
Abstract
We report a fetus at 20 + 5 weeks’ gestation with an exceptionally rare constellation of congenital anomalies. Prenatal imaging revealed left congenital diaphragmatic hernia, rightward cardiac displacement, possible tracheoesophageal fistula (TEF), transposition of the great vessels, and a large ventricular septal defect. Autopsy confirmed complete absence of tracheoesophageal separation from the larynx to the carina, consistent with Kluth type Xc2, incorporating extralobar pulmonary sequestration with esophageal communication. The findings met criteria for a communicating bronchopulmonary foregut malformation (CBPFM), Srikanth Group III. Additional anomalies included bilateral pulmonary hypoplasia, bilateral unilobar lungs with eparterial bronchi, mirror-image dextrocardia, L-looped ventricles with concordant atrioventricular and ventriculoarterial alignments, right-sided aortic arch, bilateral superior vena cava, cardiac-type total anomalous pulmonary venous connection with drainage into the morphologic right atrium, and anomalous coronary origin from the pulmonary artery. Cytogenetic studies and whole-exome sequencing were normal. To our knowledge, this is the first report of Kluth type Xc2 TEF with Srikanth Group III CBPFM since Stolte’s original description in 1952, and the first to combine this unique constellation. This case broadens the recognized spectrum of foregut and cardiopulmonary maldevelopment and underscores the importance of comprehensive evaluation in fetuses with multiple anomalies.
Keywords
Case Report
This is a case of pregnancy termination at 20 + 5 weeks gestation via induction of labor and spontaneous vaginal delivery in a 32-year-old G1P0 woman due to multiple fetal anomalies identified on prenatal imaging.
At 12 weeks US revealed a 6.5 mm nuchal translucency, which is associated with a high risk of aneuploidy and structural and heart malformations. A CVS at 13 weeks reported normal karyotype, microarray and trio whole exome sequence. Fetal ultrasounds at 16 + 6 and 19 + 5 weeks gestation revealed several abnormalities, including a left-sided congenital diaphragmatic hernia with rightward displacement of the heart and a suspected tracheoesophageal fistula. Bowel loops were herniated into the thoracic cavity, and the stomach appeared collapsed in the abdomen. A portion of lung tissue appeared to be present on the right side, behind the heart and crossing the midline. Cardiac anomalies included a large ventricular septal defect (VSD) and transposition of the great arteries. The main pulmonary artery was seen arising from the left ventricle and directed into the upper posterior right chest without visible bifurcation. The aorta originated from the right ventricle.
Autopsy examination of the phenotypically male fetus revealed multiple external dysmorphic features, including retrognathia, a short neck with bilateral webbing, and a clenched right hand with the index finger overlapping the thumb. Fetal growth was consistent with 20 weeks gestation by foot length measurement.
Internal examination revealed complex congenital anomalies. There was a left-sided hemidiaphragmatic hernia with herniation of the left liver lobe, stomach, and bowel loops into the left thoracic cavity (Figure 1A). A complete absence of tracheoesophageal separation from the larynx to the carina was noted, resulting in a continuous defect between the esophagus and the laryngotracheal complex (Figure 1B and C). Microscopic examination of cross-sections at this level demonstrated incomplete development of the tracheoesophageal septum (Figure 2B).
