
Abstract
Overall, neonatal cancer is uncommon. Because of its rarity and heterogeneity, diagnosis can be challenging. We report a unique case of a myoepithelial carcinoma in a 7 week old girl. Molecular diagnostic workup revealed a EWSR1-KLF15 gene fusion which was previously described in only six cases of myoepithelial tumors so far. All cases occurred in children and adolescents. To our knowledge, this is the first report of a congenital EWSR1-KLF15 fusion positive myoepithelial tumor in an infant.
Introduction
Cancer in newborns and infants is very rare and accounts for about 2% of all childhood cancers. Approximately one in 12 500 to 27 500 live births is affected. Compared to cancer in later childhood, neonatal cancer differs in incidence, histological subtypes, clinical behavior, anatomical site and therapy. 1 The most common congenital tumors are teratoma and neuroblastoma, though other tumor types such as soft tissue sarcomas, leukemia, kidney and brain tumors may also occur. Soft tissue tumors comprise 8–10% of neonatal tumors. Malignant soft tissue tumors in this age group are mostly rhabdomyosarcoma, infantile fibrosarcoma, rhabdoid tumors and others, some of them associated with characteristic genetic alterations. 2 Due to the rarity, the broad spectrum of subtypes and often limited availability of biopsy material, pathological diagnostics can be a challenge. 3 We report a female infant with an intramuscular myoepithelial carcinoma of the right shoulder in which an EWSR1-KLF15 gene fusion was detected.
Case Report
Clinical History
A 7-week-old, female baby presented with an indolent palpable mass ventral to the right shoulder, which had been growing rapidly since birth. Diagnostic imaging including sonography and MRI revealed an extramuscular tumor of 25 mm diameter, which was localized anterior of the coracoid processus and below the pectoralis major. The lesion was characterized by contrast enhancement and was not clearly demarcated. Core-needle biopsy showed a heterogeneous, partially necrotic tumor with a solid and trabecular growth pattern composed of epithelioid cells exhibiting nuclear atypia (Figure 1). Neuroblastoma, rhabdomyosarcoma and infantile fibrosarcoma could be ruled out by immunohistochemistry and FISH for ETV6 rearrangement. INI1 expression was retained. CD99 showed a heterogenous expression pattern. Due to foci of extracellular matrix (Figure 1(C)) and co-expression of cytokeratin (AE1/AE3), S100 and calponin (Figure 2), a myoepithelial tumor was suspected. FISH with an EWSR1 Breakapart probe showed an abnormal finding with loss of one 3′EWSR1 signal in >80% of cells (Figure 2). NGS based molecular workup (Archer FusionPlex Sarcoma Panel) revealed an EWSR1-KLF15 gene fusion (Figure 3). Necrotic foci, a high proliferation rate (50% in a mib1 staining) and nuclear atypia were worrisome for a malignant behavior. Therefore, the diagnosis of a myoepithelial carcinoma (malignant myoepithelial tumor) was established. Staging showed no evidence of metastases. Neoadjuvant chemotherapy was initiated according to the protocol proposed in the TREP project: ifosfamide, cisplatin, and etoposide (ICpE) for 4 cycles. 4 Following chemotherapy, the patient underwent tumor resection (Figure 4). Histology showed a completely viable tumor without response to therapy. Adjuvant chemotherapy was switched to 3 cycles of Ifosfamid, Vincristin and Etoposide. Nine months after chemotherapy a local relapse cranial of the scar occurred. The lesion was resected again. Histopathology confirmed relapse of a 8 mm tumor, which was completely resected with 6–22 mm safety margins. The karyotype confirmed the previous molecular findings of an EWSR1-KLF15 gene fusion (Figure 3). Surgery was followed by proton therapy with 59.4 Gy(RBE). The girl is currently disease-free two years after therapy.
