
Abstract
EWSR1::CREM gene fusions are increasingly being recognized in a diverse number of soft tissue tumors, including well-defined entities such as angiomatoid fibrous histiocytoma or clear cell sarcoma, and other unclassifiable tumors. As a group, EWSR1::CREM fused tumors often demonstrate primitive spindle or epithelioid cells, myxoid stroma, and a broad immunophenotype. Herein we present an unusual case of a child diagnosed with an intranasal malignant myxoid tumor harboring an EWSR1::CREM gene fusion. To the best of our knowledge, this is the first case of intranasal myxoid tumor with this particular fusion. Diagnosis and management of the case is discussed.
Introduction
Pediatric cancer remains one of the leading causes of death among children. 1 Global frequency of pediatric head and neck cancers ranges from 0.25 to 15% of all childhood cancers, 2 with nasal malignancies encompassing 0.7 to 3.3%3,4 of pediatric head and neck malignancies. The intranasal myxoid tumor detailed in this case report harbors a rare gene fusion. Currently there is no consensus regarding classification, prognostication, and evaluation of such cancers. The evaluation and management of this case could lend important guidance to future similar diagnoses.
Case Report
An 11-year-old boy with no significant prior medical history presented with left sided epistaxis, left nasal obstruction, and blood-stained mucus for 1 month. Family history was significant for a maternal grandfather diagnosed with lymphoma. The boy was referred to our pediatric otorhinolaryngology clinic for further assessment. Nasoendoscopic examination showed a