
Abstract
The Stickler syndromes are the leading cause of inherited retinal detachment and the most common cause of rhegmatogenous retinal detachment in childhood. The clinical and molecular genetic spectrum of this connective tissue disorder is discussed in this article, emphasising the key role the ophthalmologist has to play in the identification, diagnosis and prevention of blindness in the increasingly widely recognised sub-groups with ocular-only (or minimal systemic) involvement. Without diagnosis and prophylaxis in such high-risk subgroups, these patients are at high risk of Giant Retinal Tear detachment and blindness, especially in the paediatric population, where late or second eye involvement is common. Initially considered a monogenic disorder, there are now known to be at least 11 distinct phenotypic subgroups in addition to allied connective tissue disorders that can present to the clinician as part of the differential diagnosis.
Plain language summary
The Stickler syndromes are a group of related connective tissue disorders that are associated with short-sight and a very high risk of blindness from detachment of the retina – the light sensitive film at the back of the eye. Other features include cleft palate, deafness and premature arthritis. It is the most common cause of retinal detachment in children and the most common cause of familial or inherited retinal detachment. In contrast to most other forms of blinding genetic eye disease, blindness from retinal detachment in Stickler syndrome is largely avoidable with accurate diagnosis and prophylactic (preventive) surgery. Recent advances in the understanding of the genetic causes of Stickler syndrome mean that the diagnosis can now be confirmed in over 95% of cases and, most importantly, the patient’s individual risk of retinal detachment can be graded. Preventative surgery is hugely effective in reducing the incidence of retinal detachment in those patients shown to be at high risk. NHS England have led the way in the multidisciplinary care for patients with Stickler syndrome by launching a highly specialist service that has been free at point of care to all NHS patients in England since 2011 (https://www.england.nhs.uk/commissioning/spec-services/highly-spec-services, www.vitreoretinalservice.org)
Introduction
Although genetic eye diseases are generally considered rare, the Stickler syndromes are relatively common and yet widely under-recognised and under-diagnosed. They are the leading cause of inherited retinal detachment and the most common cause of rhegmatogenous retinal detachment in childhood.
The clinical and molecular genetic spectrum of this connective tissue disorder is discussed in this article but it should be emphasised at the outset that the ophthalmologist has a key role to play in identification, diagnosis and prevention of blindness in the increasingly widely recognised sub-groups with ocular-only (or minimal systemic) involvement.1,2 Without diagnosis and prophylaxis in such high risk sub-groups, these patients are at high risk of Giant Retinal Tear (GRT) detachment and blindness, especially in the paediatric population, where late presenting or second eye involvement is sadly all too common. 3 Prophylactic retinopexy according to a standardised protocol is hugely effective in reducing the risk of retinal detachment.
Initially considered a monogenic disorder, genetic heterogeneity was soon recognised and there are now known to be at least 11 distinct phenotypic sub-groups in addition to allied connective tissue disorders that can present to the clinician as part of the differential diagnosis (Table 1).
The Stickler syndromes and allied collagenopathies.

SEDC, spondyloepiphyseal dysplasia congenita.
Presentation
Patients can present via a wide variety of referral routes and clinical services, including self-diagnosis on the basis of personal and family medical history, but frequent triggers for referral are typically:
Neonates with Pierre-Robin Sequence (PRS) or cleft palate in association with myopia;
Infants with spondyloepiphyseal dysplasia associated with deafness and/or congenital myopia or megalophthalmos (cryptomyopia);
Patients with a family history of retinal detachment and/or cleft palate; and
Sporadic cases of retinal detachment associated with hearing loss.
Clinical features
Ocular features
The pathognomonic hallmark of all but one of the sub-groups of Stickler syndrome (the only exception being type 3 Stickler syndrome where there is no eye involvement) is a congenital abnormality of vitreous embryogenesis. The secondary vitreous is normally fully matured by 10–14 weeks of intra-uterine growth and the embryogenic abnormalities in Stickler syndrome provide the clinician with a crucial sign on which to make the diagnosis (Figure 1). This is especially important in the ocular-only sub-types where the systemic features that might alert the clinician are either mild or absent altogether. 2

Vitreous phenotypes – pathognomonic of Stickler syndrome. Schematic and slit-lamp illustrations: left, membranous congenital vitreous anomaly (haploinsufficiency mutations COL2A1); right, beaded congenital vitreous anomaly (COL11A1 dominant negative mutations).
