
Abstract
Exposure to diesel exhaust air pollutants is a key environmental threat for pulmonary and cardiovascular diseases. Oxidative stress and Transient Receptor Potential Vanilloid-1 (TRPV1) receptors exhibit pivotal contributions in mediating lung injury induced by environmental pollutants. This study investigates the histological changes in lung tissue induced by short-term particulate-free filtered diesel exhaust (FDE) exposure, with a focus on TRPV1 receptor expression and oxidative stress pathways. Male rats were allocated to four groups randomly: Non-exposed (NE), clean air exposed (CAE), FDE-exposed, and NAC pre-treated FDE-exposed groups. FDE exposure lasted 5 hours per day for five days, with histological examination and TRPV1 expression analysis conducted on day six. The NAC pre-treated group received NAC (200 mg/kg) for five days prior to each exposure. Lung tissue samples were analyzed using hematoxylin and eosin staining, and immunofluorescence for TRPV1 expression. FDE exposure caused significant histological alterations, including alveolar septal thickening, interstitial inflammation, capillary congestion, perivascular inflammation, bronchial epithelial necrosis, endothelial discontinuity, and interstitial fibrosis, as classified by a semi-quantitative INHAND (International Harmonization of Nomenclature and Diagnostic) scoring criteria for rats, visualized on a heat map. TRPV1 expression was upregulated in FDE-exposed lung tissues, particularly around congested vessels and thickened septa. NAC pre-treatment significantly reduced both histological damage and TRPV1 expression. Our study highlights a potential mechanistic relationship between TRPV1 receptor expression and oxidative stress pathways in the lung damage induced by FDE exposure, underscoring the therapeutic potential of NAC in countering these effects.
Introduction
Proximity to heavy traffic, especially in urban and industrialized areas, has been linked to detrimental effects on respiratory and cardiovascular health in humans. 1 Exposure to pollutants such as nitrogen oxides, sulfur oxides, carbon compounds, and particulate matter is linked with an increased occurrence of both non-infectious and infectious diseases, affecting the airways and lungs. 2 Diesel exhaust, in particular, contains high levels of ultrafine particles and gaseous pollutants, but the exact mechanisms through which these pollutants contribute to the development of pulmonary conditions remain poorly understood. Inflammation is widely recognized as a dominant factor in the causation of respiratory diseases related to traffic-related pollution, and it may also play a role in facilitating the transfer of harmful substances from the lungs into the bloodstream, resulting in systemic effects affecting multiple organs. 3 Furthermore, prolonged exposure to such pollutants can lead to sustained inflammation, which, in turn, may increase the risk of chronic respiratory conditions and serve as a potential cardiovascular risk factor. 3 Inflammatory mediators like histamine 4 and bradykinin 5 released in the vicinity, similar to exogenous nociceptive agents like capsaicin, 6 modulate the cardiorespiratory parameters in a reflex fashion, 7 when present in the pulmonary and systemic blood vessels.6,8
Studies examining prolonged exposure to particulate matter from diesel exhaust have reported significant histological changes in lung tissue, including diffuse alveolar damage, vascular congestion, interstitial thickening, fibrosis, and inflammatory infiltration.9,10 However, research on the manifestations of sub-acute exposure to the gaseous components of diesel exhaust remains limited. A growing hypothesis as regards to the harmful cardiorespiratory outcomes of diesel engine exhaust components suggests that triggering of Transient Receptor Potential Vanilloid-1 (TRPV1) ion channels may occupy a pivotal role. 11 Specifically, stimulation of TRPV1 receptors in pulmonary C-fibers can induce neurogenic inflammation and modulate the expression of pro-inflammatory cytokines. 12 While TRPV1 activation in response to diesel exhaust particles has been previously reported,13,14 its expression profile following short-term gaseous exposure without particulate matter remains underexplored. In our recent work, we demonstrated that the gaseous fraction of diesel exhaust affects TRPV1-mediated nociceptive cardiopulmonary reflexes in rats, potentially through mechanisms involving oxidative stress. 6 Building on these findings, the current study aims to explore the histological alterations in lung tissue induced by repeated exposure to gaseous pollutants of diesel engine exhaust, with a focus on the role of TRPV1 receptors and oxidative stress in mediating these changes. While diesel exhaust is not the only component of vehicular emissions, it remains a significant contributor to urban air pollution, especially in developing countries like India, where diesel engines are extensively used for transportation and industrial purposes. This study highlights the potential health effects of gaseous pollutants in diesel engine exhaust, which are relevant to understanding urban air quality’s respiratory impact. Furthermore, the diesel exhaust generation and filtration process used in this study mimics real-world scenarios in highly polluted urban environments with advanced particulate filtration systems, making the study findings applicable to public health scenarios. Unlike prior studies involving whole diesel exhaust or diesel exhaust particles, our study focuses exclusively on the particulate-matter free, gaseous fraction of diesel exhaust, which better represents filtered emissions in urban traffic zones. In addition, we specifically investigated short-term exposure to this gaseous phase of pollutants, in a controlled setup.