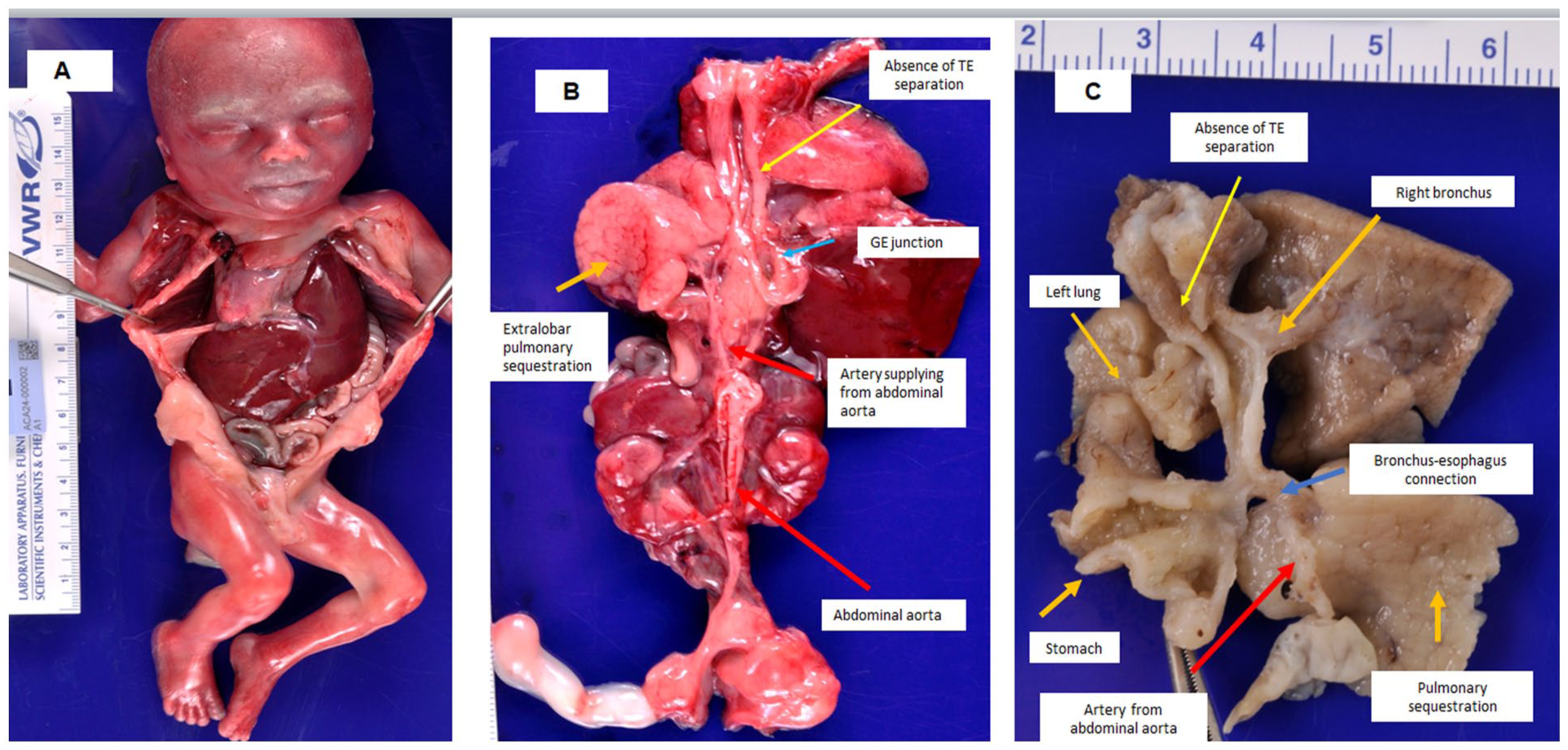
(A) Congenital left hemidiaphragmatic hernia with herniation of the stomach, small and large bowel loops, and a portion of the left liver lobe into the left thoracic cavity. (B) Tracheoesophageal fistula with complete absence of tracheoesophageal separation. An aberrant artery arising directly from the abdominal aorta supplies an extralobar pulmonary sequestration. Pulmonary sequestration. (C) Complete failure of tracheoesophageal separation extending from the larynx to the carina, resulting in a persistent common channel between the esophagus and tracheobronchial tree. An aberrant artery from the abdominal aorta supplies the extralobar pulmonary sequestration. An aberrant bronchial connection extends from the pulmonary sequestration to the lower esophagus.