smooth, fleshy intranasal mass attached to the inferolateral wall of the left nasal vestibule (Figure 1(A)). A contrast-enhanced computed tomographic (CT) scan of the paranasal sinuses showed an ovoid, minimally enhancing mass in the anterior left inferior meatus, abutting the premaxilla, and piriform aperture with no bony erosion (Figure 1(B), (C) and (D)). Endonasal biopsy and partial removal of the left intranasal mass was performed.
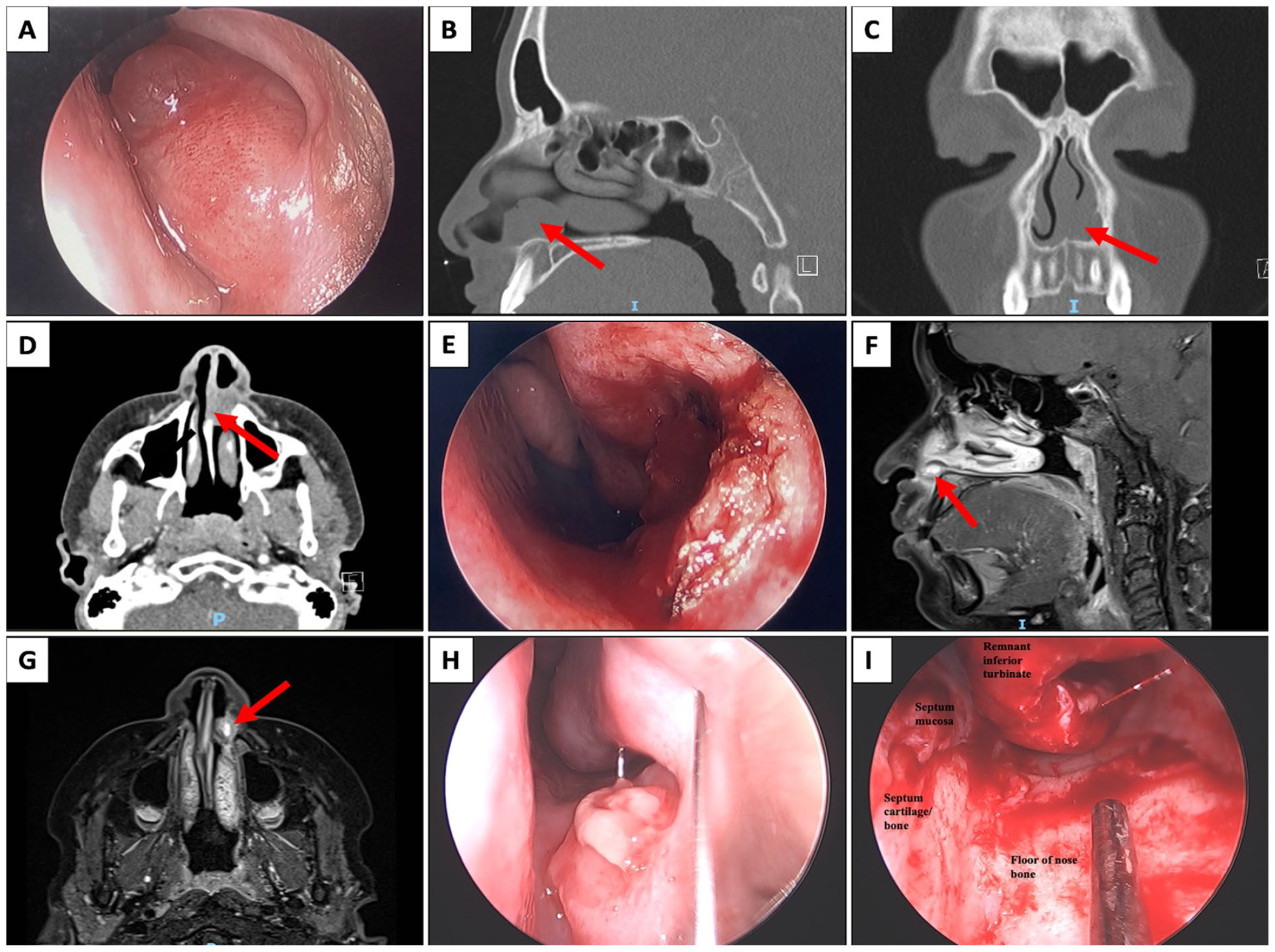
Initial appearance of left intranasal tumor on nasoendoscopic examination (A) and CT (red arrow) (B to D). Nasal cavity immediately post endonasal biopsy and partial removal of left intranasal tumor (E). MRI paranasal sinuses 3 weeks post endonasal biopsy and partial removal displayed recurrence with enhancing left nasal cavity nodule (red arrow) (F, G). Tumor regrowth noted on nasoendoscopic examination 7 weeks endonasal biopsy and partial removal (H). Left nasal cavity post subsequent endonasal excision (I).
On light microscopy, several tissue fragments of variably edematous nasal stroma (Figure 2(A)) showed a diffusely infiltrative proliferation of monotonous small round tumor cells with cellular nodules of solid sheet-like architecture (Figure 2(B)) separated by less cellular areas with abundant myxoid stroma and interspersed thin-walled blood vessels (Figure 2(C)). The tumor cells had hyperchromatic nuclei with pale amphophilic cytoplasm (Figure 2(D)), providing no morphologic clues toward the line of differentiation. The tumor cells did not form a cambium layer. Mitotic activity was brisk, numbering up to 14 per 10 high-power fields.