Materials and Methods
This study received approval from the Institutional Animal Ethics Committee at the All India Institute of Medical Sciences in New Delhi, India (Letter No. 391/IAEC-1/2022-17.03.2023). All animal procedures obeyed to the guiding principle laid down by the US National Institute of Health’s “Guide for the Care and Use of Laboratory Animals” and reported in line with ARRIVE guidelines. A schematic overview of experimental design is presented in Figure 1. Diagrammatic illustration of the experimental protocol. Rats were divided into four groups of six rats each. The first group remained non-exposed (NE) to diesel exhaust, housed in their home cages for five days (D1-D5), while the second group was exposed to clean air (CAE) for 5 hours each day over five days, as shown by the pink shaded bars. The third group was exposed to filtered diesel exhaust (FDE) for the same duration and days, indicated by the brown shaded bars. The fourth group of rats underwent the same exposure protocol as the FDE group, but in addition received five doses of N-acetyl cysteine (NAC), 30 minutes prior to each daily exposure. All the rats were taken for dissection on day 6.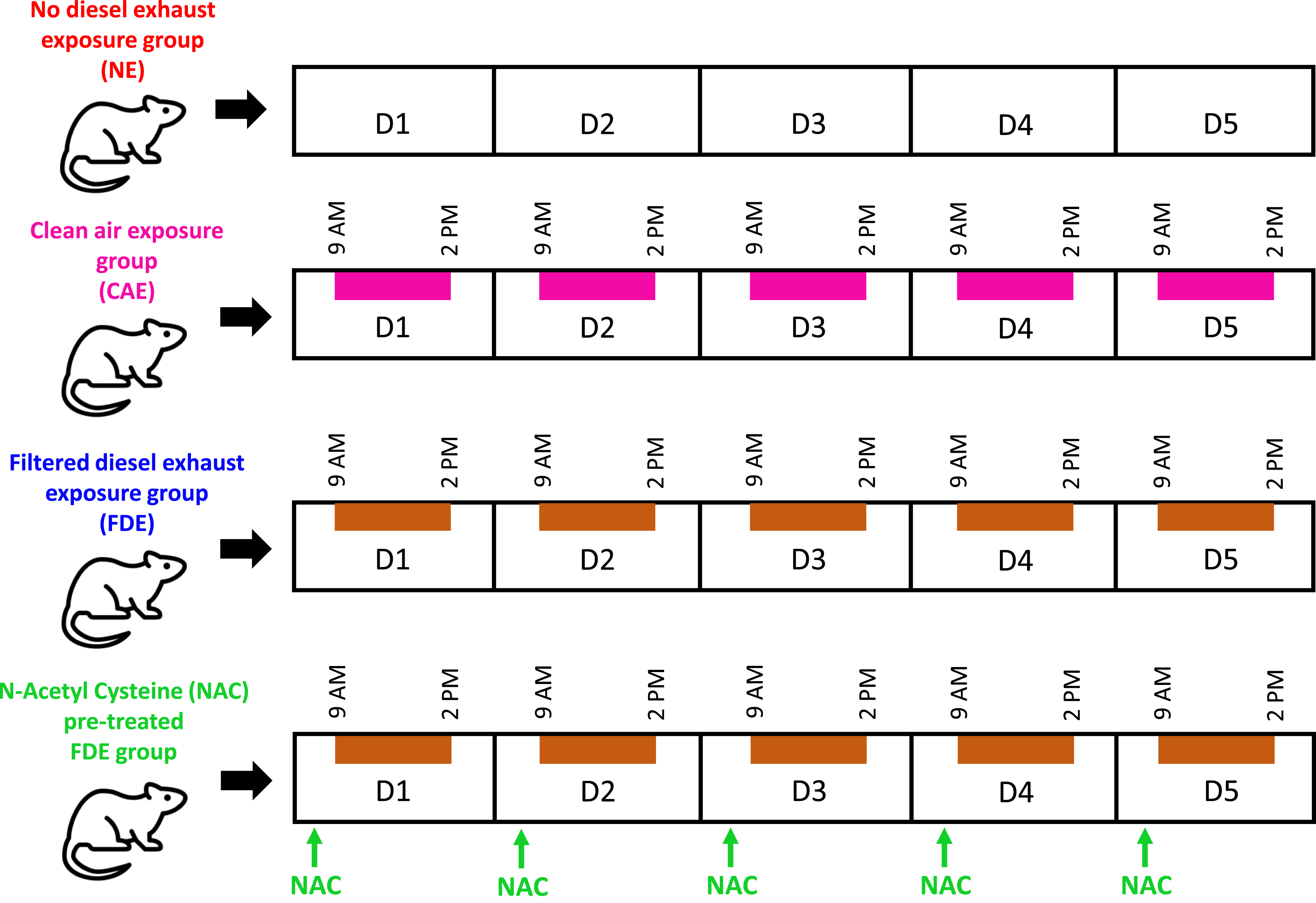
Experimental Groups and Animal Housing
Healthy adult Wistar male rats (210 ± 35 g weight) were acclimated under a light/dark cycle of 12 hours in individual cages with unrestricted access to food and water. After a 3-day acclimatization period in the departmental animal facility, the rats were allotted into four groups randomly: the Non-exposed (NE) group, the clean air exposed (CAE) group, the Filtered Diesel Exhaust-exposed (FDE) group, and the N-acetyl cysteine (NAC) pre-treated FDE group. The FDE group (n = 6) was exposed to filtered diesel exhaust for 5 hours each day over a span of five days (D1-D5), and on day six (D6), these rats were taken for dissection. In a similar