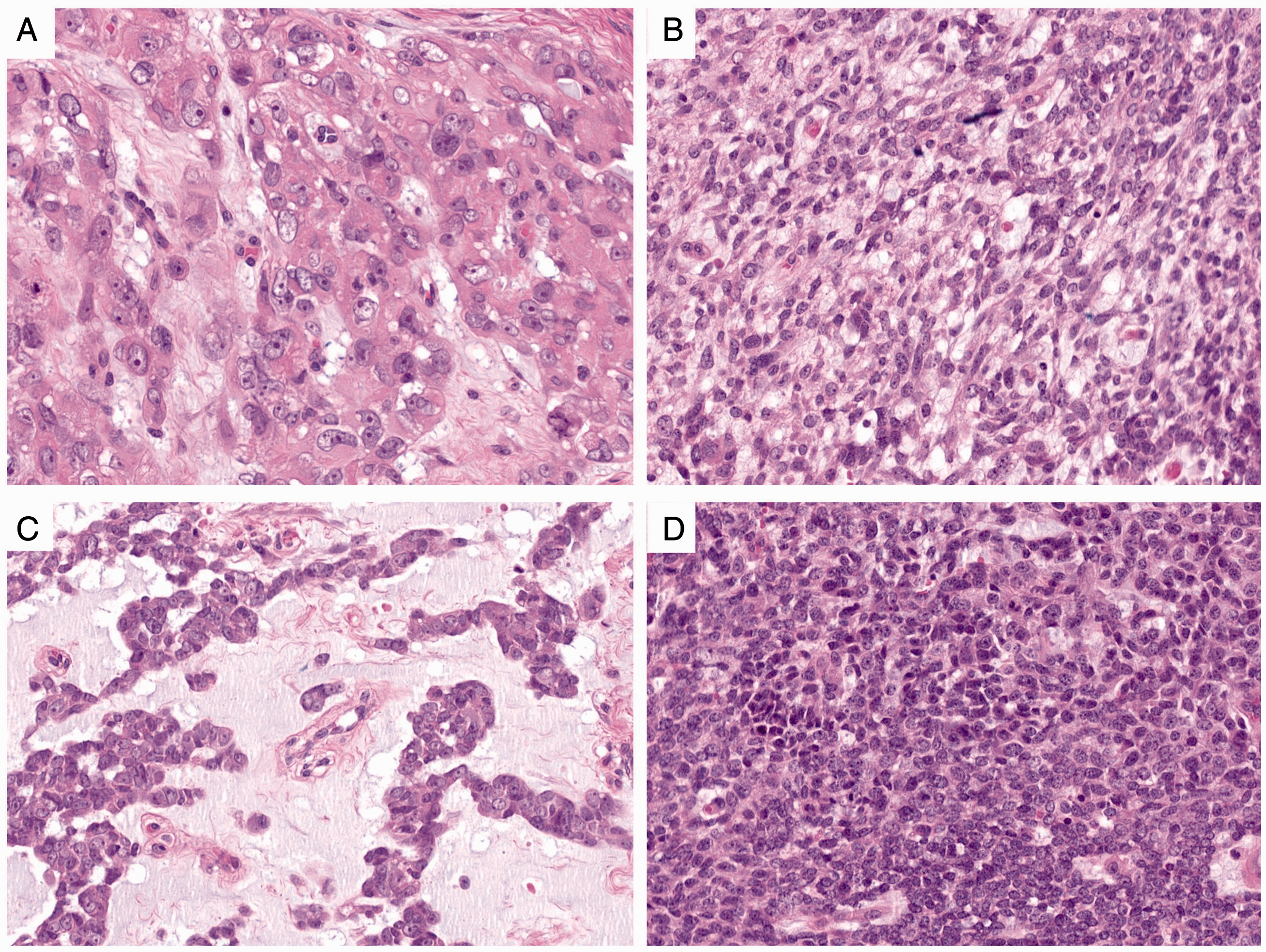
Histologic components of the MET. A, Epithelioid cells with nuclear atypia and discernible nucleoli. B, Ovoid to spindle cell morphology. C, Cords of epithelioid cells embedded in a myxoid stroma. D, Dense population of small blue round cells.