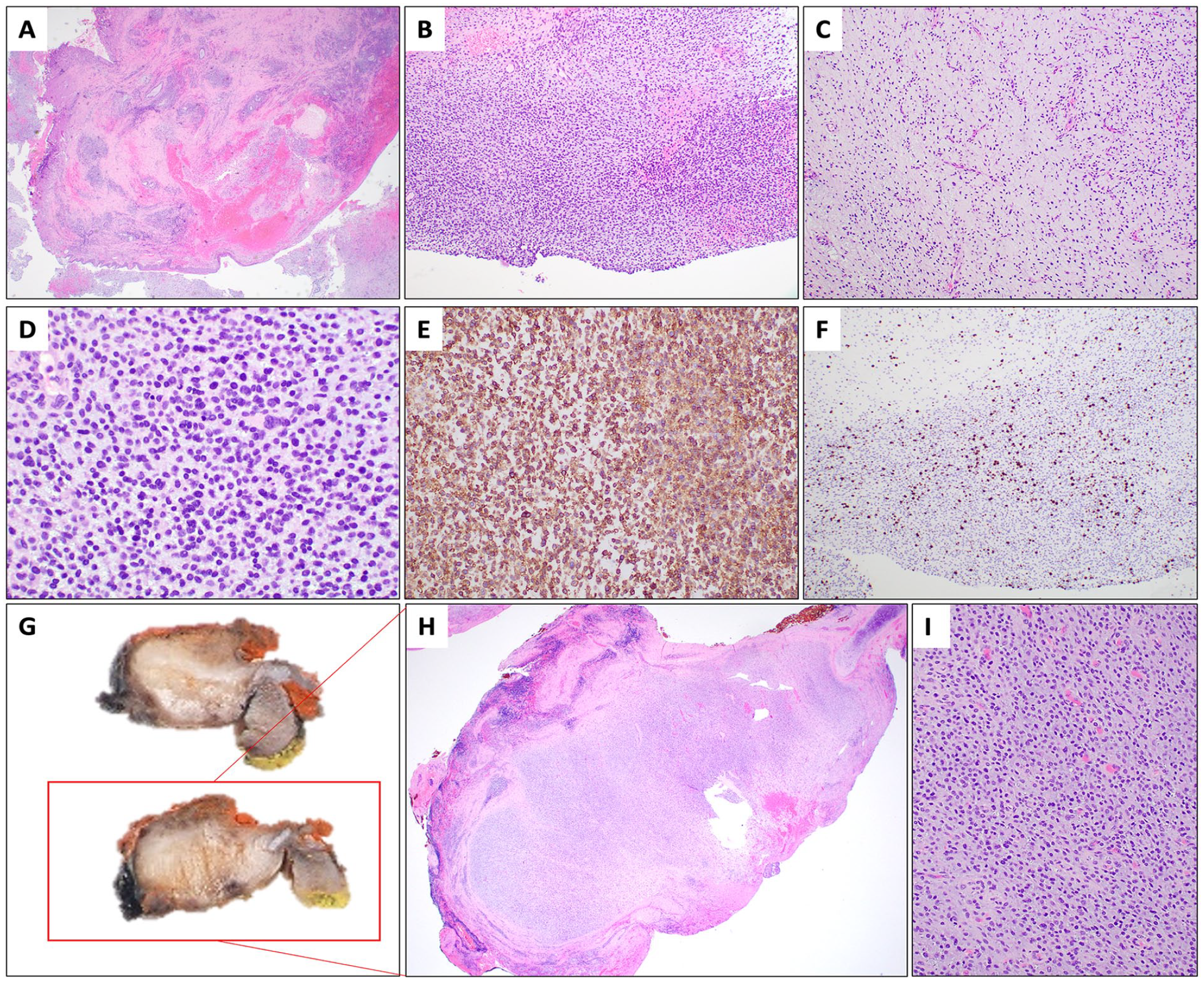
Histopathologic findings of the biopsy (A to F) and subsequent further resection (G to I). At low power examination, biopsy fragments showed stroma lined by non-dysplastic squamous and transitional mucosa. (A, hematoxylin and eosin-stained section, H&E, 20× magnification). There were cellular areas showing a proliferation of monotonous round tumor cells (B, H&E, 100× magnification) and myxoid areas showing a diffuse distribution of round cells with interspersed thin-walled blood vessels (C, H&E, 100× magnification). The tumor cells showed hyperchromatic nuclei with pale amphophilic cytoplasm (D, H&E, 400× magnification). There was membranous and cytoplasmic expression of CD99 (E, CD99 immunohistochemistry, 200× magnification). Ki67 proliferative index was 20 to 30% (F, Ki67 immunohistochemistry, 100× magnification). Macroscopic examination of the resection specimen showed a circumscribed, white to myxoid nodular tumor centered in submucosa (G). Correspondingly, light microscopy showed a circumscribed, vaguely lobular tumor surrounded by fibrous stroma with scattered lymphoid aggregates (H, H&E, 20× magnification; H is the low power light microscopic view of the section in the red box in G). The tumor on the excision specimen showed similar cytomorphologic features to the biopsy (I, H&E, 200× magnification).