fashion, the CAE group (n = 6) was exposed to clean air in the exposure chamber. The NE group (n = 6) was placed in their usual housing cages devoid of diesel exhaust exposure during the 5-day period, after which they underwent the same experimental procedures on D6. Similarly, the NAC pre-treated FDE group (n = 6) underwent the similar exposure paradigm as the FDE rats but additionally was administered five doses of NAC (200 mg/kg, intraperitoneal) from D1 to D5, 30 minutes prior to each exposure on those days (Figure 1). Further, in a separate group of preliminary experiments involving NE rats (n = 6), NAC treatment produced no effects on pulmonary histology.
Filtered Diesel Exhaust Exposure Setup
Exposure to FDE was conducted using a custom-designed whole-body exposure chamber.6,15 Diesel exhaust was generated by a 12.5-liter diesel engine operating at 3000 r/min under no-load conditions. The exhaust was diluted with ambient air at a 1:10 ratio using a blower and passed through an HEPA (H 14) filter to remove particulate matter, ensuring exposure to only gaseous pollutants. The chamber comprised six compartments, each housing one rat with free access to food and water. Conditions were maintained at 24 ± 2°C with 15 air exchanges per hour, and humidity and temperature were monitored throughout the exposure. Real-time monitoring of pollutant concentrations was performed using calibrated sensors placed within the exposure chamber, with measured levels of carbon dioxide (0.27 ± 0.07%), carbon monoxide (0.015 ± 0.008%), nitrogen oxides (6.4 ± 1.5 ppm), sulfur dioxide (0.35 ± 0.005 ppm), and volatile organic compounds (0.193 ± 0.031 ppm) comparable to those during heavy urban traffic in New Delhi (Central Pollution Control Board, Government of India). For the CAE group of rats, only the clean air was circulated within the exposure chamber, while the diesel engine remained turned off.