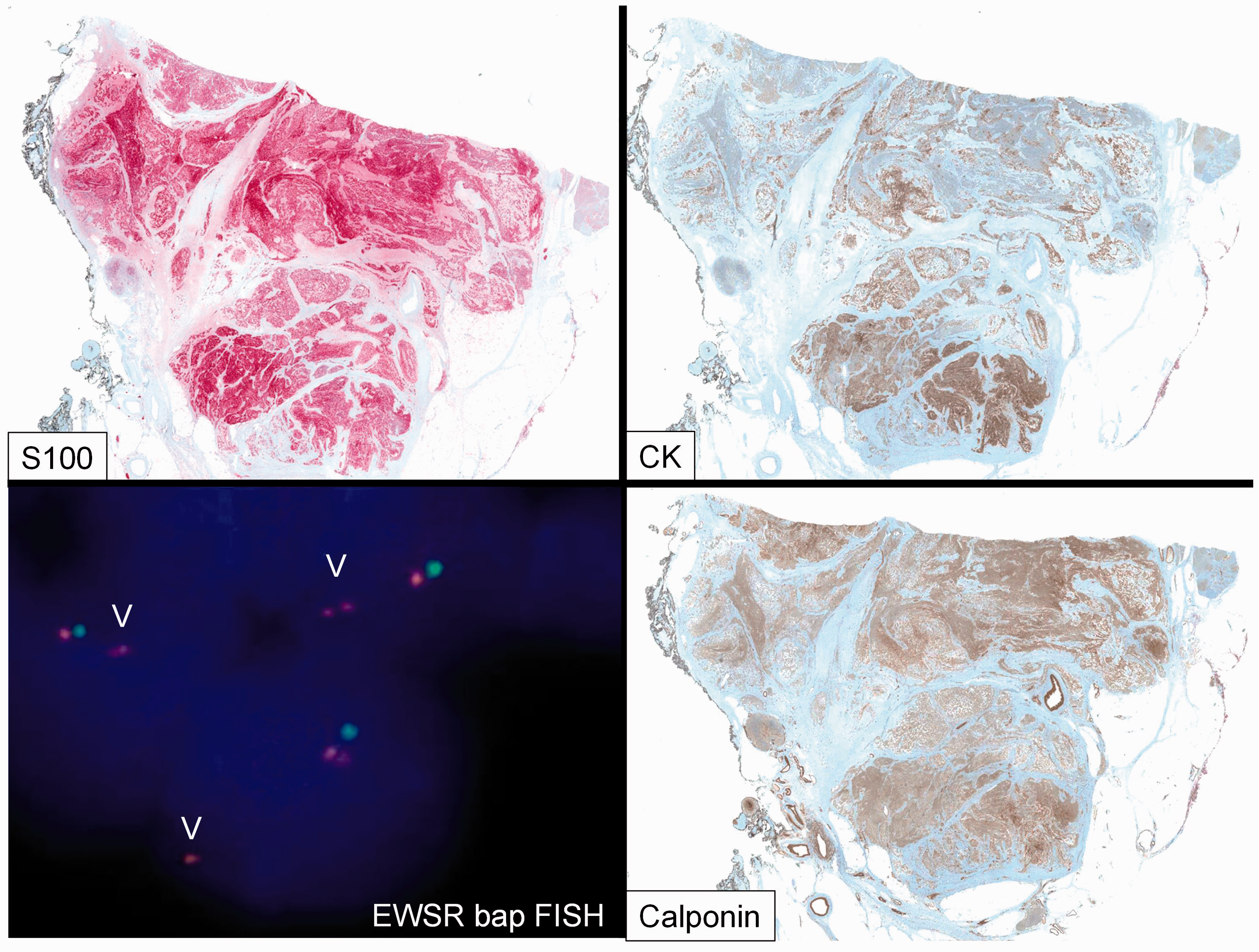
Immunophenotype: Strong S-100 expression. Heterogenous cytokeratin (AE1/AE3) expression. Homogenous Calponin expression. EWSR1 break apart FISH showing an abnormal finding with loss of one 3’EWSR1 signal.