Immunohistochemical stains were performed. The tumor cells were positive for CD99 in a variable membranous and cytoplasmic pattern, lacking the strong and diffuse membranous pattern typical of Ewing sarcoma (Figure 2(E)). Although rare scattered lesional cells showed nuclear androgen receptor expression, an absence of aberrant nuclear beta-catenin expression ruled against juvenile angiofibroma. There was patchy and variable nuclear positivity for WT1 (C-terminus). The tumor cells were negative for myogenic markers (desmin, myogenin, and MyoD1), epithelial markers (EMA, AE1/AE3), hematolymphoid markers (LCA, TDT, CD3, CD20, MPO, CD68, CD1a CD30), neuroendocrine markers (chromogranin and synaptophysin), and vascular markers (CD31, CD34). The tumor cells were also negative for p40, NUT, BCOR, PAX8, S100, MUC4, MUC6, and factor 13a. INI1 (SMARCB1) and BRG1 (SMARCA4) were retained. Ki67 proliferative index was 20 to 30% (Figure 2(F)).
Molecular studies were performed to evaluate for the possibility of a translocation-associated round cell tumor. Archer FusionPlex Pan-Solid Tumor V2 (129 genes) Next-Generation Sequencing assay detected a EWSR1 (exon 10)::CREM (exon 6) gene fusion with the following breakpoints: Chr22:29688158 and chr10:35477128, respectively (Figure 3). The number of fusion transcript reads detected was 1485, the percentage reads was 40.99%, and the number of unique start sites was 156. The diagnosis was that of a CD99-related malignant myxoid round cell tumor with EWSR1::CREM gene fusion.
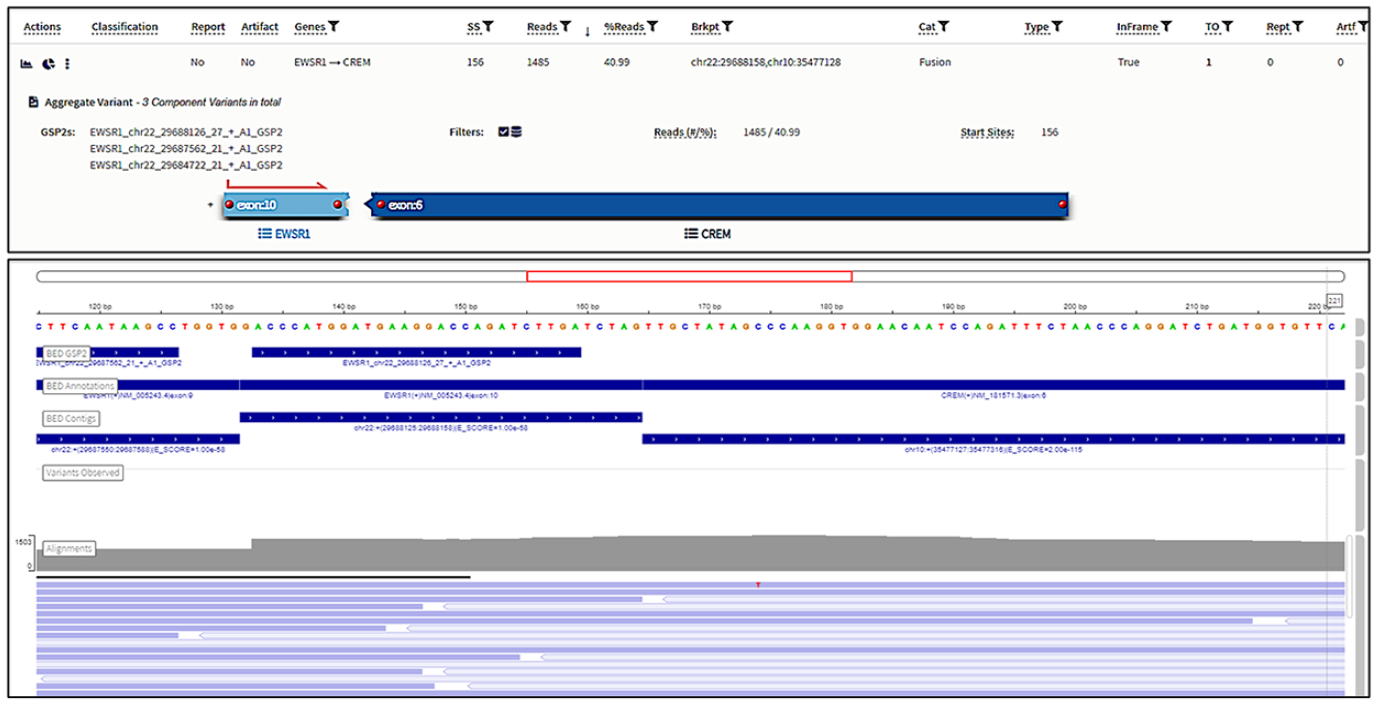
EWSR1 (exon 10)::CREM (exon 6) gene fusion detected on anchored multiplex PCR targeted sequencing assay (Archer® FusionPlex®).
Magnetic resonance imaging (MRI) of the paranasal sinuses, performed approximately three weeks after initial tumor removal, revealed residual tumor within the left nasal cavity, abutting the inferior turbinate and nasal septum. Scalloping of the maxillary bone was noted; although no definitive erosion through the cortex was noted (Figure 1(F) and (G)). Mildly