Dissection and Lung Tissue Processing
On the day of the experiment (D6), anesthesia of rats was performed by injecting freshly prepared urethane solution (0.5 g/mL) intraperitoneally, at a dosage of 1.5 g/kg. Once sufficient anesthesia was achieved, the thoracic cavity was accessed through parasternal incisions made bilaterally upward from the xiphisternum, and the sternum was retracted to expose the heart and lungs. Both lungs were carefully detached en bloc from their attachments and removed from the thoracic cavity. The lungs were then fixed at total lung capacity by instilling cold 4% paraformaldehyde through a tracheal cannula under controlled pressure. The trachea was then secured with a suture, and the lungs were submerged in the same fixative for 6 hours. 16 The lungs were then sectioned into different lobes, and 2-3 cm tissue samples from each lobe were placed into labeled glass bottles containing cold 4% paraformaldehyde, for fixation at 4°C, overnight. The next day, the fixed tissues were thoroughly rinsed with tap water and underwent gradual dehydration through serial ethanol dilutions (50%, 70%, 90%, and 100%). Subsequently, the samples were immersed in cedar wood oil for 10-15 days to ensure complete clearing, after which they were embedded in paraffin blocks, following standard protocols. Using a microtome, the blocks were then sliced into 5 μm sections. For each lung lobe, ten slides were prepared, with five slides designated for histological assessment and five for immunofluorescence analysis.
Lung Histology and Immunofluorescence
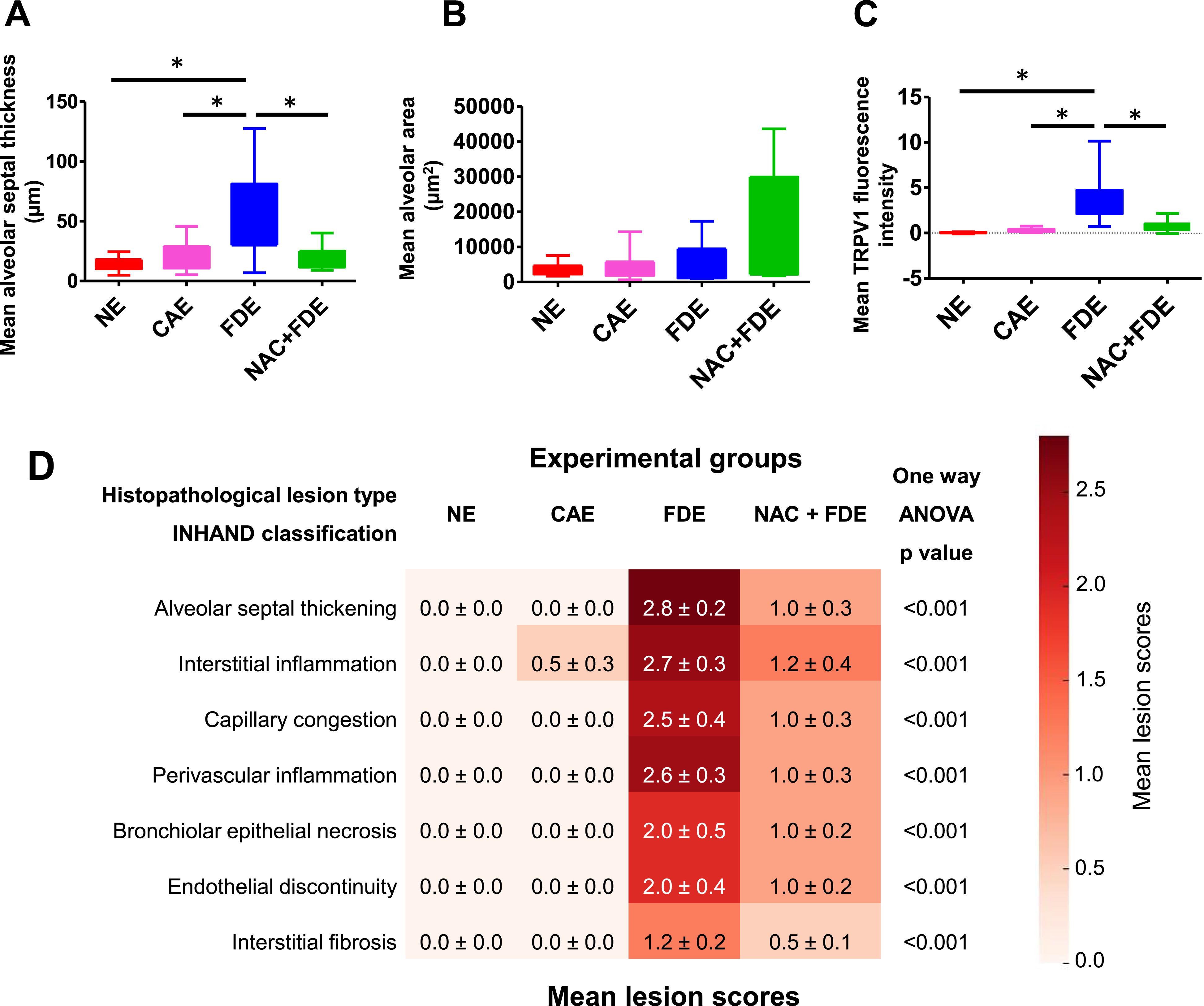
Slides intended for histological analysis were rehydrated through a series of ethanol dilutions and subsequently stained with hematoxylin and eosin, following standard protocols. After staining, the sections were dehydrated, mounted with DPX, and examined using a light microscope. The entire slide was scanned at low magnification (×10), and for analysis approximately 10-12 photomicrographs were captured at ×20 magnification to ensure comprehensive coverage of the slide. Semi-quantitative scoring of lung lesions (alveolar septal thickening, interstitial inflammation, capillary congestion, perivascular inflammation, bronchial epithelial necrosis, endothelial discontinuity, and interstitial fibrosis) was performed using INHAND (International Harmonization of Nomenclature and Diagnostic) criteria for the rat respiratory system, by the Society of Toxicological Pathology. 17 Mean ± SEM scores were compiled and represented as a heat map for intergroup comparison.
