
Abstract
The reconstruction of large phylogenetic trees from data that violates clocklike evolution (or as a supertree constructed from any m input trees) raises a difficult question for biologists–how can one assign relative dates to the vertices of the tree? In this paper we investigate this problem, as suming a uniform distribution on the order of the inner vertices of the tree (which includes, but is more general than, the popular Yule distribution on trees). We derive fast algorithms for computing the probability that (i) any given vertex in the tree was the j–th speciation event (for each j), and (ii) any one given vertex is earlier in the tree than a second given vertex. We show how the first algorithm can be used to calculate the expected length of any given interior edge in any given tree that has been generated under either a constant- rate speciation model, or the coalescent model.
Introduction
A fundamental task in evolutionary biology is constructing evolutionary trees from a variety of data. These constructed trees show the ancesteral relationship between the species.
Not only the relationship between species is of interest, but also the time between speciation events. When constructing an evolutionary tree from a set of molecular data which satisfies the molecular clock, the edge lengths can be interpreted as a time scale. In many cases, no time scale is obtained when constructing a tree though:
Often, molecular data does not satisfy the molecular clock and so the edge lengths do not represent a time scale. Trees can be constructed from morphological data or non -standard molecular data like gene order. This does not provide any edge lengths. Having several different trees, one can combine them and construct a ‘supertree.’ Even though there may have been time scales on the original trees, most supertree methods return a tree without a time scale.
For those trees, we still want to find edge lengths representing the time between speciation events. In this paper, we will estimate the edge lengths from the shape of the tree. The method works for trees which evolved under the Yule model [Yule, 1924; Edwards, 1970; Harding, 1971; Page, 1991]. Under the Yule model, in each point of time, each species is equally likely to split. Minor changes to the method for the Yule model give us an edge length estimation for trees under the popular coalescent setting [Nordborg, 2001].
An example for a tree with unknown edge lengths is the primate supertree Part of the primate supertree. Figure 4–13 are some subtrees, for details see [Vos and Mooers]. recently published in [Vos and Mooers]. Figure 1 shows a part of
recently published in [Vos and Mooers]. Figure 1 shows a part of 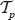 . The primate tree is a supertree on 218 species and was constructed with the MRP method (Matrix Representation using Parsimony analysis, see [Baum, 1992; Ragan, 1992]). Since for most of the interior vertices, no molecular estimates were available, the edge lengths for the tree were estimated. In [Vos and Mooers], 106 rank functions on
. The primate tree is a supertree on 218 species and was constructed with the MRP method (Matrix Representation using Parsimony analysis, see [Baum, 1992; Ragan, 1992]). Since for most of the interior vertices, no molecular estimates were available, the edge lengths for the tree were estimated. In [Vos and Mooers], 106 rank functions on  were drawn uniformly at random. For each of those rank functions, the expected time intervals, i.e. the edge lengths, between vertices were considered (the expected waiting time after the (n – 1)th event until the nth event is 1/n). The authors of [Vos and Mooers] concluded their paper by asking for an analytical approach to the estimation of the edge length, which we will provide below.
were drawn uniformly at random. For each of those rank functions, the expected time intervals, i.e. the edge lengths, between vertices were considered (the expected waiting time after the (n – 1)th event until the nth event is 1/n). The authors of [Vos and Mooers] concluded their paper by asking for an analytical approach to the estimation of the edge length, which we will provide below.