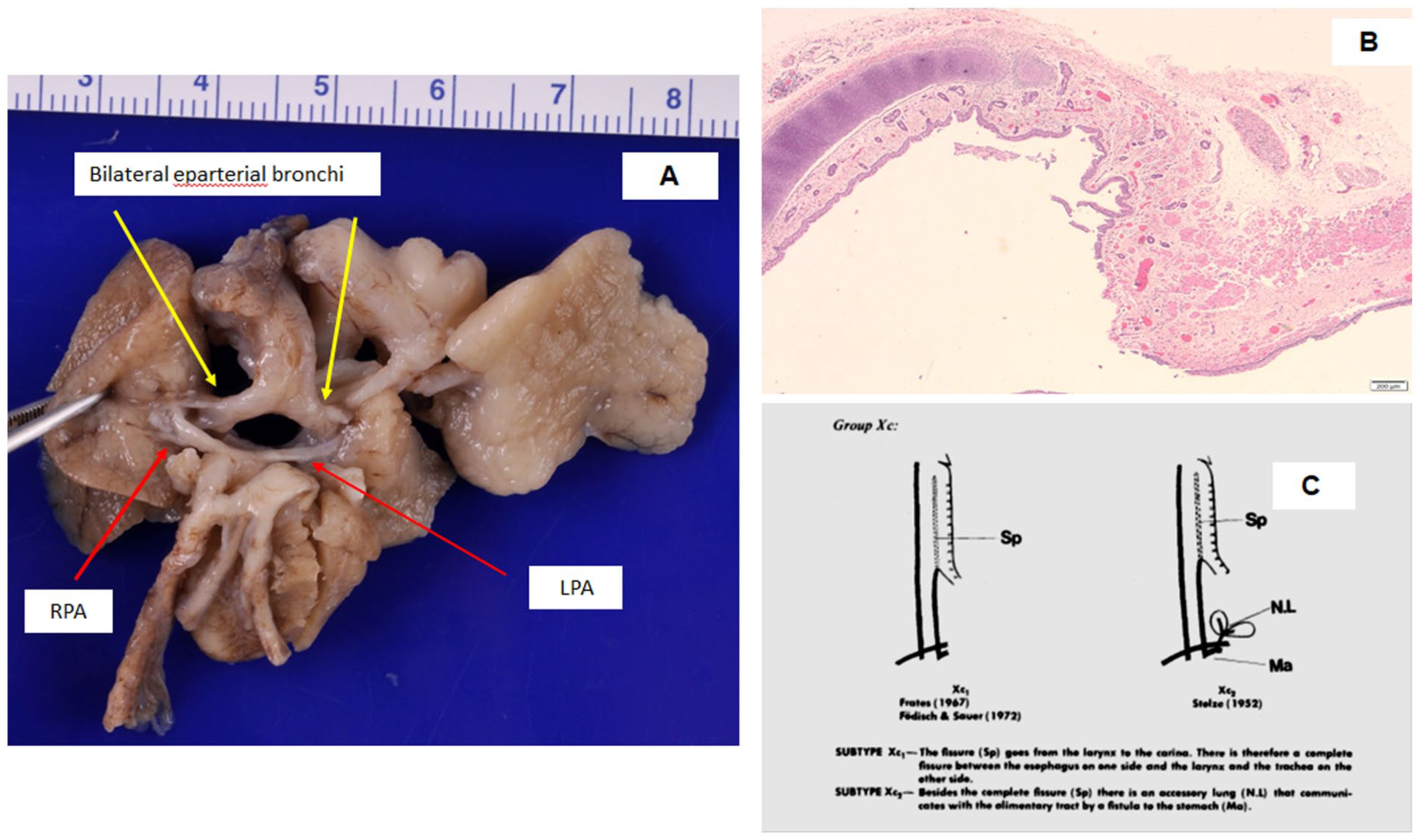
(A) Bilateral eparterial bronchi, with both main bronchi located above their respective pulmonary arteries. In normal anatomy, only the right upper lobe bronchus arises above the right pulmonary artery (eparterial), while the bronchi to the right middle and lower lobes pass below (hyparterial). The left main bronchus is typically hyparterial, lying entirely below the left pulmonary artery. (B) Cross-sectional view of a tracheoesophageal fistula, demonstrating complete absence of tracheoesophageal separation. There is evidence of an attempted but incomplete formation of the tracheoesophageal septum, consistent with a severe developmental anomaly. (C) Schematic diagram illustrating Kluth subtype Xc2, showing a complete tracheoesophageal fistula (Sp) and an accessory lung (N.L) that communicates with the gastrointestinal tract via a fistula to the stomach. Adapted from Kluth D. Atlas of Esophageal Atresia. Journal of Pediatric Surgery, Vol. 11, No. 6, December 1976.
