
Abstract
Persistent right aortic arch (PRAA) in cats is an uncommon vascular anomaly with clinical signs referable to oesophageal obstruction. To our knowledge no reports of axial skeletal malformations concomitant to PRAA have been reported in cats. The aim of this study is to depict a new clinical feature in cats affected by PRAA. In the study six cats with a diagnosis of vascular ring anomaly were enrolled. A complete physical examination, a neurological examination and a total body radiograph were performed on each animal. Four of the six cats showed contemporary PRAA and skeletal malformations. Additionally, for the first time, a genetic test was performed on one subject to detect DNA alterations in the homologous DiGeorge region of cat. The percentage of skeletal malformations reported in the normal population was compared with animals with PRAA and showed a higher frequency. Genetic testing failed to demonstrate a correlation between PRAA and DiGeorge genomic deletion. A review of veterinary and human diseases that presented both conditions was assessed. The few animals enrolled do not allow definitive conclusions. Further studies are required to corroborate the correlation between PRAA and axial skeletal malformations in cats.
Introduction
During embryogenesis mammals develop six pairs of aortic arches. From the left aortic arch normally develop the aorta, ligamentum arteriosum and left subclavian artery; from the right one develops the right subclavian artery. All vascular ring anomalies result from abnormal development of arches III, IV or VI. 1 These anomalies have been reported in a wide variety of animal species, including the domestic cat,2,3 cougars, 4 dogs, 5 horses,6,7 elephant seals, 8 cattle,9 lamas and alpacas. 10 The most common anomaly in dog is the persistent right aortic arch (PRAA) with a left ligamentun arteriosum (95%). 11 No comparable data exist for PRAA in cat.
Clinical findings of vascular ring anomalies are usually referable to oesophageal obstruction. Dilation of the oesophagus cranial to the base of the heart and left displacement of the trachea are common radiographic signs. 11 Definitive diagnosis often requires exploratory surgery 1 and prognostic factors are not completely clear. Some authors suggest that early surgical correction and the presence of mild dilation of the oesophagus are indicative of a better prognosis, 12 but more recent studies do not confirm this.13,14
PRAA seems to be hereditary both in dogs and humans.15,16 Heritability of heart defects in dogs, based on epidemiological studies, indicates that purebred dogs are more prone to develop heart defects than mongrels. Breeding studies involving dogs with similar heart defects showed a high frequency of congenital heart disease in offspring. The type of heart defect detected in those puppies was identical or closely related to the one of the parents. 15 In humans 80% of conotruncal heart defects, including aortic arch abnormalities, have been associated with deletion of the chromosome 22q11 region, 17 also known as the DiGeorge region. DiGeorge syndrome is caused mainly by genomic deletions: 90% are 3 Mb deletion, and 7% are 1.5 Mb genomic deletions. The other 3% are represented by other caused as TBX1 gene mutations. 18 Deletion on this region in humans can lead to concomitant skeletal and vascular malformations. Comparative mapping of this region failed to demonstrate its linkage to conotruncal heart defects in dogs, 19 but a more recent study on inheritance of a specific form of PRAA in German Pinschers showed a chromosome-wide significantly-linked genomic region on CFA26, which corresponds to the human DiGeorge critical region (DGCR). However, despite linkage and association between this form of PRAA and the canine DGCR, none of the 13 genes and their 36 polymorphisms appear responsible for it, suggesting that chromosomal aberrations might be causal. 20
Reports of multiple malformations in dogs21–23 and cats24–27 have been published, but no skeletal malformations in animals affected by PRAA have been reported with the exception of one case of cribriform plate aplasia in an elephant seal. 8 Additionally, no searching for deletions in the DiGeorge region in cats has been performed. Our aim is to describe the presence of a new clinical feature in cats affected by PRAA and to attempt to correlate this malformation with gross genomic alteration involving the DGCR.