In order to estimate the edge lengths, we developed the algorithms RANKPROB and COMPARE. Those algorithms answer questions like:
Was speciation event with label 76 in the primate tree (see Fig. 1) more likely to be an early event in the tree or a late event? What is the probability that 76 was the 6th speciation event? Was it more likely that speciation event 76 happened before speciation event 162 or 162 before 76?
The algorithms work for trees where every labeled history is equiprobable. This class of model, which includes the Yule model and the coalescent model, has been popular in macroevolutionary studies [Nee and May, 1997; Zhaxy-bayeva and Gogarten, 2004]. Note that the algorithms here are the same for the Yule model and the coalescent model, whereas the edge length estimation has minor differences for the two models.
The algorithms RAMKPROB, COMPARE and an algorithm for obtaining the expected rank and variance for a vertex were implemented in Python, see [Gernhard, 2006].
Let  be a rooted phylogenetic tree [Semple and Steel, 2003] with |V| = n leaves. The set of interior vertices of
be a rooted phylogenetic tree [Semple and Steel, 2003] with |V| = n leaves. The set of interior vertices of  shall be
shall be  into
into  . A vertex ν with r(ν) = i is said to have rank i. Note that r induces a linear order on the set
. A vertex ν with r(ν) = i is said to have rank i. Note that r induces a linear order on the set  := {r : r is a rank function on
:= {r : r is a rank function on  }. We are interested in the distribution of the possible ranks for a certain vertex, i.e. we want to know the probability of r(ν) = i for a given
}. We are interested in the distribution of the possible ranks for a certain vertex, i.e. we want to know the probability of r(ν) = i for a given 
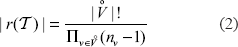
Examples of stochastic models on phylogenetic trees where each rank function is equally likely include:
The Yule model has the probability distribution The coalescent model has the same probability distribution on rooted binary ranked trees as the Yule model. So For some sets of trees (e.g. those drawn from the uniform model [Pinelis, 2003], also known as PDA model), no rank function is induced. If one assumes that all rank functions are equally likely on these trees, one can apply Equation 1 to such trees as well.
 which is the uniform distribution [Edwards, 1970; Brown, 1994].
which is the uniform distribution [Edwards, 1970; Brown, 1994]. is the uniform distribution [Aldous, 2001].
is the uniform distribution [Aldous, 2001].
The following algorithm calculates the probability distribution of the rank of a vertex ν in a rooted binary phylogenetic tree  . The idea of the algorithm is the following (cf. Figure 2). Label the vertices on the path from ν to the root ρ by ν = x1 …, xn = ρ. Let
. The idea of the algorithm is the following (cf. Figure 2). Label the vertices on the path from ν to the root ρ by ν = x1 …, xn = ρ. Let  be the subtree of
be the subtree of  containing the vertex xm and all its descendants. Let
containing the vertex xm and all its descendants. Let  be the number of rank functions on the tree
be the number of rank functions on the tree  where ν has rank i. The values
where ν has rank i. The values  are calculated iteratively for m = 1,…, n. The probability
are calculated iteratively for m = 1,…, n. The probability  equals
equals  . The α-values in the fraction have a lot of factors in common which cancel out. In the following algorithm, we calculate α-values without the unnecessary terms instead,
. The α-values in the fraction have a lot of factors in common which cancel out. In the following algorithm, we calculate α-values without the unnecessary terms instead,  . We have
. We have  .
.
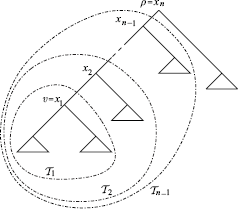
Labeling the tree for the algorithm RANKPROB.


Labeling the tree for the recursion in RANKPROB.
Proving the correctness and runtime of RANKPROB makes use of the following two observations.

Proof. Let  . We first show that
. We first show that  for
for 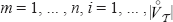 . That implies
. That implies