For immunofluorescence, following rehydration, blocking, and antigen retrieval as per the manufacturer’s instruction manual, the slides were placed in a humidified chamber and washed three times with PBS and PBS-Triton X-100. They were then incubated for 10-12 hours at room temperature with a rabbit anti-rat TRPV1 primary antibody (1:200) overnight. The rabbit anti-rat TRPV1 primary antibody (0.8 mg/mL) was acquired from Alomone Labs (Catalog No: ACC-030), Thermo Fisher Scientific, USA, with further dilutions prepared in bovine serum albumin (1%) in PBS and stored at −20°C. After further washing with PBS and PBS-Triton X-100, the slides were incubated in the dark for 4°hours at room temperature, with a goat anti-rabbit fluorescent secondary antibody (1:100). The goat anti-rabbit Alexa Fluor 568 fluorescent secondary antibody (2 mg/mL) was purchased from Invitrogen (Catalog No: A-11011), Thermo Fisher Scientific, USA, and subsequent dilutions were also made in bovine serum albumin (1%) in PBS and stored at 4°C, shielded from light.
The slides were subsequently fixed using a mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) and visualized using fluorescence microscopy, the next day. A total of 10-12 photomicrographs were captured at ×20 magnification, covering the entire slide area, utilizing blue and red light filters for DAPI and TRPV1 visualization, respectively. Preliminary experiments with different dilutions of primary and secondary antibodies revealed that optimum immunostaining was observed with 1:200 and 1:100 dilutions of primary and secondary antibodies, respectively. Therefore, these dilutions of antibodies were used for subsequent immunostaining of slides.
Statistical Analysis
The alveolar septal thickness, alveolar cross-sectional area, and TRPV1 fluorescence intensity were measured using ImageJ software and expressed as mean ± SEM values. The statistical significance was evaluated by comparison of the above parameters in the FDE group with the NE and CAE groups. Further, the effect of NAC pre-treatment on FDE-induced pulmonary changes was analyzed by comparison of the above parameters in the NAC pre-treated FDE group with the FDE group. The comparisons among different groups were made by ANOVA, followed by post hoc correction using Dunnett’s test, using GraphPad Prism software. P value of <0.05 was considered significant.