Assessing the vitreous phenotype is particularly important in the sub-group who do not exhibit refractive myopia, leading to the diagnosis being erroneously discounted. The majority (85%) of patients do exhibit congenital myopia but, of those who are not refractively myopic, many have congenital megalophthalmos in association with cornea plana (‘cryptomyopia’). Refractive error may be high but, in contrast to secondary developmental myopia, is often relatively stable and not associated with secondary peri-papillary myopic atrophy. Radial paravascular pigmented lattice develops as a late secondary phenomenon and, as with myopia, its absence should not eliminate the diagnosis. In the neonate, the fundus examination may appear disarmingly normal before retinal detachment ensues, although post-oral retinal breaks are a relatively common finding with indirect ophthalmoscopy and scleral depression at examination under anaesthesia.
The association with congenital quadrantic lamellar cataract (Figure 2) is well recognised and can be a useful diagnostic marker although it can be present in both type 1 and type 2 Stickler syndrome and does not therefore distinguish between sub-groups in the way that the differing vitreous phenotypes allow.
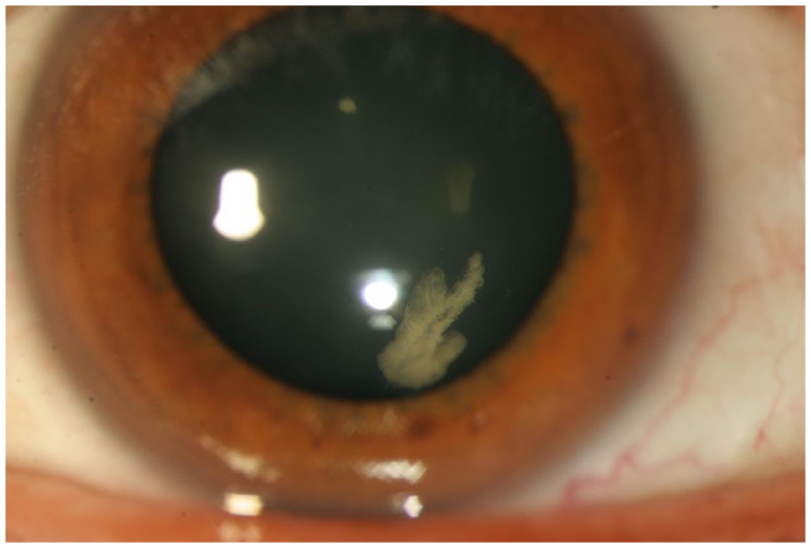
Congenital quadrantic lamellar cataract is well recognised and can be a useful diagnostic marker although it can be present in both type 1 and type 2 Stickler syndrome and does not therefore distinguish between sub-groups in the way that the differing vitreous phenotypes allow. Reproduced with permission from Snead MP, et al. 2
Prophylaxis
In addition to confirming the diagnosis, molecular genetic analysis is key to stratifying the risk of blindness from retinal detachment and GRT to which both type 1 and type 2 sub-groups are particularly prone (Figure 3). The risk of visual loss from retinal detachment in the more recently identified recessive sub-groups (Table 1) is more difficult to quantify due to the paucity of numbers and long-term follow-up data.

GRT – child with undiagnosed type 1 Stickler syndrome, already blind in the fellow eye from retinal detachment.
In the largest published study on prophylaxis against GRT, Fincham et al. reported results from 487 patients with genetically confirmed type 1 Stickler syndrome. 3 Time to retinal detachment was compared between patients who received bilateral prophylaxis and those that did not, with and without individual patient matching. Matching was blinded to outcome events and individual patient matching protocols purposely weighted bias against the effectiveness of treatment.
The results showed that the bilateral untreated control group (n = 194) had a 7.4-fold increased risk of retinal detachment compared with the prophylaxis group (n = 229, p < 0.001); and the matched bilateral control group (n = 165) had a 5.0-fold increased risk compared with the matched prophylaxis group (p < 0.001).