The lungs were bilaterally unilobed with severe pulmonary hypoplasia, more marked on the left. Both main bronchi appeared symmetric and of epiarterial type (Figure 2A). An extralobar pulmonary sequestration, measuring 3.0 cm × 1.6 cm × 1.5 cm, was identified in the left lower chest and upper abdominal cavity (Figure 1C). These findings correspond to Kluth’s subtype Xc2, characterized by a complete tracheoesophageal fistula with an accessory lung communicating with the gastrointestinal tract (Figure 2C). The sequestrated lung received arterial supply from the abdominal aorta (Figure 1B and C) and drained venously into the hemiazygos vein. A bronchial connection to the lower esophagus was observed at the level just above the gastroesophageal junction (Figure 1C), consistent with Srikanth Group III communicating bronchopulmonary foregut malformation (CBPFM). 1 The stomach was abnormally small with a diverticular outpouching.
The cardiac examination showed atrial situs inversus, dextrocardia, L-looped ventricles (left-sided right ventricle and right-sided left ventricle), corresponding to mirror-image dextrocardia, concordant atrioventricular and ventriculoarterial alignment, situs inversus of the great arteries (I.L.I.), a right-sided aortic arch and bilateral superior vena cava (Figures 3A–C and 4B). An anomalous origin of the coronary arteries from the pulmonary artery (ACAPA) was present, with 2 coronary orifices opening into the pulmonary valve sinuses (Figures 3C and 4C). The right-sided superior vena cava drained into the coronary sinus. The inferior vena cava connected to the right atrium located on the left. There was total anomalous pulmonary venous connection (TAPVC), the left and right pulmonary veins drained separately into the morphologic right atrium on the left side (Figure 4A) without forming a common confluence or vertical vein. The abdominal organs were in situs solitus, without structural abnormalities. Contrary to the prenatal suspicion of ventricular septal defect and possible transposition of the great arteries, postmortem examination showed an intact ventricular septum and concordant ventriculoarterial alignment, thereby excluding both lesions.
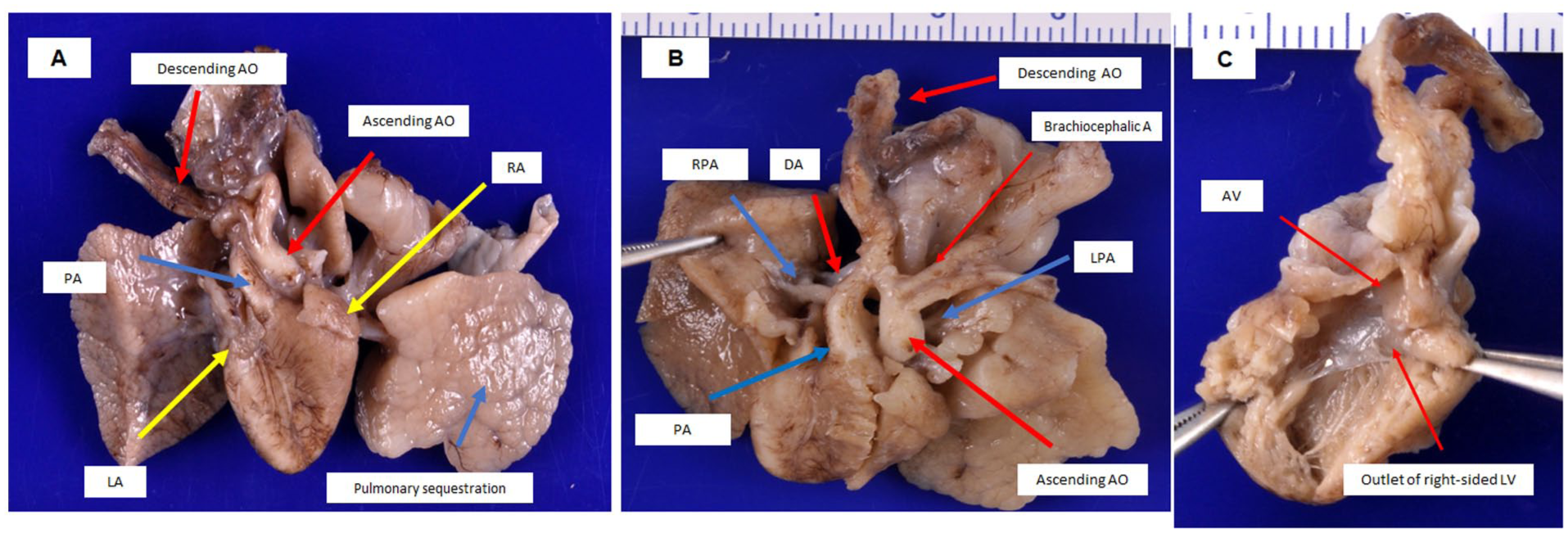
(A) Mirror-image arrangement of the heart (situs inversus of the heart), demonstrating dextrocardia with L-looped ventricles, and bilaterally unilobar lungs, consistent with abnormal thoracic laterality. (B) Mirror-image configuration of the great arteries, showing the anatomy of the pulmonary artery (PA), right and left pulmonary arteries (RPA, LPA), ductus arteriosus (DA), and ascending and descending aorta. (C) The right-sided left ventricle (L-looped) gives rise to the aorta with a concordant ventriculoarterial connection. Notably, there is absence of visible coronary arterial orifices in the aortic root.