Results
The present study investigates the effects of FDE exposure on lung histology and TRPV1 expression in NE, CAE, and NAC pre-treated FDE-exposed groups of rats. Histological and immunofluorescence analyses were performed to assess structural and molecular changes in lung tissue. A heat map was generated based on the INHAND-compliant lesion scoring, highlighting histopathological changes across all groups. FDE exposure led to significant pulmonary histological alterations, including alveolar septal thickening, interstitial inflammation, capillary congestion, perivascular inflammation, bronchial epithelial necrosis, endothelial discontinuity, and interstitial fibrosis, as compared to the NE and CAE groups. The FDE-exposed group exhibited a marked increase in alveolar septal thickness and widespread infiltration of neutrophils and lymphocytes, with occasional interstitial fibrosis and endothelial discontinuity in pulmonary capillaries. Additionally, FDE exposure significantly elevated TRPV1 expression, which was predominantly localized near congested pulmonary vessels and inflamed alveolar septa. Notably, NAC pre-treatment effectively mitigated these pathological changes, reducing alveolar septal thickening and inflammatory infiltration while attenuating the increased TRPV1 expression observed in the FDE-exposed group.
FDE Exposure-induced Changes in Lung Histology
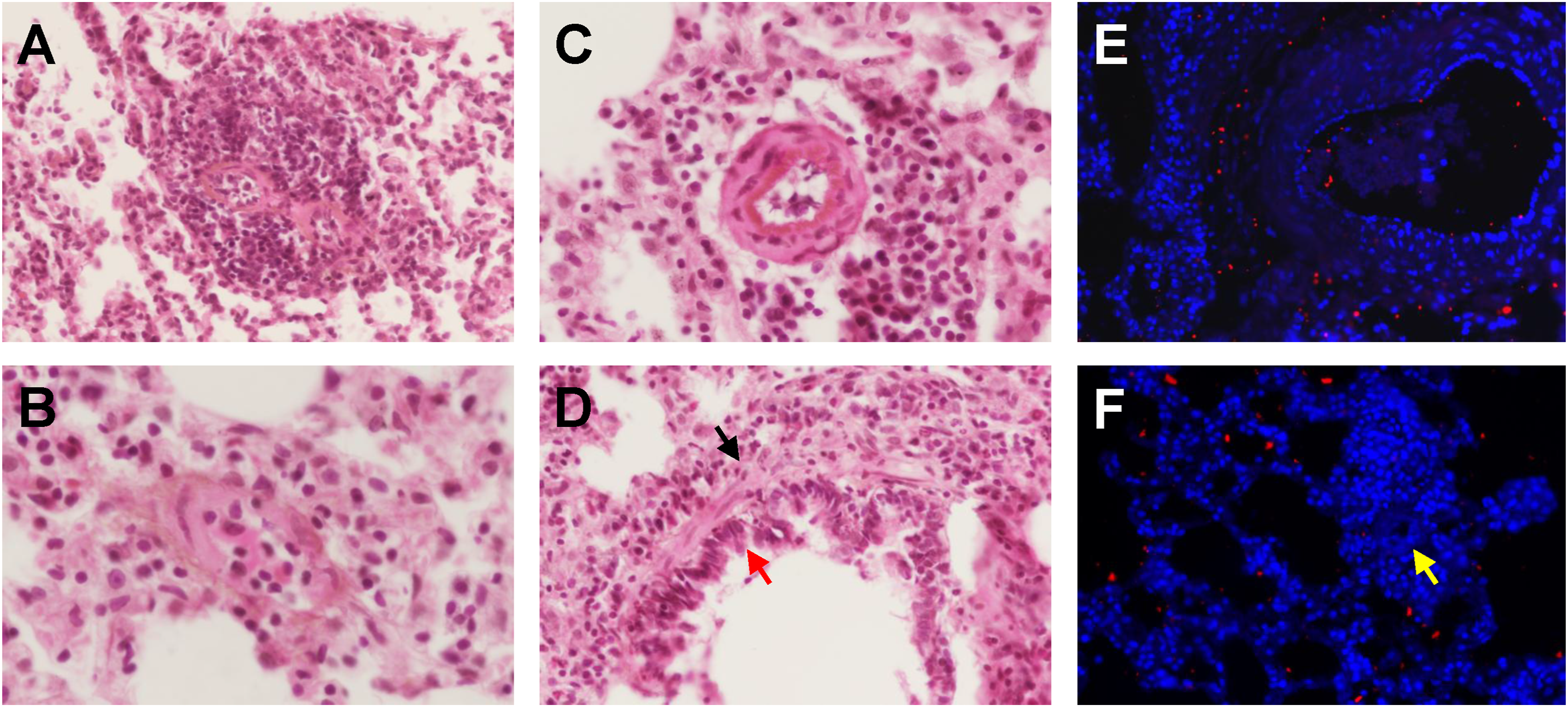
The histological changes in the hematoxylin and eosin-stained lung sections, associated with FDE exposure, are shown in Figures 2 and 5A-D, contrasting with the NE, CAE, and NAC pre-treated FDE-exposed groups of rats. The FDE-exposed group displayed notable thickening of the alveolar septa with interstitial inflammatory infiltration, compared to the NE and CAE groups (Figure 2). The mean ± SEM alveolar septal thickness was significantly increased in the