For those patient groups presenting with retinal detachment in their first eye (and therefore undergoing prophylaxis only in their fellow eye), untreated eyes had a 10.3-fold increased risk of retinal detachment compared with eyes receiving prophylaxis (p < 0.001). Matched untreated eyes had an 8.4-fold increased risk compared with eyes receiving prophylaxis (p < 0.001). Alarmingly, of those patients with genetically confirmed type 1 Stickler syndrome who suffer retinal detachment, 50% suffer retinal detachment in their second eye within 4 years of the first eye (Figure 4a, b). 3

Auditory features
Despite the early observation that hearing loss was evident in individuals with Stickler syndromes, 10 until recently sparse attention has been paid to detailed description of the auditory phenotypes involved. There are several reasons why this may be a valuable endeavour; an understanding of the prevalence, severity, and pathophysiological basis of hearing loss in Stickler syndromes may lead to clinical interventions and hearing rehabilitation. Untreated hearing loss can lead to social isolation and loneliness, and is a major modifiable risk for cognitive decline. 11 Knowledge about processes of hearing loss in Stickler syndromes might also shed light on the syndromes in general.
Hearing loss in Stickler Syndrome can be conductive (middle ear), sensorineural (cochlear and potentially auditory nerve) or mixed. There is an increased incidence of middle ear effusions and serous otitis media. The auditory phenotypes in Stickler syndrome sub-types vary, although information available for each is limited. A meta-analysis of hearing loss in Stickler Syndrome indicated that of 313 patients reported in 46 studies, 63% of patients with type 1 were reported as having hearing loss, compared with 82.5% in type 2, and in this latter group hearing loss was more severe. 12 Sensorineural or mixed hearing loss was most common in both groups, and pure conductive loss was evident in only a minority (type 1 10.4%, type 2 5.3%). In the largest reported study of individuals with genetically confirmed type 2 Stickler syndrome, 69% reported hearing loss. 13 In the majority of cases (77%) this was sensorineural, though a higher proportion of individuals (24%) had hypermobile tympanic membranes on tympanometry testing than had previously been reported.
Given the varied auditory phenotypes described in the literature, no definitive information is available on the pathophysiology of hearing loss in Stickler syndromes. Collagen abnormalities have been associated with tympanic membrane, middle ear (ossicular) and cochlear dysfunction. Audiological techniques to determine site of lesion are developing rapidly, including wideband tympanometry, which assesses middle ear function across a wide range of frequencies, and auditory techniques to distinguish cochlear from neural dysfunction hold much promise.
Recent research has shown that both recessive and biallelic mutations affecting a specific locus of the gene for type 2 Stickler syndrome can result in profound or total sensorineural hearing loss. The major Stickler genes COL2A1, COL11A1 and COL11A2 are all subject to alternative splicing, and the natural alternative splicing of exon 9 in COL11A1 modifies the effect of mutations occurring at that locus reducing the severity of the skeletal dysplasia. However, since exon 9 is expressed in Meckel’s cartilage (which gives rise to the malleus and incus of the inner ear and the anterior ligament of the malleus tympanic plate), recessive or biallelic mutations affecting this locus result in a phenotype with very severe hearing loss.14,15
Management
The fact that the hearing loss associated with Stickler Syndromes commonly contains an element of conductive loss has led to an incidence of surgical interventions, including ventilation tube (grommet) insertion in children and middle ear surgery. Whilst there is no published evidence in this regard, anecdotal reports indicate that this may not be beneficial, and may lead to sequelae such as long-term tympanic membrane perforations. Prosthetic management of hearing loss, such as with digital hearing aids, or cochlear implantation in the case of severe-profound sensorineural hearing loss, is low-risk and high benefit.
The integration of Audiology and Otology services in the multidisciplinary management of patients with Stickler Syndromes is advised, and this was pioneered by the NHS England Highly Specialised Stickler Syndrome Diagnostic Service. Hearing tests (audiometry and tympanometry) at diagnosis and regularly at follow up are beneficial.