enlarged left cervical lymph nodes measuring up to 10 × 21 mm were noted on MRI scan. The cervical lymph nodes showed minimal to mild F-fluorodeoxyglucose (FDG) uptake on whole body positron emission tomography and computed tomography (PET/CT) scan, otherwise no other FDG-avid distant metastases were noted. Fine needle aspiration cytology, and core biopsy of left cervical lymph nodes showed no malignancy.
The case was discussed in multiple multidisciplinary tumor boards from both pediatric and adult tertiary institutions. A decision was made to proceed with endonasal excision of the left intranasal tumor, with the aim of achieving surgical resection with clear margins. Intraoperatively, the tumor was noted to be attached to the inferior-lateral wall of the nasal vestibule, extending anteriorly to the mucocutaneous junction of the left nasal vestibule, posteriorly to just under the head of the inferior turbinate. Grossly the septum and incisive foramen did not appear to be involved. The tumor was dissected cleanly off the floor of the nasal cavity, with further dissection of the septal mucosa off the underlying septal cartilage, bone of the vestibule floor, lateral nasal wall, and inferior turbinate. Fourteen frozen sections were sent with the goal of obtaining clear margins. Initially frozen sections of mucosa from the head of inferior turbinate, left superior septal margin and left posterior nasal floor were positive for myxoid stroma. Further resection of these areas yielded benign mucosa.
The excision specimen was a circumscribed gelatinous white nodular tumor (0.8 cm maximal dimension), centered in the submucosa (Figure 2(G)). Light microscopy showed a circumscribed, vaguely lobular tumor surrounded by fibrous stroma with scattered lymphoid aggregates (Figure 2(H)). The excision specimen demonstrated morphologic features similar to those seen in the biopsy specimen (Figure 2(I)) and a similar pattern of CD99 expression. There was no necrosis or lymphovascular invasion. The deep resection margin was involved by tumor.