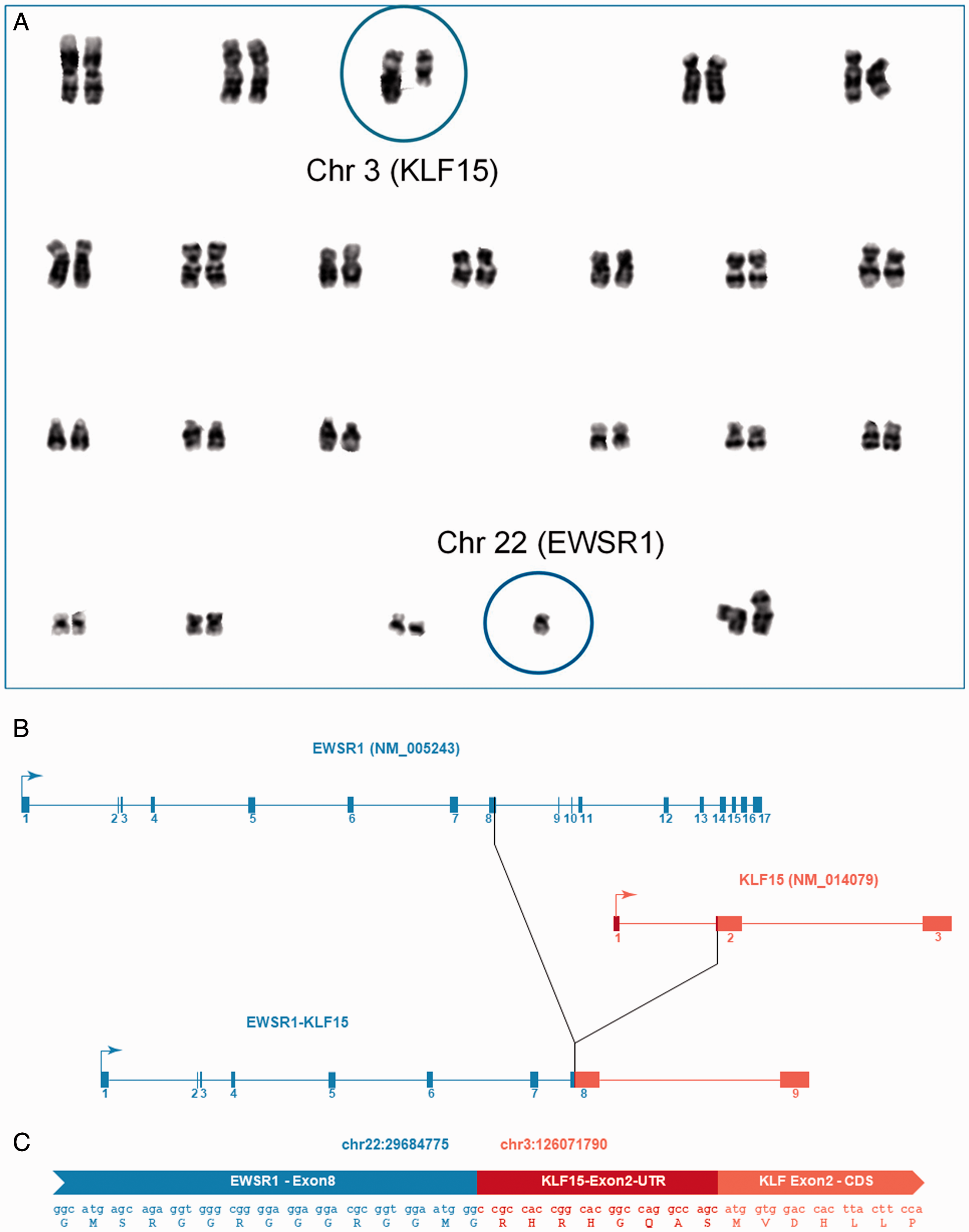
A, Karyogram representing the 10 metaphases analyzed that contained an unbalanced translocation between chromosome 3 and 22 leading to loss of 3q and partial loss of 22q. The karyotype was described as 45,XX,der(3)(3pter→3q11.2::22q11.1→22q11.1::22q12.2), −22 [10]. The circled chromosome 3 shows the normal copy on the left and the derivative chromosome on the right. The circled chromosome 22 shows the normal copy. B. Intron-exon structures of the EWSR1 and KLF15-transcripts as well as of the EWSR1-KLF15 fusion transcript described in the text. Boxes with numbers represent exons, lines represent introns and arrows indicate the direction of transcription. C, The sequence of a sample read containing the EWSR1-KLF15-fusion is displayed along with the in-silico translated amino acid sequence. The part of the read that originated from Exon 8 of EWSR1 is coloured blue. The part of the read that originated from Exon 2 of KLF15 is coloured red. The 5’-untranslated region (UTR) of KLF15 is coloured dark red, whereas the coding sequence (CDS) of KLF15 appears light red. The fusion is in-frame.
