
Abstract
This clinical report describes a family with both Marfan and ocular-only Stickler syndromes. We report 2 cases of ocular-only Stickler syndrome and 2 cases of Marfan syndrome concurrent with ocular-only Stickler syndrome. Type 1 Stickler syndrome and Marfan syndrome share many clinical similarities, and it can be difficult to differentiate them solely based on clinical presentation. Vitreous phenotyping allows for the identification of vitreous anomalies pathognomonic of Stickler syndrome, which can guide future gene sequencing. Having the accurate diagnosis of Marfan or type 1 Stickler syndrome is important, as patients with type 1 Stickler syndrome have higher rates of retinal detachment and will benefit from prophylaxis.
Introduction
Marfan [MIM154700] and Type 1 Stickler syndrome [MIM 108300, 609508] are connective tissue disorders that may share many clinical similarities – short sight, astigmatism, retinal detachment, palate deformity and musculoskeletal abnormalities. Differential diagnosis on clinical grounds can be challenging given that both disorders exhibit dominant inheritance and patients with Stickler syndrome may sometimes exhibit a “Marfanoid” habitus and many patients with confirmed Marfan syndrome, do not.
This report describes and highlights the importance of the vitreous phenotype as a key feature to aid differential diagnosis in a single pedigree in which both disorders are featured in different family members.
Case description
Index case (individual III-2)
The proband was referred for cataract surgery and vitreoretinal assessment in the context of unilateral poor vision, a history of high myopia and a family history of Marfan syndrome. An assessment was sought on the basis of possible lens instability with relation to Marfan syndrome and secondly with regard to the need or otherwise for prophylaxis to prevent retinal detachment. The proband's younger brother (Figure 1, III-4) had suffered retinal detachment in both eyes.
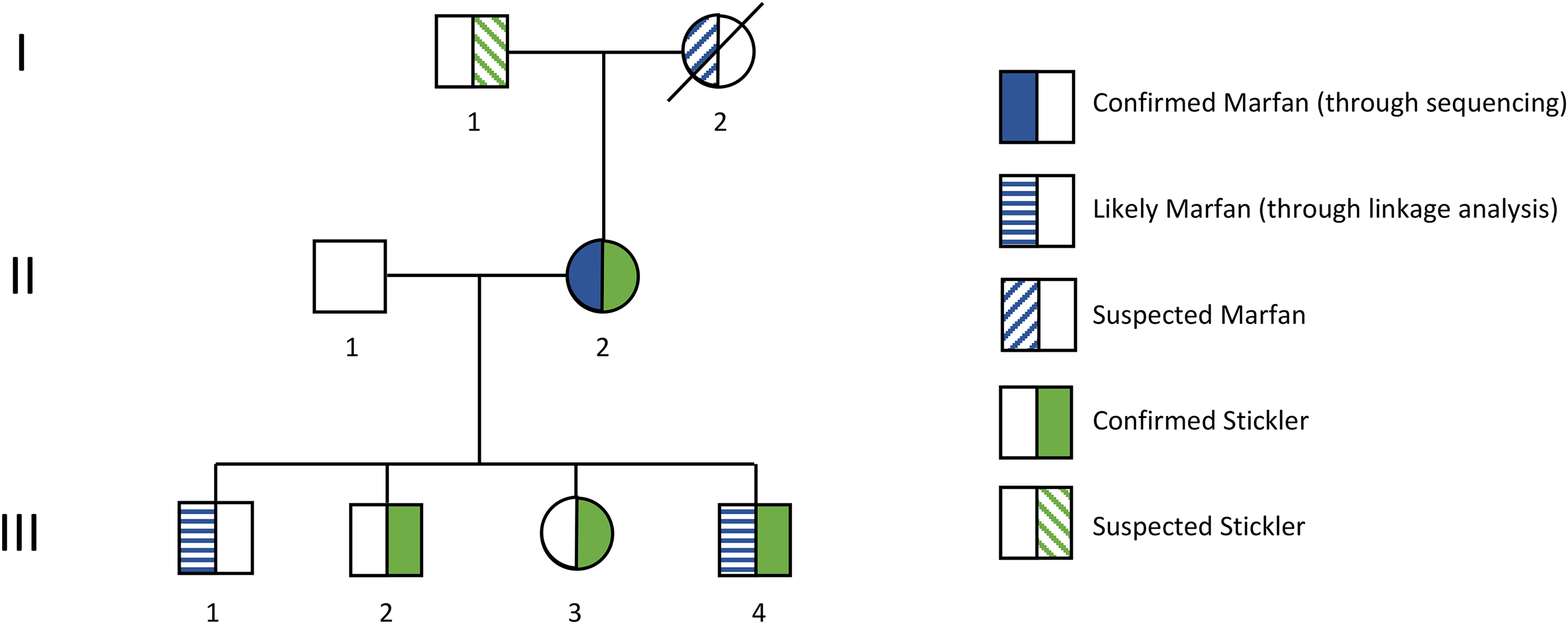
Pedigree drawing showing individuals with suspected and genetically proven Stickler syndrome and Marfan syndrome.