Musculo-skeletal features
Most patients have skeletal changes, and musculoskeletal complications often present at different times of life but the phenotypic impact on the musculoskeletal system in the various sub-groups of Stickler syndrome are heterogenous and vary both within and between families in tandem with age-related changes.
Infants and young children commonly have joint hypermobility, with a range of movement greater than that expected for age (Figure 5). When severe, this can be associated with delayed gross motor milestones and children can be appear less coordinated and clumsy. Children often have flexible flat feet and muscular discomfort. Pain with prolonged activity is most commonly reported in the fingers, wrists and legs, and together with nocturnal idiopathic leg pains are common presentations to medical professionals. Hypermobility commonly reduces during childhood and is rarer in adults. 1 Some children with Stickler syndrome have short stature in comparison with unaffected siblings, 16 but most are of normal height. A slim marfanoid habitus with arachnodactyly is occasionally present in some but is not universal.

Stickler syndrome: joint hypermobility. Reproduced with permission from Snead. 9
Spondyloepiphyseal dysplasia (SEDC) is a more severe phenotype affecting the spine and long bones. 10 Radiographs may reveal widened metaphyses, enlarged joints with prominent epiphyses. Flattened vertebrae (platyspondyly) are also seen and fusion of carpal centres have also been described. 17 In type 1 Stickler syndrome, the failed fusion of growth plates is due to abnormal collagen II being distributed in the physis.
Hip changes similar to those seen in Perthe’s disease are a recognised finding in middle childhood, and are identified when children present with pain or as an incidental finding on radiographs requested for another reason. 18 It is likely that the changes in the hip in Stickler’s syndrome are caused by femoral head failure from a severe delay in ossification, rather than avascular necrosis. 1 Protrusio acetabuli and Coxa vara (deformity of the hip joint) are other common findings with reduced range of movement detected on clinical examination. 19 In the knees genu valgum alignment, Osgood Schlatters disease and osteochondral defects are seen in late childhood and early adolescents, 20 but the frequency is likely similar to the general population. Older children commonly present with stiff joints and may be referred to a rheumatologist with a concern regarding an inflammatory arthropathy.
During adolescence, additional spinal changes may present, including curvatures. Both scoliosis and a Scheuermann’s disease like kyphosis develop due the failed ossification and the presence of ovoid vertebral bodies. 21 These spinal abnormalities can be asymptomatic but in some may present with localised back pain. Regular review is important to assess the angle of the curvature and ensure prompt referral to spinal surgeons.
In adulthood a combination of mild skeletal dysplasia, hypermobility, scoliosis and early onset osteoarthritis produce significant changes to the quality of life for patients (Figure 6). There is little difference between the musculoskeletal impact of type 1 and type 2 Stickler Syndrome. Weight-bearing joints and the spine feature prominently although all peripheral joints and spinal segments are affected. The knee tends to be the joint that is most commonly reported as being painful, followed by the hip, lower back, ankle and shoulders.

Structural bone abnormality in the knees of a patient with the Stickler Syndrome (note the asymmetry in the femoral condyles with the medial condyle being abnormally small).
Management
In childhood, management includes a combination of screening, education regarding the condition and prognosis, and supportive therapy of individual problems.
Flat feet and hypermobility often improve with age and milder cases do not need any specific therapy. However, maintaining activity, including encouraging low impact sports such as swimming and cycling and ensuring children wear well-fitted supportive footwear, are often beneficial. Moderate-to-severe cases require formal paediatric musculoskeletal assessment to exclude structural bone abnormalities followed by individualised physical and occupational therapy plans. Orthotics may also be prescribed if additional plantar arch support is required.
Hand writing difficulties are common in younger children due flexible finger joints, and supportive aids in the form of thickened pencil grips and writing slopes often assist children in the education setting. An occupational therapy review of handwriting in the school environment is recommended when significant difficulties are present. Referral to therapy professionals experienced in the management of children with hypermobility, rheumatological conditions and skeletal dysplasias is important with the expectation that intermittent long-term review will be needed to adapt exercise programmes as biomechanics change with age. An understanding of the natural course of the condition is helpful for therapy professionals, and school staff should also be educated regarding the impact of all aspects of the condition.