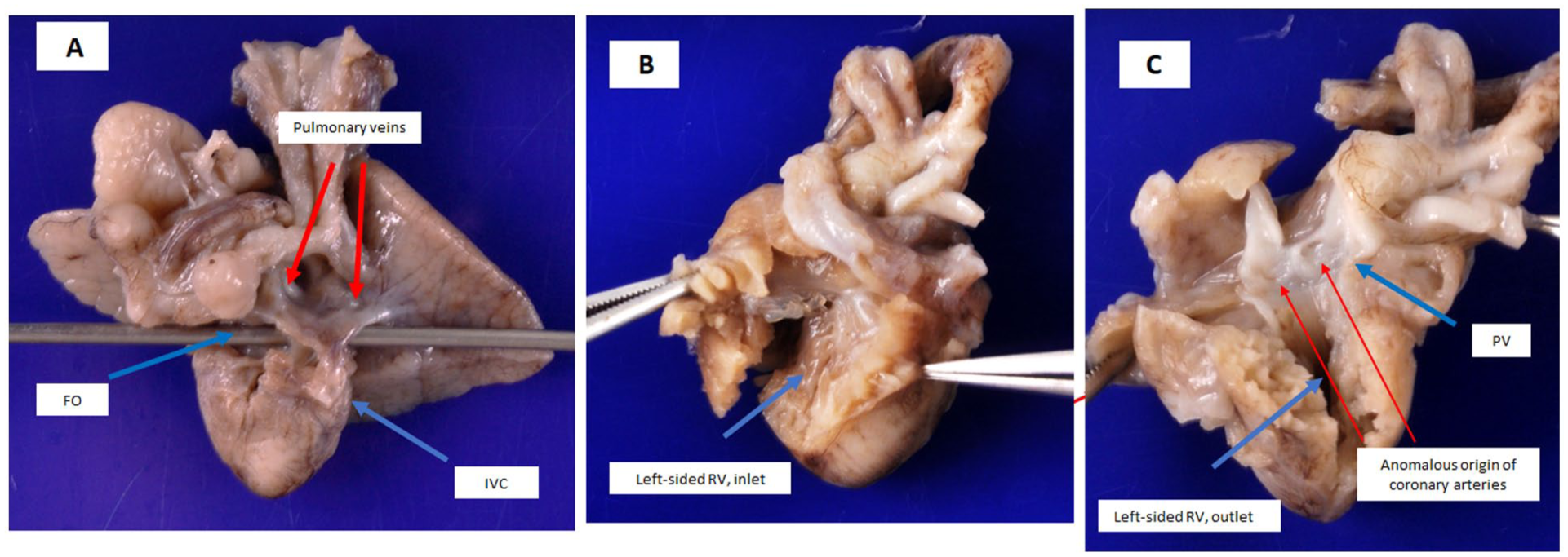
(A) The inferior vena cava (IVC) drains into a left-sided right atrium. A probe is positioned through the foramen ovale. Bilateral pulmonary veins drain separately into the left-sided right atrium without a vertical vein, consistent with total anomalous pulmonary venous connection (TAPVC). (B) Mirror-image arrangement of the heart, showing a left-sided right atrium and right ventricle, inlet with tricuspid valve. (C) Outlet portions of the right ventricle is visualized. There is anomalous origin of the coronary arteries from pulmonary artery (ACAPA), with identifiable coronary arterial orifices located within the pulmonary artery.