The proof is by induction over m.
For  . Vertex ν is the root of
. Vertex ν is the root of 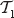 , so
, so  .
.
Let m = k and  holds for all
holds for all  clearly holds for all
clearly holds for all  since
since 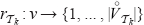 . So it remains to verify that the term (*) returns the right values for
. So it remains to verify that the term (*) returns the right values for 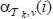 . Assume that the vertex ν is in the (i – j – l)-th position in
. Assume that the vertex ν is in the (i – j – l)-th position in 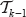 (with i – j – 1 > 0) for some rank function
(with i – j – 1 > 0) for some rank function 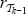 and ν shall be in the i-th position in
and ν shall be in the i-th position in  .
.
Now combine the linear order in the tree 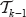 induced by
induced by  with a linear order in
with a linear order in  induced by
induced by  to get a linear order on
to get a linear order on  . The first j vertices of
. The first j vertices of 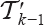 must be inserted between vertices of
must be inserted between vertices of  with lower rank than ν so that ν ends up to be in the i-th position of the tree
with lower rank than ν so that ν ends up to be in the i-th position of the tree  . Count the number of possible way to do this as follows. The tree
. Count the number of possible way to do this as follows. The tree  has
has  possible rank functions. Combining a rank function
possible rank functions. Combining a rank function  with a rank function
with a rank function  to get a rank function
to get a rank function 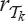 with
with 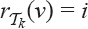 means inserting the first j vertices of
means inserting the first j vertices of  anywhere between the first (i - j - 2) vertices of
anywhere between the first (i - j - 2) vertices of 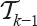 . There are
. There are
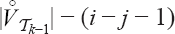 vertices of rank bigger than ν in
vertices of rank bigger than ν in 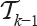 with the remaining
with the remaining  vertices in
vertices in  , there are
, there are

 with
with 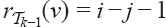 is
is 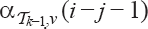 by the induction assumption. Multiplying all those possibilities gives
by the induction assumption. Multiplying all those possibilities gives

 . The value
. The value  is then the sum over all possible j which establishes the correctness of the algorithm.
is then the sum over all possible j which establishes the correctness of the algorithm.

All that remains is to verify the runtime. Note that the combinatorial factors
The most time consuming part of the algorithm is line 13. Adding up all calculations needed for obtaining  comes to:
comes to:
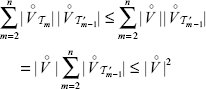
The last inequality holds since the vertices of the 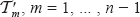 , are distinct. Therefore, the runtime is quadratic.
, are distinct. Therefore, the runtime is quadratic.
 from Theorem 3, the expected value
from Theorem 3, the expected value 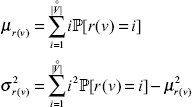
The Yule model
Avery common stochastic model for rooted binary phylogenetic trees with edge lengths is the continuous-time Yule model [Edwards, 1970]. As in the discrete Yule model, at every point in time, each species is equally likely to split and give birth to two new species. The expected waiting time for the next speciation event in a tree with n leaves is 1/n. That is, each species at any given time has a constant speciation rate (normalized so that 1 is the expected time until it next speciates).
Assume that the primate tree 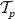 evolved under the continuous-time Yule model. In [Gernhard, 2006], the tree shape of
evolved under the continuous-time Yule model. In [Gernhard, 2006], the tree shape of  (i.e. the tree without edge lengths) under the discrete Yule model is tested against the uniform model and accepts the Yule model.
(i.e. the tree without edge lengths) under the discrete Yule model is tested against the uniform model and accepts the Yule model.
Here, we describe how to estimate the edge lengths for a tree which is assumed to have evolved under the continuous-time Yule model.
Let (u, ν) be an interior edge in  with u the immediate ancestor of ν. Let X be the random variable ‘length of the edge (u, ν)’ given that
with u the immediate ancestor of ν. Let X be the random variable ‘length of the edge (u, ν)’ given that 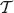 is generated according to the continuous-time Yule model.
is generated according to the continuous-time Yule model.
The expected length 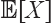 of the edge (u, ν) is given by
of the edge (u, ν) is given by


It remains to calculate the probability Labeling the tree for estimating the edge lengths.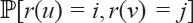 . This is equivalent to counting all the possible rank functions where r(u) = i and r(ν) = j. The subtree
. This is equivalent to counting all the possible rank functions where r(u) = i and r(ν) = j. The subtree 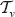 consists of ν and all its descendants. The tree
consists of ν and all its descendants. The tree  equals the tree
equals the tree  where all the descendants of ν are deleted, i.e. ν is a leaf in
where all the descendants of ν are deleted, i.e. ν is a leaf in 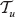 , see Figure 4.
, see Figure 4.