FDE-exposed group (53.82 ± 7.27 μm), as compared with the NE (13.58 ± 1.10 μm) or the CAE (20.47 ± 2.65 μm) group of rats (Figures 2 and 4; P < 0.05, Dunnett’s test). Lungs of the FDE-exposed rats demonstrated widespread infiltration of inflammatory cells, majorly neutrophils and lymphocytes (Figure 5), as well as occasional areas of interstitial fibrosis. The pulmonary arterioles and capillaries exhibited congestion, thickened walls with or without fibrosis, and perivascular inflammatory infiltration, in contrast to the normal pulmonary capillaries observed in the NE and CAE groups (Figures 2 and 5). In addition, FDE-exposed lungs focal showed areas of endothelial discontinuity in the pulmonary capillaries, through which extravasation of inflammatory cells was observed (Figure 5). Furthermore, focal areas of bronchial epithelial damage accompanied by inflammatory cell infiltration around the bronchioles were also evident (Figure 5). The mean ± SEM alveolar cross-sectional area was not significantly different in the FDE-exposed group (5642 ± 1747 μm2) as compared with the NE (3821 ± 557 μm2) or the CAE (4265 ± 1402 μm2) group of rats (Figures 2 and 4; P > 0.05, Dunnett’s test). Semi-quantitative scoring of lung lesions using INHAND criteria showed a significant increase in histopathological lesion scores of alveolar septal thickening, interstitial inflammation, capillary congestion, perivascular inflammation, bronchial epithelial necrosis, endothelial discontinuity, and interstitial fibrosis in the FDE-exposed rats as compared with the NE and CAE exposed group of rats (Figure 4D; P < 0.001, one-way ANOVA). Photomicrographs (20x) of hematoxylin and eosin-stained lung sections illustrating the histological alterations linked to exposure to filtered diesel exhaust (FDE), contrasting with the non-exposed (NE), clean air exposed (CAE), and N-acetyl cysteine (NAC) pre-treated