The case was further discussed during tumor board. Intra-operatively, the deep aspect of the tumor was mobile over the bone and elevated easily off the underlying bone with no apparent gross invasion. Hence no gross involvement of the deep margins was suspected, and the main concern was that results indicated microscopic involvement of the deep margins. However, further clearance of deep margins would require a significantly wider resection with inferior maxillectomy and possibly inferior septectomy, and give rise to significant morbidity and reconstructive issues. The tumor board opted for close post-operative surveillance, as the location of the tumor allowed for easy visualization of potential recurrence, and following discussion between the patient’s family and the radiation oncology team, surveillance was chosen over adjunctive radiation therapy. Post-operatively, the child experienced significant pain that has been slowly resolving over the past 12 months. Monthly nasoendoscopic surveillance has not shown any recurrent disease for the past 12 months post operatively. A 1-year post-operative MRI scan did not show any residual disease.
Discussion
EWSR1::CREM fusion gene has been discovered in a wide variety of tumors. To our knowledge, this is the first known case of intranasal EWSR1::CREM gene fusion tumor. Nasal cavity tumors are furthermore rare among the pediatric population. The most common pediatric malignant intranasal tumor is rhabdomyosarcoma. 5 Presentation can often be non-specific, with physicians misdiagnosing nasal symptoms as benign sinonasal disease. 6 As such, patients may present with advanced local destruction due to delayed diagnosis. 7
Even with a high clinical index of suspicion, many intranasal tumors cannot be differentiated by clinical or radiological features, 8 and require surgical biopsy to evaluate morphologic, immunohistochemical, and/or molecular features to attain diagnosis. 5
On the basis of morphology, this tumor may be categorized as a myxoid neoplasm with round cell features. In the pediatric population, this generates several differential diagnoses in the head and neck location, including primitive myxoid mesenchymal tumor of infancy, nasal chondromesenchymal hamartoma, and myxoid liposarcoma with round cell change. Primitive myxoid mesenchymal tumor of infancy is a locally aggressive tumor, preferentially affecting soft tissue of infants. Histologically, it is represented by primitive stromal cells set in a myxoid stroma with delicate blood vessels. In recent years, these tumors have been associated with BCOR internal tandem duplication. 9 Nasal chondromesenchymal hamartoma is a rare benign tumor of the sinonasal tract, usually presenting in infants. Histologically, it is represented by a proliferation of cartilaginous and mesenchymal elements, which may show myxoid appearance. This tumor may be the presenting tumor in DICER1 familial tumor susceptibility syndrome. 10 Myxoid liposarcoma typically presents in the deep soft tissue location, and is the most common liposarcoma of children and adolescents. It may show low or high grade histological appearance, the latter morphologically indistinguishable from other round cell sarcomas. Myxoid liposarcoma is characterized by DDIT3 rearrangements. 11
This is a diagnostically challenging and rare malignant neoplasm with immunohistochemical staining for CD99, harboring a EWSR1::CREM gene fusion. CREM is a member of the cyclic AMP response element-binding protein (CREB) family of transcription factors. The CREB family of transcription factors encompasses activating transcription factor-1 (ATF-1), CREB1, and CRE modulator (CREM). These genes have been implicated as fusion partners of EWSR1 in tumors of diverse lineages with different clinicopathologic characteristics. This includes angiomatoid fibrous histiocytoma, 12 clear cell sarcoma of soft tissue, 13 clear cell sarcoma-like tumor of the gastrointestinal tract, 14 primary pulmonary myxoid sarcoma, 15 and hyalinizing clear cell carcinoma of salivary gland. 16 In recent years, descriptions of other distinctive and rare tumors bearing these fusions have emerged, including myxoid mesenchymal tumor with predilection for intracranial location, 17 a subset of malignant epithelioid mesothelioma with retained BAP1 expression in young adults, 18 and malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. 19