Examination of the proband revealed a dense cataract in his right eye obscuring any view of the fundus and the eye was hypotonous (low pressure). The fellow eye had clear media and exhibited the type 1 vitreous anomaly pathognomonic of type 1 Stickler syndrome. Visual acuities were reduced to counting fingers in the right eye and maintained at 6/5 in the left eye. He had a high arched palate.
Molecular genetic analysis confirmed a heterozygous base change in the COL2A1 gene NC_000012.12(NM_001844.5):c.192C > A p.(Cys64Ter). The COL2A1 gene is subject to tissue dependent alternative splicing and this pathogenic variant falls within exon 2. Since this exon is spliced out of mature cartilage, expression would be expected to result in the ocular-only variant of type 1 Stickler syndrome. 1 Prior linkage analysis had showed that he was at low risk of carrying the familial FBN1 pathogenic variant NC_000015.10(NM_000138.5):c.6661T > C p.(Cys2221Arg).
The proband underwent routine cataract surgery under general anaesthesia, and with a clear view to the posterior pole was found to have a chronic 200 degree giant retinal tear in association with Grade B proliferative vitreoretinopathy (PVR) which was repaired with vitrectomy and silicone oil tamponade under the same anaesthetic together with 360 prophylactic retinopexy to the fellow (left) eye. The silicone oil tamponade was safely removed 5 months later and 3 years later he underwent uncomplicated cataract surgery to his left eye. Upon last review, his visual acuities were stable at 6/18 right eye, 6/5 left eye.
Individual II-2
The proband's mother had a history of myopia and left amblyopia and was diagnosed with Marfan syndrome in her 20s during her first pregnancy. Molecular genetic analysis confirmed a FBN1 pathogenic variant c.6661T > C p.(Cys2221Arg). She underwent bilateral cataract surgery in her 30s, and had a history of joint hypermobility and progressive hearing loss. She underwent an aortic valve replacement at the age of 42. Her father (Figure 1, I-1) had a history of bilateral retinal detachments in childhood and was blind. Her mother (Figure 1, I-2) died at the age of 40 of an aortic dissection and had also been diagnosed with Marfan syndrome based on clinical presentation.
Corrected visual acuities were 6/12 right eye and 6/7.5 left eye. Ocular examination demonstrated bilateral aphakia (absent lenses), with both eyes exhibiting the type 1 vitreous anomaly, pathognomonic for type 1 Stickler syndrome. Although she had a confirmed clinical and molecular genetic diagnosis of Marfan syndrome, her vitreous phenotype was consistent with type 1 Stickler syndrome and predictive analysis confirmed she also carried the same COL2A1 c.192C > A p.(Cys64Ter) pathogenic variant identified in the proband.
Individual III-4
As part of family screening the proband's brother (Figure 1, III-4) was examined. He had previously been diagnosed with Marfan syndrome through linkage analysis and suffered retinal detachment in his left eye aged 13 which was not successfully repaired. Visual acuities were 6/36 right eye and no perception of light in the left eye. Ocular motility examination revealed nystagmus with right iris transillumination and a quadrantic lamellar lens opacity with nuclear sclerosis. His right eye also exhibited the type 1 vitreous anomaly pathognomonic of type 1 Stickler syndrome and predictive analysis confirmed he also carried the same COL2A1 c.192C > A p.(Cys64Ter) pathogenic variant identified in the proband.
Individual III-3
As part of family screening the proband's sister (Figure 1, III-3) was examined. She has a past medical history of pulmonary embolism and deep vein thrombosis, as well as tick borne encephalomyelitis resulting in raised intracranial pressure and papilloedema. Ocular examination revealed she exhibited the type 1 vitreous anomaly pathognomonic of type 1 Stickler syndrome in both eyes and predictive analysis confirmed she also carried the same COL2A1 c.192C > A p.(Cys64Ter) pathogenic variant identified in the proband. Subjectively, she has generally reduced hearing, for which she had middle ear surgery. She has no joint abnormalities.