Note that  if
if  . Therefore, assume
. Therefore, assume  in the following.
in the following.
The number of rank functions on  is
is  . The probability
. The probability  can be calculated with RANKPROB(
can be calculated with RANKPROB( ). So the number of rank functions in
). So the number of rank functions in 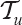 with
with 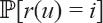 is
is  .
.
The number of rank functions on 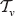 is
is  . Let any linear order on the trees
. Let any linear order on the trees  and
and  be given. Combining those two linear orders into an order, r, on
be given. Combining those two linear orders into an order, r, on  with r(ν) = j means that the vertices with rank 1, 2,… j – 1 in
with r(ν) = j means that the vertices with rank 1, 2,… j – 1 in  keep their rank. Vertex ν gets rank j. The remaining
keep their rank. Vertex ν gets rank j. The remaining  vertices in
vertices in 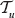 and
and  vertices in
vertices in  have to be shuffled together. According to Remark (1), this can be done in
have to be shuffled together. According to Remark (1), this can be done in
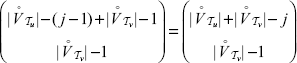
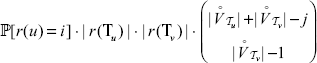
 with r(u) = i and r(ν) = j. For the probability
with r(u) = i and r(ν) = j. For the probability 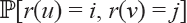 :
:

Since  and
and  are independent of i and j, those factors cancel out, giving
are independent of i and j, those factors cancel out, giving


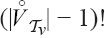 is independent of i and j, this factor cancels out, and so
is independent of i and j, this factor cancels out, and so

 . With this notation, the expected edge length
. With this notation, the expected edge length 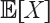 is
is

Suppose that the growth process is stopped as soon as the n – 1-st speciation event occurs. In this case the expected length X of a pendant edge below an interior vertex ν is:

The expected depth of vertex ν from the first branchpoint is:
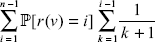
So the depth Y of the leaf in question from the first branchpoint has expectation independent of ν:

In other words, assigning to each edge of a given tree topology its expected length gives a tree which obeys a molecular clock.
However, the expected edge length may be calculated for each possible binary resolution of the supertree. Assume the supertree 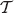 has the possible binary resolutions
has the possible binary resolutions  . For an edge (u, ν) in
. For an edge (u, ν) in  where u is the immediate ancestor of ν, the expected edge length is calculated in the trees
where u is the immediate ancestor of ν, the expected edge length is calculated in the trees  for i = 1, …, m. The expected edge length in
for i = 1, …, m. The expected edge length in  is denoted by ei for i =1, …, m. Note that if u is a vertex with more than two descendants in
is denoted by ei for i =1, …, m. Note that if u is a vertex with more than two descendants in  then ν is in general not a direct descendant of u in
then ν is in general not a direct descendant of u in  . The value ei in resolution
. The value ei in resolution  is then the sum of all expected edge lengths on the path from u to ν in
is then the sum of all expected edge lengths on the path from u to ν in  .
.
Calculate the expected edge length  of (u, ν) in the supertree
of (u, ν) in the supertree  by
by

 under the Yule model is [Brown, 1994]
under the Yule model is [Brown, 1994]

Again, once the expected length of pendant edges is included the resulting tree obeys a molecular clock, meaning that all leaves are at the same depth.
The edge length estimation in the previous section works for the continuous-time Yule model. By changing the method above slightly, we get an edge length estimation for the coalescent process. In the coalescent setting, we have
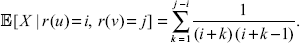