Routine radiographic screening is not required in childhood. Targeted investigations including radiographs should be requested based on clinical review by paediatricians with an interest in rheumatology or orthopaedic surgeons with paediatric expertise. Referral to additional specialists including spinal surgeons and paediatric orthopaedic surgeons are appropriate when treatable abnormalities are identified, for example, leg alignment abnormalities, severe scoliosis, kyphosis or spinal column instability.
Pain-relieving medications can be used as required for muscle and joint pain, especially to support engagement with physical activity but are rarely required regularly in young children. Simple analgesia in the form of paracetamol and nonsteroidal anti-inflammatory drugs (NSAIDs) is all that is recommended in the paediatric population. Opiate-based medications are often unhelpful for bone and muscle pain, with side effects outweighing any benefit. Codeine-containing medicines should not be prescribed to children under 12 years old due to their varied speed of hepatic metabolism.
The NHS England Highly Specialised Service offers all children and young people a formal paediatric rheumatology assessment, including provision of advice and education regarding the condition and the potential prognosis. This specifically includes discussion regarding appropriate supportive measures and recommendations regarding physical activity during the different stages of childhood.
Adults
Management of pain in adults with the Stickler Syndrome is important. NSAIDs are more effective than paracetamol although carry more co-morbidity with long-term use and renal, cardiovascular and gastrointestinal co-morbidities should be evaluated carefully before committing to using such medication. Weak opioids are rarely helpful and strong opioids should be avoided. Other pain medications such as tricyclics and gabapentinoids may have a role. Selective serotonin and noradrenergic reuptake inhibitors have some benefit in pain management. Topical approaches to pain management including topical NSAIDs, heat-based treatment, t
The mainstay of conservative pain management is an exercise programme, focussing usually on core stability. It is unknown whether aerobic or strengthening forms of exercise are more effective for pain management. Splinting and orthotics have a role where necessary. Pain Management Programmes with psychological support using cognitive behavioural therapy should also be considered and can be helpful.
Surgical management may be required to replace or re-align joints. Sometimes the dysplasia, particularly with femoral head failure, requires tailored specialist Orthopaedic evaluation; however, most patients with the Stickler Syndrome will not require specialist evaluation. There are no particular peri-operative anaesthetic requirements with regards to clotting risk nor cardiac evaluation. Reports of patients with the Stickler Syndrome having a higher incidence of mitral valve prolapse have not been substantiated on subsequent study.22,23 Anaesthetic risk in patients with jaw, chest wall and spinal abnormalities need to be evaluated on an individual case basis (see below).
The impact of the musculoskeletal complications of the Stickler Syndrome extend into all aspects of patients’ lives. Workplaces must recognise the impairments that the condition may cause and are required by law to make reasonable assessments and modifications for their employees. Often, patients with Stickler Syndrome will find physical work in their fifth and sixth decades to be difficult and will need to have career trajectories to take this into account. The importance of educating families and those in the community of the likely impact of the Stickler Syndrome can help reduce the distress associated with this long-term condition.
Oro-facial features
Classical facial changes are often seen at birth but can become more pronounced in early childhood. Changes include malar hypoplasia, flattened or broad nasal bridge and, in some, micro/retrognathia as part of a Pierre–Robin-sequence-associated cleft palate. 1 Stickler syndrome is the cause of over 30% of all Pierre–Robin sequence cases. Some people with Stickler syndrome have milder facial features, which makes identifying the condition more challenging when using diagnostic strategies that rely on facial phenotype. In those with collagen II mutations, facial features can become less prominent in adulthood, whereas those with collagen XI mutations usually persist but can be milder.17,24 In general, reliance on facial phenotype for diagnosis is unwise, and slit-lamp examination for the pathognomonic vitreous phenotypes is far more dependable as well as indicating Stickler sub-groups in clinically challenging phenotypes, such as those complicated by double heterozygosity. 25
The association with Pierre–Robin sequence and cleft palate is well recognised in all sub-groups of Stickler syndrome, although interestingly cleft lip is rare (2/1604, 0.12%). In a series of 502 cases of Stickler syndrome, a history of cleft repair was present in almost 40% of type 1 Stickler syndrome patients and 25% of type 2 Stickler syndrome patients, Pierre–Robin sequence being present in 13% and 1.5% of type 1 and type 2 patients, respectively. 26 Examination of the palate by inspection and palpation is important as some patients will be unaware of their high palate or sub-clinical soft cleft (Figure 7).