Conclusions
Marfan and Stickler syndromes have overlapping features such as joint hypermobility, palate deformities, myopia, astigmatism, and retinal detachment that can make differentiating between the two diseases based on clinical presentation alone challenging. However, the members of this family presented with the ocular-only variant of type 1 Stickler syndrome which does not typically manifest systemically. This makes the differential diagnosis even more difficult, since patients with concurrent Marfan and ocular-only Stickler syndrome (such as individual II-2) also present with systemic features due to Marfan syndrome. To differentiate reliably between both conditions in a clinical setting, familial history may be useful as both Marfan and type 1 Stickler syndrome are inherited in an autosomal dominant fashion. However, diagnosis based on clinical presentation and familial history alone may lead to missed diagnoses or even misdiagnosis in certain patients, especially in older patients who were diagnosed before mutation analysis was available. As individual I-1 had bilateral retinal detachments during childhood, it is highly likely that he suffered from undiagnosed Stickler syndrome. Additionally, there is a significant proportion of type 1 Stickler syndrome cases arising from de novo mutations in the COL2A1 gene, making them the first in their family to have type 1 Stickler syndrome. 2
The clinical presentation of patients with Stickler and Marfan syndrome may vary greatly. However, all patients with ocular-only Stickler syndrome exhibited type 1 vitreous anomaly resulting from abnormal embryonic development of vitreous, which is pathognomonic for type 1 Stickler syndrome. Type 1 vitreous anomaly consists of vestigial vitreous gel occupying the retrolental space bordered by a convoluted membrane (Figure 2a). 3 In contrast, patients with type 2 Stickler syndrome exhibit type 2 vitreous anomaly, consisting of irregular and thickened lamellae resembling beads in the vitreous (Figure 2b). 3
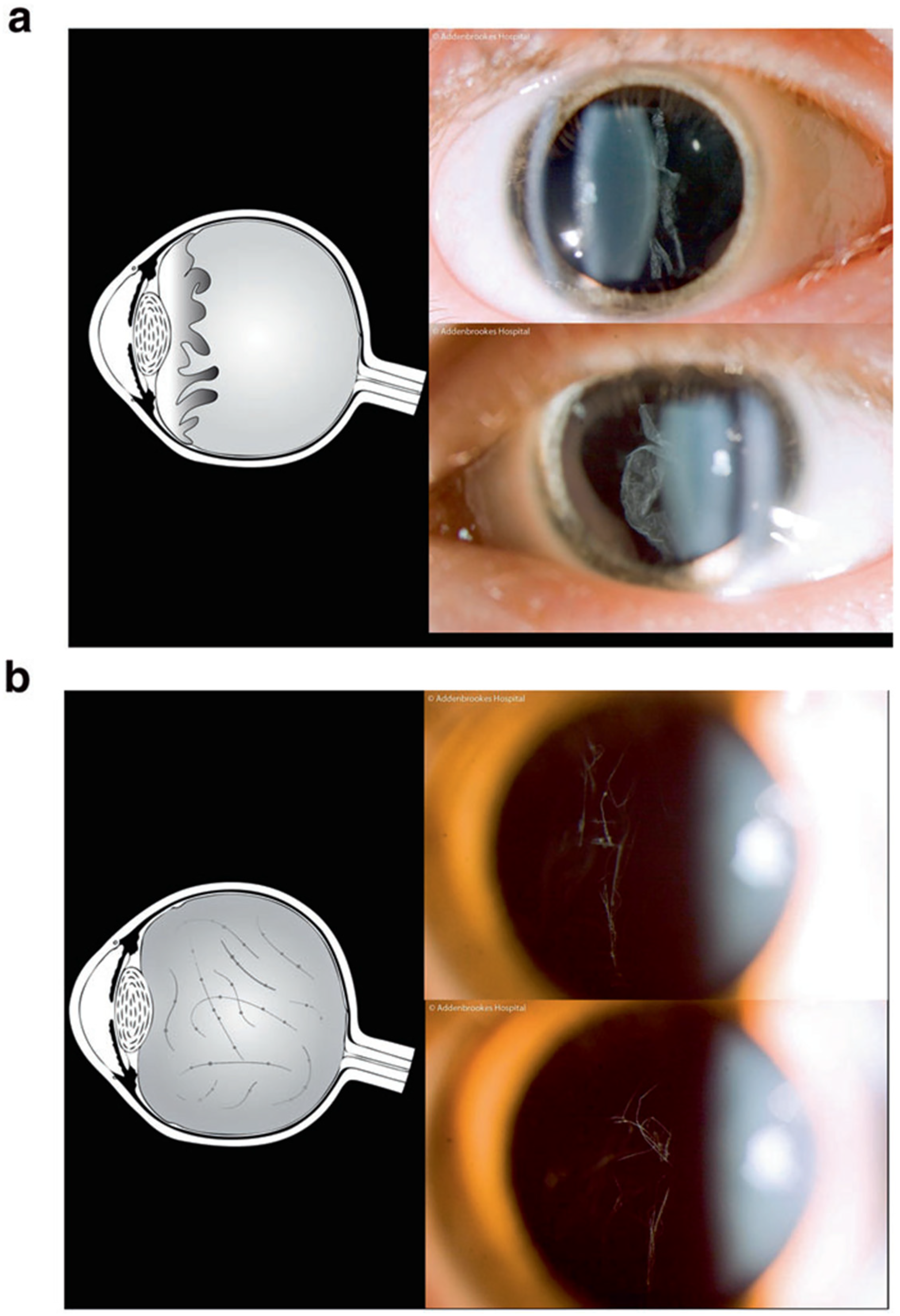
Vitreous anomalies in Stickler syndrome. (a) Membranous congenital vitreous anomaly seen in type 1 Stickler syndrome, (b) Beaded congenital vitreous anomaly seen in type 2 Stickler syndrome. Reproduced with permission from Snead et al. 4
Vitreous phenotyping is the process of identifying vitreous anomalies through slit lamp examination, and is of key importance in differentiating Marfan from Stickler syndrome because type 1 vitreous anomalies have not been observed in patients with pure Marfan syndrome. Instead, patients with Marfan syndrome usually exhibit a different set of vitreous changes, including posterior vitreous detachment, early vitreous liquefaction and abnormal vitreous attachments at the edges of lattice degeneration. 5 Vitreous phenotyping thus has key importance in the initial diagnosis of Stickler syndrome and guiding gene sequencing, which is the gold standard of diagnosis but may be slower and expensive. Without the findings from vitreous phenotyping, screening for COL2A1 variants would not have been done in this family as their ocular symptoms would have been attributed to Marfan syndrome.