Within this group, EWSR1::ATF1 and EWSR1::CREB1 gene fusions are more common, while EWSR1::CREM fusions are less characterized. To date, EWSR1::CREM gene fusions have been uncommonly described in several morphologically diverse neoplasms, including clear cell sarcoma of soft tissue,20,21 hyalinizing clear cell carcinoma, 22 myxoid angiomatoid fibrous histiocytoma, 21 and intracranial myxoid mesenchymal tumors (which some authors believe could represent myxoid angiomatoid fibrous histiocytoma in the intracranial location).17,23 The tumor in this case is morphologically distinct from clear cell sarcoma of soft tissue and hyalinizing clear cell carcinoma. While this tumor showed a fibrous periphery with lymphoid aggregates, features of (myxoid) angiomatoid fibrous histiocytoma, including deposition of amianthoid fibers, pseudoangiomatous spaces, expression of EMA, and expression of desmin were absent. In the same vein, this tumor also showed a degree of morphologic overlap with primary pulmonary myxoid sarcoma (PPMS) which typically harbors the EWSR1::CREB1 fusion; however this tumor did not demonstrate pronounced reticular morphology or EMA expression, which are characteristic of PPMS. 24
Recently, unclassifiable tumors harboring EWSR1::CREM gene fusions have been reported. These include an aggressive spindle cell tumor in the abdominal cavity of a child, and a MUC4-positive sclerosing epithelioid fibrosarcoma-like tumor in the chest wall of an adult. 21 The unusual morphology and location of the tumor of our current case further expands the spectrum of neoplasms associated with EWSR1::CREM gene fusions.
For management purposes, this tumor was broadly grouped with non-rhabdomyosarcoma soft tissue tumors (NRSTS). The biological potential of this rare tumor remains uncertain. If graded following the French Federation of Cancer Centers Sarcoma Group (FNCLCC) system, it may be considered grade 2. Current best evidence of management of pediatric NRSTS can be derived from a Children’s Oncology Group (COG) ARST 0332 study, 25 a prospective study which developed a risk stratification system for young NRSTS patients and assessed risk-adapted treatment outcomes. Of the 550 patients involved in this study, 22 patients had low-grade tumors with malignant cells evident microscopically at the resection margin. 4 out of those 22 patients (18%) had local events or progression. Given the low local recurrence rate and high rate of effective salvage for local recurrence, the study suggested that omission of radiotherapy should be considered in this subgroup of patients after maximal resection, with the caveat that local recurrence should not create substantial risks. As both radiotherapy and further surgical excision would create significant risks and morbidity for this patient, extensive discussion was needed among the multidisciplinary team and with the parents on further management. Ultimately, the decision was made for close surveillance, contributed by the location of the tumor being amenable to direct visualization for surveillance of recurrent disease.
Due to the rarity of this malignant tumor, multiple multidisciplinary discussions were needed throughout the course of evaluation and management of this patient. Continued surveillance of this patient and analysis of tumors with similar gene fusions are needed to further characterize such tumors including their metastatic potential and best management, before general guidelines can be established.
In a pediatric oncology patient, heavier weight is placed on any treatment decision due to the potential long-term complications and morbidity. Consideration of the reconstructive challenges and effects on craniofacial growth is needed when deciding to proceed with surgical resection. 26 Chemoradiotherapy also confers significant additional risk of developing toxicity and secondary malignancies. 27
Conclusion
This report details an extremely rare nasal myxoid tumor harboring an EWSR1:: CREM gene fusion. Comprehensive genomic sequencing allows for improved understanding of pathogenesis, prognosis and treatment response for such malignancies. Given the rarity of these tumors, discussion in a panel of multidisciplinary experts is encouraged to ensure all treatment modalities and options are considered. The true prevalence and behavior of nasal myxoid round cell tumors with EWSR1::CREM gene fusion, and their relation to tumors harboring fusions between EWSR1 and the CREB family of transcription factors requires further investigation.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.