Materials and methods
All cats with a diagnosis of PRAA presented to the Department of Veterinary Clinical Sciences, Section of Surgery of the Università degli Studi di Milano from November 2006 to December 2009 were included. Informed client consent was obtained from each owner. Each animal underwent a complete physical and neurological examination. Blood samples were collected for haematological and biochemical analysis. Diagnosis was based on clinical signs and on the presence of a dilation of the oesophagus cranial to the base of the heart on contrast oesophagram. Given the finding of a malformed thoracic vertebra in the first cat a total body radiograph of each patient was assessed.
A lithium–heparin blood sample was collected from one cat for genetic studies of the DiGeorge region using CGH array technology — a high-resolution tool for human genome-wide DNA copy number variation profiling (Human Genome CGH Microarray, 44K, Agilent) — and cytogenetic techniques. Both analyses were performed as reported in literature. 28
All animals except one underwent a standard thoracotomy to confirm the diagnosis and correct the vascular anomaly.
Results
Six animals with PRAA were included in the study, including four males (all domestic shorthair) and two females (Maine Coon and domestic shorthair). Their age ranged from 2 to 11 months. Two cats (cats 2 and 3) belonged to the same litter. In 4/6 cats we detected at least an axial skeletal malformation. Malformations reported were: supernumerary thoracic and lumbar vertebrae, the presence of 15 ribs in the left side of the thorax, anomalies of thoracic and caudal vertebrae and a supernumerary sternebra (Figures 1–3). No alterations of cervical vertebrae were detected. For a detailed list of the malformations see Table 1.

X-ray showing 15 ribs in the left hemithorax. The arrowhead shows the two ribs articulated within the same vertebra

Presence of a thoracic block vertebra (T8-T9), a butterfly vertebra (T12) and 6 lumbar vertebrae
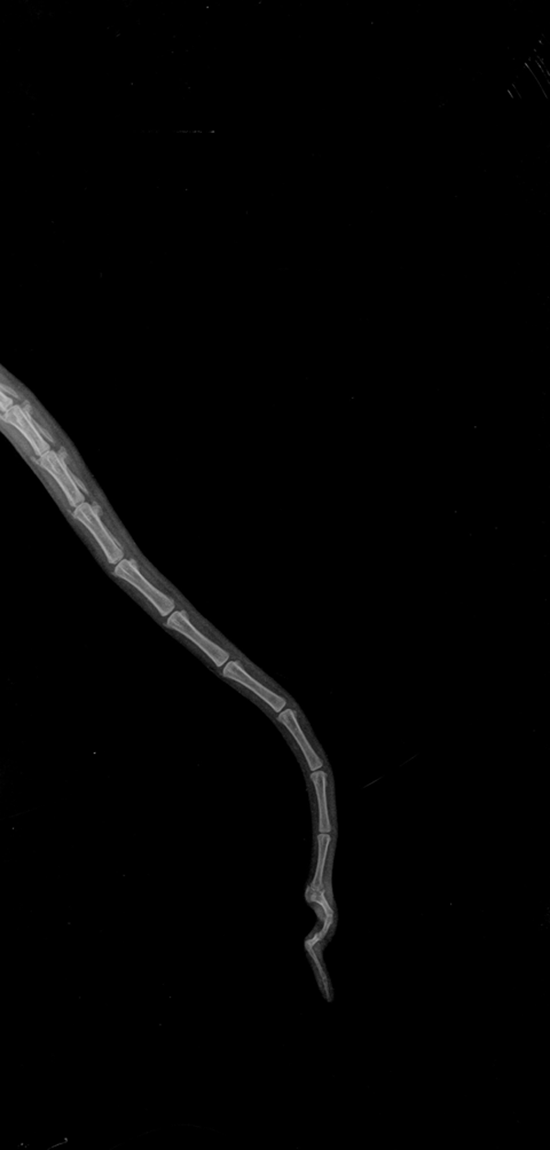
Malformation of a caudal vertebra
Animals included in the study and associated skeletal malformations

Cat 1 did not undergo surgery because the owner declined. All other subjects had the diagnosis of PRAA confirmed. In cat 2 concomitant persistence of the cranial left vena cava was detected. After surgery animals were fed with a liquid diet for 1 month. Then, they were fed four times a day with small amounts of commercial diet. All animals stopped to regurgitate after the correction of the PRAA with the exception of cat 6, which maintained the same clinical signs.