Examination of the palate is important in suspected Stickler syndrome – this patient reported no palate abnormality when questioned.
Ancillary investigations
(a) Given the dominant inheritance in the vast majority of cases of Stickler syndrome, it is imperative to examine both parents and all siblings of affected children. This can be particularly valuable for neonates where vitreous phenotyping can be challenging or for index cases who have already undergone previous vitrectomy surgery.
(b) Audiometry assessment is useful to distinguish conductive hearing loss (secondary to mid-line cleft abnormalities Stickler or non-Sticker) from sensorineural loss (see above) and may be combined with tympanometry for assessment of tympanic membrane hypermobility.
(c) Radiology imaging may be normal even in the presence of obvious joint hypermobility. Any joint(s) may be affected but most common involved are hips, knees and lumbar spine (see above). Imaging can reveal evidence of epiphyseal dysplasia, particularly of the hips, and patients may volunteer a past historical diagnosis of Legg-Perthes disease, although this probably represents dysplastic development of the femoral head rather than true avascular necrosis.
Differential diagnoses
Spondylo-epiphyseal dysplasia congenita (SEDC), Kniest dysplasia and Czech dysplasia are all allelic with type 1 Stickler syndrome but usually result from dominant negative rather than haploinsufficiency mutations resulting in more severe skeletal dysplastic changes.27–29 As with type 1 Stickler syndrome, the risk of blindness from retinal detachment is high and variations in pre-mRNA splicing of COL2A1 can in some instances blur the clinical differential between these allied type II collagenopathies. 30
Marfan syndrome is also associated with an abnormal vitreous architecture, but more usually progressive rather than congenital myopic astigmatism. No case of myopia was found under 3 years of age in one large series, in contrast to the congenital myopia found in type 1 Stickler syndrome. Retinal detachment is a recognised association reported in 8–50% of cases; approximately 75% of these occur before 20 years of age and patients may exhibit joint hypermobility and a crowded, high-arched (but rarely cleft) palate. Marfan syndrome may be associated with ectopia lentis (which is uncommon in Stickler syndrome) but the vitreous phenotype is key to the clinical differential diagnosis between the two disorders. Cardiovascular associations include mitral valve prolapse, mitral regurgitation, dilation of the aortic root, and aortic regurgitation, with aneurysm of the aorta and aortic dissection being the major life-threatening complications. 9
Wagner vitreoretinopathy (WGVRP)/erosive vitreoretinopathy (ERVR) were historically considered to be variants of Stickler syndrome without ocular involvement. 31 The two disorders are now known to be genetically and phenotypically separate and the term ‘Wagner–Stickler’ should be abandoned as inaccurate and confusing. The ocular features of Wagner and ocular-only Stickler can also be differentiated on vitreous phenotype.31,32
Donnai-Barrow syndrome (DBS)/facio-ocualo-acoustico-renal (FOAR) syndrome reports have suggested some similarities between DBS and Stickler syndrome on the basis of myopia and retinal detachment, but the absence of vitreous anomaly, hearing loss and the associated renal abnormalities should aid in differentiating the two disorders. 33
Molecular genetic diagnosis
The great majority of cases of Stickler syndrome (type 1) are inherited in an autosomal dominant fashion and the majority will harbour heterozygous COL2A1 loss-of-function variants resulting in haplo-insufficiency of type II collagen – the major structural protein of the vitreous body.34–36 Type II procollagen exists in two alternatively spliced forms. A short form, which is expressed in cartilage, has exon 2 spliced out and so loss-of-function variants on exon 2 result in ocular-only Stickler syndrome without systemic involvement. 32
Type 2 Stickler syndrome is caused by variants in COL11A1 encoding the alpha-1 chain of type XI collagen, with patients exhibiting a different vitreous phenotype.37,38 Typical molecular changes result in either substitution of an obligate glycine within the Gly-Xaa-Yaa amino acid sequence repeat region of the molecule, mRNA missplicing or deletions/duplications, any of which typically leaves the message in-frame.24,37–40