However, vitreous phenotyping may not always be possible, as slit lamp examination may not be possible in children aged 4 or younger (unless under anaesthesia) or in patients with previous retinal surgery. Other clinical features, including cardiovascular abnormalities such as aortic root regurgitation are unique to Marfan syndrome, and may aid in differentiating between Marfan and Stickler syndrome. However, there is variable disease severity among patients with Marfan and Stickler syndrome, so vitreous phenotyping remains the most reliable diagnostic technique to distinguish between the two conditions.
Correctly diagnosing Stickler and Marfan syndrome is important as patients with type 1 Stickler syndrome are at much higher risk of developing retinal detachments than patients with Marfan syndrome.6,7 Prophylactic argon laser photocoagulation or 360 degree retinal cryotherapy aim to prevent the development of new retinal tears as well as the progression of giant retinal tears to retinal detachment, and have been found to reduce risk of retinal detachment by 4.4 fold to 12 fold in patients with Stickler syndrome,8,9 whereas the benefit of prophylaxis in Marfan syndrome is more uncertain. Patients who have had a retinal detachment in one eye may also benefit from prophylaxis on the fellow eye as a large proportion of patients with Stickler or Marfan syndrome suffer from bilateral retinal detachments.8,10 As in the case of members in this family, one member was incidentally found to have retinal pathology when being treated for their cataract. This emphasises the importance of careful fundal examination in these patients to ensure sight threatening pathology is not missed.
In summary, the incidence of both Marfan and Stickler syndrome in the same family highlights the difficulty of differentiating between the two conditions clinically, and the importance of vitreous phenotyping in differential diagnosis. Having the correct diagnosis has important implications in implementing prophylactic measures and the management of the patient.
Footnotes
Acknowledgements
Not applicable.
Author contributions
Conceptualization, M.P.S.; data collection, H.M.; writing—original draft preparation, Z.S., H.M., M.P.S.; writing—review and editing, H.M., Z.S., A.J.R., A.M., P.A., T.R.W.N, H.M., and M.P.S. All authors have read and agreed to the published version of the manuscript.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Disclosure of conflict of interest
None declared.
Ethics approval
Not applicable.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article
Patient consent
Informed consent for the patients’ information to be published was obtained.