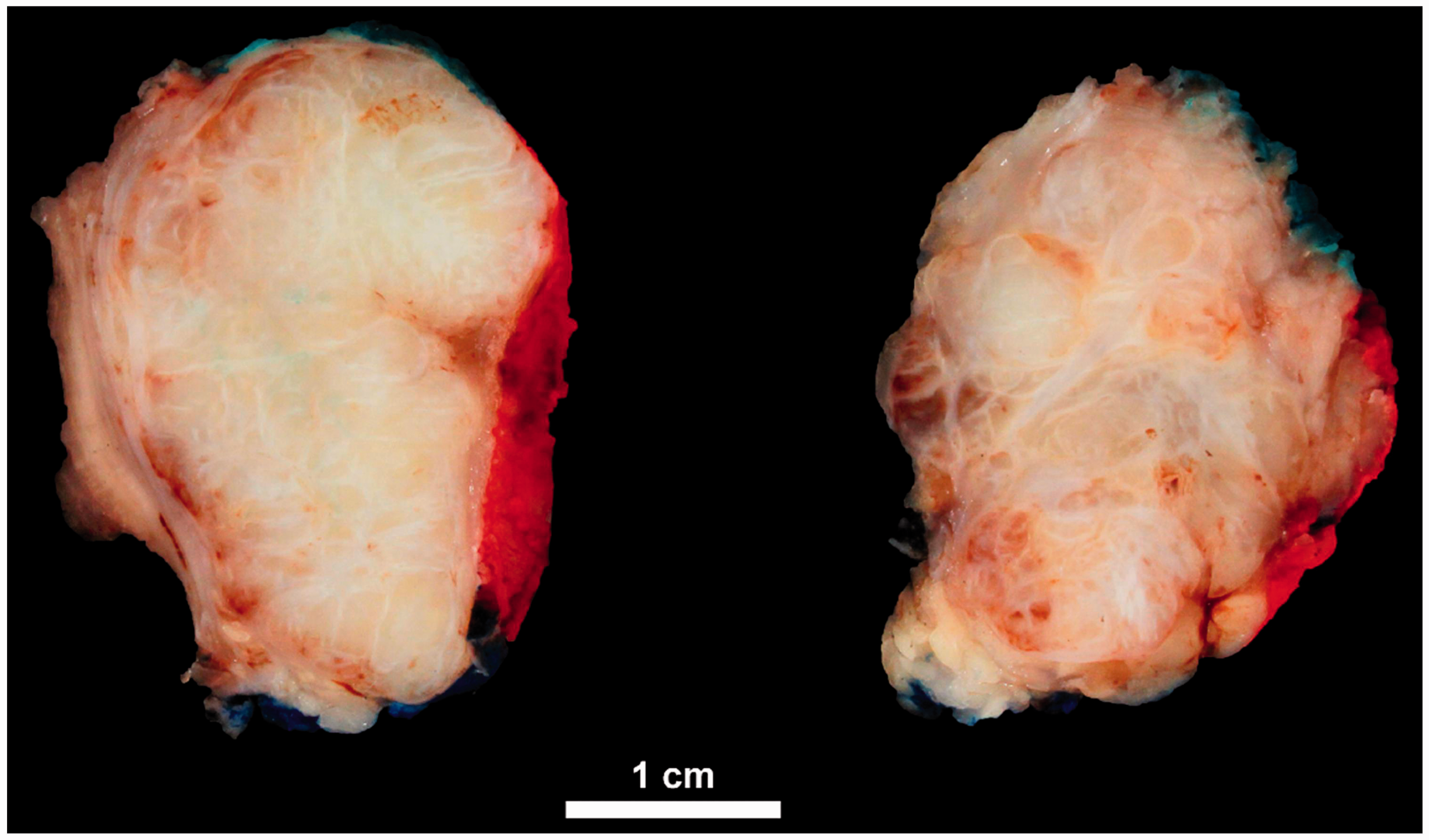
Macroscopic appearance of the resected tumor after chemotherapy: Infiltrative margins and a lobular growth pattern.