The calculations in Section 3.1 and 3.2 provide exact values for the expected length of an interior edge under the Yule or coalescent process as an alternative to simulations. However simulations also provide some indication of the variability in the estimate of edge lengths, and it may be of interest to also investigate analytically the variance (or even the distribution) of the edge length in future work, rather than just its mean.
Comparing Two Interior Vertices
The algorithm RANKPROB can also be used for comparing two interior vertices. Assume again that every rank function on a rooted binary phylogenetic tree  is equally likely. The aim is to compare two interior vertices u and ν of
is equally likely. The aim is to compare two interior vertices u and ν of  . Was u more likely before (of lower rank than) ν or ν before u? In other words, what is the probability
. Was u more likely before (of lower rank than) ν or ν before u? In other words, what is the probability

 . Note that it does not hold
. Note that it does not hold  even with the uniform distibution on the rank functions. The probability
even with the uniform distibution on the rank functions. The probability  is equivalent to counting all the possible rank functions on
is equivalent to counting all the possible rank functions on 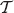 in which u has lower rank than ν and divide that number by all possible rank functions on
in which u has lower rank than ν and divide that number by all possible rank functions on  . One idea is to sum up the probabilities
. One idea is to sum up the probabilities  in Equation 3 for all i < j which yields to a runtime of O(|V|4). The following algorithm COMPARE solves the problem in quadratic time. In the following, for a vertex ν, the subtree
in Equation 3 for all i < j which yields to a runtime of O(|V|4). The following algorithm COMPARE solves the problem in quadratic time. In the following, for a vertex ν, the subtree  of
of  consists again of ν and all its descendants.
consists again of ν and all its descendants.


The runtime of COMPARE is O
Proof. Note that the probability of u having smaller rank than ν in tree  equals the probability of u having smaller rank than ν in tree
equals the probability of u having smaller rank than ν in tree 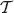 , since for any rank function on
, since for any rank function on  there is the same number of linear extensions to get a rank function on the tree
there is the same number of linear extensions to get a rank function on the tree 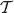 .
.
So it is sufficient to calculate the probability 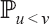 in
in 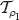 . If ρ1 = u then u is an ancestor of ν in
. If ρ1 = u then u is an ancestor of ν in  , so return
, so return  . If ρ1 = ν then ν is an ancestor of u in
. If ρ1 = ν then ν is an ancestor of u in  so return
so return  .
.
Now assume that ρ1 ≠ u and ρ1 ≠ v. The run of RANKPROB calculates the probability  in the tree
in the tree 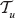 and
and 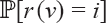 in
in  for all i. Next, combine those two linear orders. Assume that r(ν) = i and that j vertices of
for all i. Next, combine those two linear orders. Assume that r(ν) = i and that j vertices of  are inserted before ν. Inserting j vertices of
are inserted before ν. Inserting j vertices of 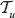 into the linear order of
into the linear order of  before ν is possible in
before ν is possible in  ways. The probability that the vertex u is among the j vertices which have smaller rank than ν is
ways. The probability that the vertex u is among the j vertices which have smaller rank than ν is  . There are
. There are  possible linear orders on
possible linear orders on  and
and  possible linear orders on
possible linear orders on  . The number of linear orders where vertex ν has rank i in
. The number of linear orders where vertex ν has rank i in  , ν has rank i + j in
, ν has rank i + j in  and r(u) < i + j therefore equals
and r(u) < i + j therefore equals
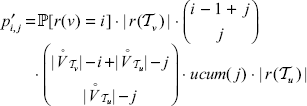
Combining a linear order on  with a linear order on
with a linear order on  is possible in
is possible in

 linear orders on
linear orders on  and
and  linear orders on
linear orders on 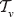 , so on
, so on  , there are
, there are


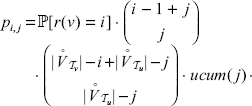
This shows that COMPARE works correct.
Since RANKPROB has quadratic runtime, COMPARE also has quadratic run time.
Footnotes
Acknowledgements
We thank Arne Mooers for very helpful comments and suggestions on earlier versions of this manuscript and the two anonymous referees for a very careful report. The Second author's work is partially supported by grant NSF-DMS-0241246