DNA analysis on cat 5 was carried out by both conventional cytogenetic techniques and CGH array technology. In this case, owing to the absence of a cat-specific copy number variation profiling array, we used a human CGH array. The cytogenetic analysis did not reveal any gross genomic alterations (Figure 4). Despite the difference between the human and cat genome the analysis worked well. The results obtained did not highlight any deletion in the DGCR (Figure 5).

Karyotype obtained from cat 5. The chromosomes were Giemsa stained and arranged following standard cat karyotype

CGH array result. In this case the critical region on HSA22 for DiGeorge syndrome is displayed in the yellow box. No duplications or deletions are present in this genomic region
Discussion
The normal spine of cats consists of seven cervical, 13 thoracic, seven lumbar, three sacral and 24 caudal vertebrae. Usually, the sternum is composed of seven or eight sternebrae. Any difference is considered to be abnormal. 29 From the results of a retrospective radiographic study performed on vertebral segments of 200 cats affected by various pathologies, 23% presented congenital axial skeletal abnormalities. Thoracicisation of L1 and the malformation of the sacrocaudal junction were the most notable malformations. 30 No animal in this study had concomitant skeletal and vascular malformation.
In our study 66% of the animals presented a skeletal alteration located in the thoracic, lumbar, caudal spine or in the sternum. No animals showed clinical signs related to those anomalies as reported by Newitt. 30 Even though the number of subjects involved in our study is poor, cats affected by PRAA seem to be more prone than the normal population in presenting this condition.
Of the two animals from the same litter only one had vertebral malformations (male). This cat had persistence of the left cranial vena cava. The heritability of PRAA has not been described in cats, but has been demonstrated in dogs.20,31 In dogs and cats some breeds are reported to be prone to PRAA. 32 However, we found this pathology only in one declared pure breed cat (Maine Coon) whose pedigree was not available. This variance should be due to a larger presence of mixed breed with respect to pure breed cats in the local population. To our knowledge this is the first report of a Maine Coon affected by PRAA.
As there is a suspicion of genetic mutations in the DiGeorge region in dogs affected by PRAA, we postulated similarities between DiGeorge syndrome and PRAA in cats.
In humans DiGeorge syndrome is caused by a microdeletion involving chromosome 22. The deletion occurs near the middle of the chromosome at a location on the long arm designated as q11.2. It has a prevalence estimated at 1:4000 live births 33 and it is thought to play a role in as many as 5% of all congenital hearts defects. 34 Symptoms consist of cleft palate, heart defects, learning difficulties, vertebral malformations, renal anomalies, thymic aplasia and hypocalcaemia.16,35 Butterfly vertebrae were noted more frequently than in the normal population. The combination of this anomaly with conotruncal heart defects should heighten suspicion for a deletion in the DiGeorge region. 36 Alterations in the cervical spine are described and are considered an important marker of the 22q11.2 microdeletion syndrome. 37 The pathogenesis of skeletal malformations remains unclear in this syndrome at present, but the haploinsufficency of the gene TBX1 may be implied. 36 We did not report cervical malformations in the cats in our study. To determine if cats should have a similar syndrome all animals were evaluated for the presence of other macroscopic anomalies. No concomitant alterations were noticed at physical and neurological examination. In humans the diagnosis of DiGeorge syndrome is made by fluorescence in situ hybridisation (FISH) using DNA probes from the 22q11.2 chromosomal region. DiGeorge’s minimal critical region is comprised between base pair (bp) 17,398,178 and bp 19,779,416 on chromosome 22 (refer to hg17). This region is a homologue to cat chromosome D3 43,587,024–45,877,480 bp (National Human Genome Research Institute/Genome Technology Branch (NHGRI/GTB) V17e/felCat4 genome assembly). The analyses of the gene content on this cat genome region confirm that it is the homologue to human DGCR. The negative results of the test cannot exclude DiGeorge syndrome in cats because it was only performed in one animal. However, this finding should show that, as in dogs, PRAA is inherited in a complex manner consistent with a polygenic basis 15 or chromosomal aberrations. 