Discussion
Myoepithelial tumors (MET) are rare in neonates and infants. In the largest case series of 29 pediatric MET, only four occurred in children younger than one year. 5 Approximately 20% arise in the pediatric population, although the age range is very broad. They are equally distributed between females and males.6,7
MET, also known as myoepitheliomas or mixed tumors, were originally described in salivary glands, but they can also arise in soft tissues and less frequently in bone and visceral organs where the cell of origin to date remains unknown.6,8,9 Although most of them are benign, some MET show a malignant clinical course. They are also called myoepithelial carcinomas and affect children more often than adults. 5
MET are characterized by a broad range of cytological and architectural heterogeneity. Macroscopically, a multinodular or lobular appearance may be evident. Frequently, the tumor cells are epithelioid, but round or spindle cell morphologies have also been observed. They may be embedded in an extracellular matrix (myxoid, hyalinized or chondroid). However, they all have in common a distinctive, but not specific immunophenotype with co-expression of epithelial markers (cytokeratins, EMA), calponin and S100. 6,10 SOX10 might also be helpful in the immunohistochemical workup. 11 As mentioned before, some MET are malignant. Various histological criteria such as necrosis, mitotic rate or atypia were evaluated as indicators of an aggressive behavior. However, only the presence of nuclear atypia with discernible nucleoli has been reported as reliable criterion for malignancy so far. 6
In contrast to their counterparts in the salivary glands, MET of soft tissue share a different molecular phenotype. In almost half of the cases, EWSR1 rearrangements can be detected. A large number of fusion partners are described so far, e.g. POU5F1, PBX1, ZNF44, ATF1, PBX3, KLF15 and KLF17. 8,12 Alternative molecular aberrations are less frequent and include homozygous deletion of the SMARCB1 gene 13 , FUS12 and PLAG114 rearrangements.
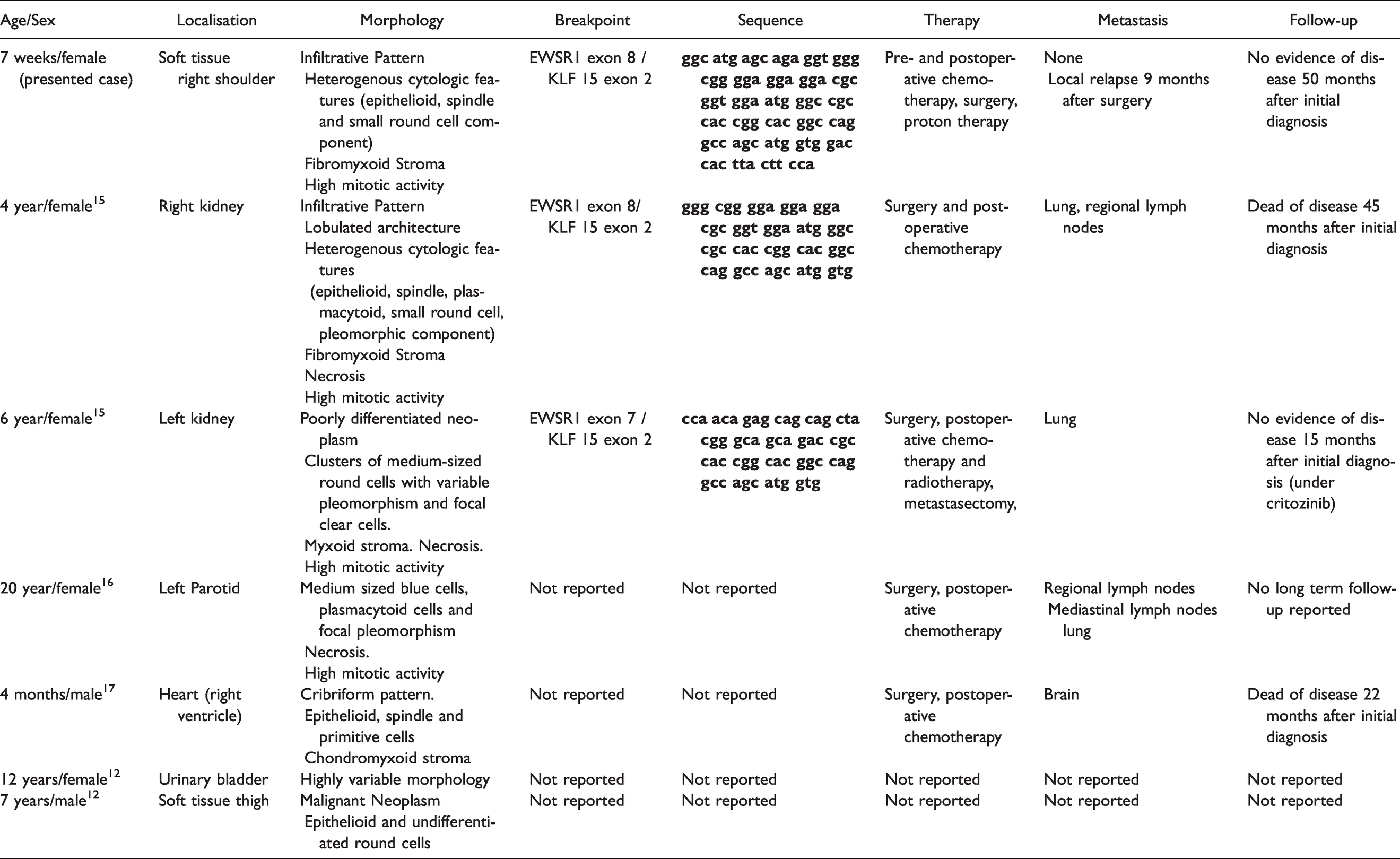
The EWSR1-KLF15 gene fusion is very rare in MET and was first described in two renal myoepithelial carcinomas. The patients were two girls, 4 and 6 years old. Both presented with a huge kidney mass and lung metastases. 15 Another EWSR1-KLF15 positive myoepithelial tumor was reported in the parotid of a 20-year-old female patient. At the time of diagnosis, pulmonary metastases were already present. 16 Recently, an intracardiac MET with EWSR1-KLF15 gene fusion in a 4-month-old male infant was published. After resection the patient had a local relapse and developed brain metastasis. 17 In the very latest case series, two more cases with EWSR1-KLF15 gene fusion were described: One case was a thigh mass in a 7-year-old male patient. The second developed in the urinary bladder of a 12 year-old female. 12 All cases in the literature have certain similarities with our case. First, they show a heterogeneous morphology with evidence of an undifferentiated round cell component. Second, all cases occurred in children and adolescents. Third, some of them share features of malignant behavior. Four of them manifested with metastases at the time of diagnosis. One showed an aggressive round cell morphology. Only the tumor in the urinary bladder was reported as benign. The information about the so far reported ESWR1-KLF15 fusion positive myoepithelial tumors are summarized in Table 1.