Recessive Stickler syndrome is much less common but has been reported in association with homozygous variants in genes for type IX collagen, COL9A1, COL9A2 and COL9A3 (see Table 1).4–6,41–44 Compound heterozygous COL11A1 variants where alternative splicing can modify the effect of mutations in COL11A1 can result in recessive type 2 Stickler syndrome with unusually profound hearing loss rather than a more severe (or lethal) skeletal dysplasia.14,15
NHS England highly specialised Stickler syndrome diagnostic service
Diagnostic service sequence analysis of genes generally examines and reports on variations within a designated region 5′ and 3′ of each exon, typically 30 bp up- and down-stream. However, because of the degenerate nature of the splice sites, intronic variants outside the AG and GT dinucleotides of the acceptor and donor splice sites are most often classified as being of unknown clinical significance unless there is some functional evidence of their pathogenicity. It is now becoming clear (particularly in type 1 Stickler syndrome) that mutations deep within introns can also interfere with normal processing of pre-mRNA and result in pathogenic effects on the mature transcript.30,39 In diagnostic laboratories, these deep intronic variants most often fall outside of the regions analysed and so are rarely identified or reported.
With next generation sequencing identifying more of these unclassified variants, it may be necessary to perform additional studies to determine the pathogenicity of such sequence anomalies as recent research has shown that deep intronic mutations are regularly identified in Stickler syndrome, 39 with resulting variable effects on phenotype as a result of differing transcripts that result in either haploinsufficiency or a dominant negative effect.
The most common approach for next generation sequencing utilises a capture approach to enable large numbers of genes and patients to be sequenced simultaneously. However, this rarely enables sequence deep within the intronic regions to be determined. A cost-effective alternative in those patients exhibiting the membranous type 1 vitreous anomaly has been to sequence the whole COL2A1 gene, using PCR amplification of the complete COL2A1 genomic region followed by conversion of these large sections of DNA into smaller, more readily sequenced fragments.34–36,39 Deep intronic variants can then be identified and in silico analysis performed to predict the consequence of such variants. However, accurately determining the in vivo splicing of the variant transcript requires a functional assay to be performed. To achieve this, we clone the variant intron into a bespoke construct and express the novel transcript in a mammalian cell line. This enables the effect of the variant to be determined and compared with both the in silico predictions and the patient’s phenotype. This approach has resulted in the ability to reclassify variants of ‘unknown clinical significance’, and provide the certainty to enable predictive/pre-symptomatic testing in family members and future pregnancies.
As the whole genome sequencing of patients with rare disease becomes more common, the population frequency of such variants will become an additional tool to guide the determination of variant pathogenicity.
Summary
The Stickler syndromes are the most common cause of familial retinal detachment and the most common cause of rhegmatogenous retinal detachment in children. Half of all patients with genetically confirmed type 1 Stickler syndrome who experience retinal detachment and who do not receive prophylaxis will suffer retinal detachment in their second eye within 4 years of the first eye. There are now known to be at least 11 distinct phenotypic sub-groups, and molecular genetic analysis has a key role to play in risk stratification for prevention of blindness and duel sensory (auditory) impairment.
Footnotes
Author contributions
Martin Snead: conceptualization; formal analysis; funding acquisition; methodology; project administration; supervision; writing – original draft; writing – review and editing;
Howard Martin: writing – review and editing;
Peter Bale: writing – original draft; writing – review and editing;
David Baguley: writing – original draft; writing – review and editing;
Nick Shenker: writing – original draft; writing-review and editing;
Philip Alexander: writing – review and editing;
Annie McMinch: writing – original draft; writing – review and editing;
Arabella Poulson: writing – original draft; writing – review and editing
Conflict of interest statement
The authors declare that there is no conflict of interest.
Ethics statement
Ethical approval and informed consent was not required for this review.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.