20 Another cause of association between skeletal and vascular malformations can be the deficiency of glypican 3, a heparan sulphate suspected to be responsible for Simpson–Golabi–Behmel syndrome. This syndrome is characterised by a wide range of developmental anomalies, including vertebral malformations, vascular anomalies, macroglossia, dysplastic and cystic kidneys, hernias and supernumerary nipples. 38 The vascular anomalies more commonly represented are patent ductus arteriosus, atrial septal defect, ventricular septal defect, preductal coarctation of the aorta and pulmonary stenosis. No aortic arch malformations were described in humans. Our animals did not show macroscopic anomalies and a different cardiac malformation was recognised. It cannot be excluded that cats present a syndrome similar to that of humans because mice with deletion of glypican 3, a validated animal model of Simpson–Golabi–Behemel syndrome, showed several of the clinical signs, but no vascular anomalies. 38 . Looking for mutations of the gene encoding for glypican 3, as in Simpson–Golabi–Behmel syndrome, would be important to demonstrate the correlation between these pathologies.
In dogs and cats few accounts of concomitant malformations involving different organs have been described. In dogs the ocular-chondrodysplasia of Labrador Retrievers and Samoyeds is a syndrome characterized by short-limbed dwarfism and ocular abnormalities.21,39 Differently from Labradors, Samoyeds can also present concomitant haematological alterations. 40 Kartagener’s syndrome is another pathology in which multiple malformations are present. Animals affected present situs inversus totalis, rhinosinusitis and brochiectasia. The latter two signs refer to the presence of ciliary dyskinesia. The pathology has also been seen in the Cavalier King Charles spaniel, Dachshund, Chow Chow, Border Collie and Doberman Pinscher. 41
In cats multiple anomalies mostly affect the cardiac, 24 skeletal42,43 and urogenital 25 systems. A recent case report described the presence of vertebral abnormalities, anal atresia, radial agenesis, and cardiovascular and renal defects in a kitten. The authors supposed that this animal was affected by a syndrome present in humans called VACTERL (vertebral abnormalities, anal atresia, cardiovascular defects, tracheoesophageal fistula/oesophageal atresia, renal abnormalities, limb defects). 42 In order for a case to be considered a VACTERL association, two or more core defects must be present, ie, oesophageal atresia, anal atresia, upper pre-axial limb reduction defects and costovertebral abnormalities. 44 None of those signs were present in the animals in the current study with the exception of costovertebral abnormalities. Moreover, some authors do not include cardiovascular malformations in the diagnosis of this syndrome. 45 For this reason we discount the possibility that our animals were affected by VACTERL syndrome.
Owing to the presence of concomitant multiple anomalies in animals, we suspected that skeletal malformations and PRAA could be related. PRAA in cats is reported as not being a common condition. 46 Collecting a large number of animals from a single centre to determine the real incidence of the skeletal malformations is difficult. The retrospective analysis of radiographs of animals affected by PRAA from different centres should aid in determining it. A larger number of subjects are mandatory to confirm the correlation between PRAA and axial skeletal malformations in cats. The genetic test performed did not highlight deletion in the DGCR. The test was only performed in one cat. Therefore, we cannot consider it to be definitive data.
Conclusion
This study can be considered as the starting point for further investigations to correlate the presence of these two malformations and to evaluate the possible genetic correspondence to human syndromes.
Footnotes
Acknowledgements
We are grateful to Stefania Gimelli (Geneve University Hospital, Suisse) for help with the CGH array analysis and to Professor Mauro Di Giancamillo (Università degli Studi di Milano, Italy) for the radiographic images.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not for profit sectors.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.