Summary of Reported EWSR1-KLF15 Fusion Positive Myoepithelial Tumors in Comparison to the Presented Case.
Since no robust criteria for assessing tumor behavior have yet been established, other parameters would be desirable in this respect. It would be interesting to know whether the different gene fusions could provide insight on the biologic behavior of tumor. Recently, Suurmeijer et al. evaluated possible associations between translocation subtypes and clinicopathologic and morphologic features in a cohort of 66 fusion positive MET. Indeed, they found some emerging genotype-phenotype correlations. In particular, the rare myoepithelial tumors with EWSR1-KLF15 gene fusions seem to be characterized by an undifferentiated (small blue) round cell component, which might explain a more aggressive clinical course. 12
Due to their broad heterogeneity and overlapping morphology to other soft tissue tumors, the diagnosis of MET can be a challenge. Depending on the dominant cell component, several differential diagnosis should be considered: For example, epithelioid cells are typical for epithelioid sarcoma, (extrarenal) rhabdoid tumor, epithelioid malignant peripheral nerve sheath tumor and sclerosing epithelioid fibrosarcoma. The latter shows additionally not only a fibrotic stroma, but also EWSR1 rearrangements, which are also characteristic for MET. However, SEF is strongly MUC4 positive and has a different fusion partner (CREB3L1 or CREB3L2). The first three mentioned tumors with epithelioid morphology are also characterized by loss of INI1 expression, which can also be found in a subset of MET, as mentioned above. In addition, they express S100 (epithelioid MPNST) and cytokeratin (epithelioid sarcoma), but none of them should show a co-expression of S100 and cytokeratin, In addition, both do not frequently occur in the pediatric population.
The undifferentiated round cell component opens the broad differential diagnosis of round cell sarcomas (Ewing sarcoma, Ewing-like sarcoma, desmoplastic round cell tumor, undifferentiated synovial sarcoma, etc), whereby the detailed discussion of each entity would go beyond the scope of this article. However, it should be stressed how important a molecular workup is, since many round-cell sarcomas show EWSR1 rearrangements. Hence, the fusion partners differ from those of MET. For completeness, extraskeletal myxoid chondrosarcoma (EMC) should also be mentioned. It is probably one of the closest mimics due to the strands of round to oval cells embedded in myxoid stroma. Furthermore, EMC may also show an EWSR1 rearrangement, but the typical fusion partner is NR4A3. However, only rare cases have been reported in children.
In conclusion, we report a rare MET of soft tissue with malignant features in a female infant. The molecular workup showed a rare EWSR1-KLF15 gene fusion, which supported the diagnosis. The girl is in complete remission after surgery and radiotherapy of the tumor relapse.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.