Cytogenetic analysis including RAD, microarray, and karyotyping yielded normal results. Villous tissue was submitted to an external laboratory (Blueprint Genetics) for whole exome family plus analysis, which is primarily focused on established disease genes that have been previously associated with genetic disorders. The test did not detect any known disease-causing or rare variants that could explain the patient’s phenotypes.
Discussion
The Gross classification system is the most widely used clinical framework to categorize EA/TEF into 5 morphologic subtypes. 2 In contrast, Kluth’s classification, proposed in 1976, provides a more comprehensive anatomical categorization, with 10 broad types and 96 subtypes based on detailed morphological features, including esophageal stenosis, atresia, agenesis, pouch configuration, gap distance, and bronchial-alimentary communications. 3 In this case, the anomaly is best classified as a Kluth type Xc2 variant, characterized by total tracheoesophageal fistula from the larynx to the carina without esophageal atresia. This subtype also involves a communicating lung component. A similar anomaly was described by Stolze in 1952, involving an accessory lung that communicated with the stomach. In our case, however, the bronchus of the sequestrated lung communicated with the lower esophagus instead of the stomach.
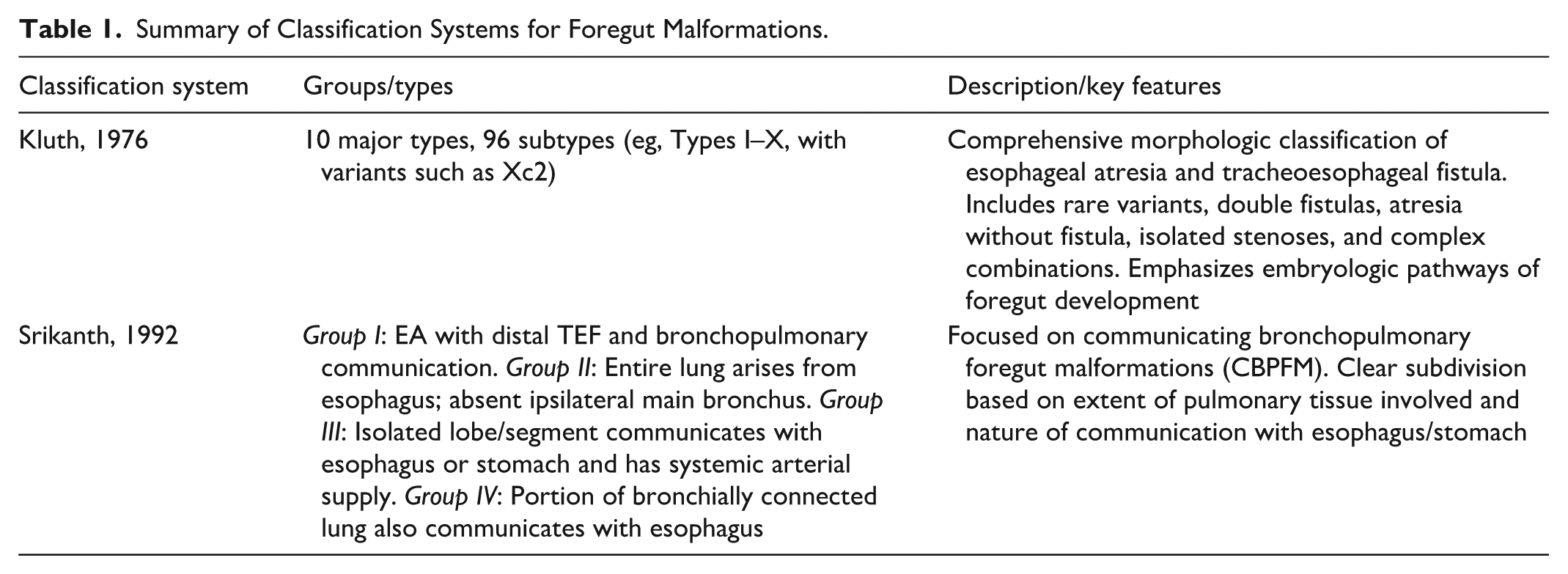
The presence of extralobar pulmonary sequestration with a bronchial connection to the gastrointestinal tract represents a rare congenital defect known as communicating bronchopulmonary foregut malformation (CBPFM), a term coined by Gerle et al. 4 Pulmonary sequestration refers to a non-functioning mass of lung tissue that lacks a normal connection to the tracheobronchial tree and receives systemic arterial supply. While the bronchus of most extralobar sequestrations terminate blindly at the parenchyma, a patent communication with the gastrointestinal tract, as seen in this case, is extremely uncommon. Srikanth et al. 1 proposed a classification system for CBPFM in 1992, dividing it into 4 groups (Table 1). Our case fits Group III, characterized by an isolated pulmonary segment with a bronchial connection to the lower esophagus and systemic arterial supply from the abdominal aorta.
