-Wen Duan in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (31660109).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
ZQC and ZLZ performed the laboratory experiments and statistical analyses. LLW and YWD collected plant materials in the field and cultivated plants in the green house. LHM designed the research and wrote the manuscript. All authors read and reviewed the final manuscript.
Data Availability
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.