Summary of Classification Systems for Foregut Malformations.
Total anomalous pulmonary venous connection (TAPVC) is an uncommon congenital cardiac defect, classified into 4 types: supracardiac, cardiac, infracardiac, and mixed. In the cardiac type, pulmonary venous drainage typically enters the coronary sinus. However, in this case, bilateral pulmonary veins drained each directly into the morphologic right atrium positioned on the left side (Figure 4A) without forming a common confluence or vertical vein which is a rare variant.
This case represents an exceptionally rare constellation of congenital anomalies, including Kluth type Xc2 tracheoesophageal fistula, Srikanth Group III communicating bronchopulmonary foregut malformation (CBPFM), extralobar pulmonary sequestration, mirror-image dextrocardia with situs inversus of the atria, L-looped ventricles, situs inversus of the great arteries, bilateral eparterial bronchi, anomalous coronary artery origin from the pulmonary artery, and total anomalous pulmonary venous connection with cardiac drainage. Each of these malformations is uncommon in isolation; to our knowledge, their concurrence has not previously been reported in the literature.
During embryologic development, both the trachea and esophagus arise from the primitive foregut. Between the 4th and 6th weeks of gestation, the tracheoesophageal septum forms, dividing the primitive foregut into the ventral laryngotracheal tube and the dorsal esophageal tract. Disruption in this process, particularly due to defective mesenchymal proliferation, can result in congenital anomalies such as esophageal atresia (EA) and tracheoesophageal fistula (TEF). On a developmental level, foregut separation relies on precise coordination of signaling pathways including FGF, Wnt, and BMP, which regulate interactions between mesenchyme and epithelium. The homeobox gene BARX1, expressed in the mesenchyme at the site of separation, plays a critical role by inhibiting Wnt signaling. Disruption in this regulation—such as BARX1 deletion—has been implicated in failed foregut separation, resulting in EA/TEF. 5
The association of laterality disturbances (eg, situs inversus, bronchial isomerism) with foregut partitioning defects (such as tracheoesophageal fistula) and complex cardiovascular anomalies (including TAPVC and ACAPA) suggests a disruption of embryologic left–right patterning pathways that simultaneously influence foregut and cardiopulmonary development. Such clustering of anomalies supports the concept that abnormal signaling during early embryogenesis can result in defects spanning multiple organ systems. In our case, whole-exome family-plus molecular genetic test did not identify any pathogenic or likely pathogenic variants in established disease-associated genes, underscoring the possibility of either novel genetic mechanisms or non-genetic developmental perturbations. Taken together, these considerations highlight that the concurrence of cardiac situs inversus and a bronchopulmonary foregut malformation likely reflects a shared disturbance of early embryonic signaling pathways, underscoring the exceptional rarity and clinical significance of this case. This case highlights the intricate interrelationship of early foregut, pulmonary, and cardiac development, and underscores the value of detailed anatomical classification in delineating rare fetal anomalies. By documenting this constellation of malformations, we expand the recognized phenotypic spectrum of combined laterality, foregut, and cardiovascular anomalies, and provide a reference point that may inform future genotype–phenotype correlation studies.
Footnotes
Acknowledgements
The authors would like to thank Charlotte Monroe for her assistance with formatting and submitting our article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.