FDE-exposed groups of rats. The FDE-exposed group displayed notable thickening of the alveolar septa with interstitial inflammatory infiltration, compared to the NE and CAE groups. Arrow in the FDE image highlights a pulmonary capillary exhibiting congestion, thickened walls, and surrounding inflammatory infiltration, in contrast to the normal pulmonary capillaries observed in the NE and CAE groups. In the NAC pre-treated FDE-exposed group (NAC + FDE), the interstitial, capillary, and peri-capillary changes observed in the FDE-exposed group were significantly reduced. Scale bars in each image represent 100 μm.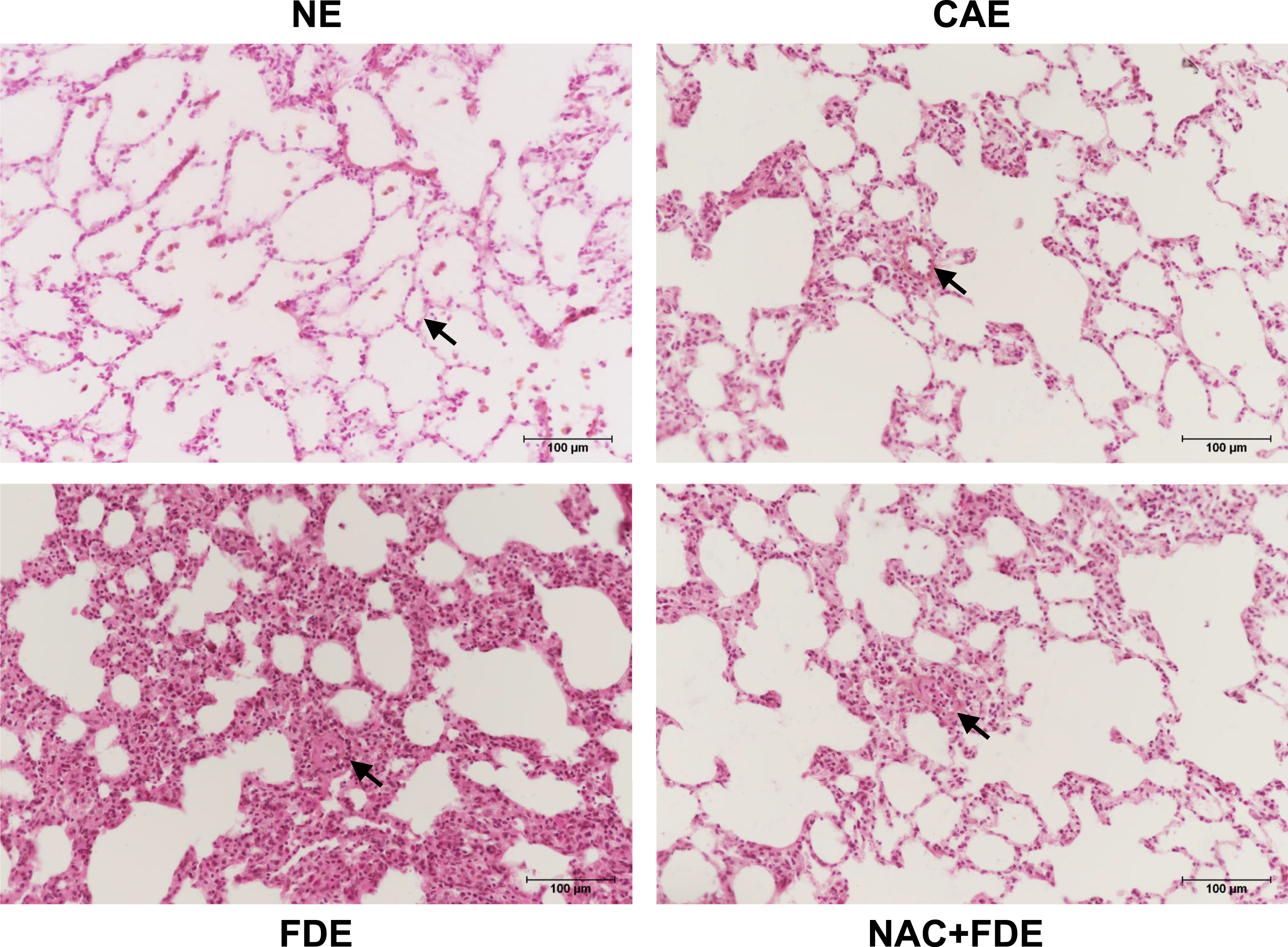
FDE Exposure-induced Alterations on TRPV1 Expression and Localization
The fluorescent microscopic images of lung sections stained with rabbit anti-rat TRPV1 primary antibody (1:200), followed by goat anti-rabbit fluorescent secondary antibody (1:100) and fixed with 4′,6-diamidino-2-phenylindole (DAPI) containing mounting medium, revealed an increased expression of TRPV1 in the FDE-exposed group in comparison with the NE and CAE groups (Figure 3). The mean ± SEM TRPV1 fluorescence intensity (pixels) was increased in the FDE-exposed group (3.524 ± 0.493) significantly, as compared with the NE (0.045 ± 0.013) or the CAE (0.298 ± 0.040) group of rats (Figures 3 and 4; P < 0.05, Dunnett’s test). Further, in the FDE-exposed lungs the increased TRPV1 expression was majorly localized near congested pulmonary vessels, and along thickened alveolar septa with inflammatory cell infiltration (Figure 5D, E). Fluorescent microscopic images (20x) of lung sections stained with rabbit anti-rat TRPV1 primary antibody (1:200), followed by goat anti-rabbit fluorescent secondary antibody (1:100) and fixed with 4′,6-diamidino-2-phenylindole (DAPI) containing mounting medium, reveal an increased expression of TRPV1 in the filtered diesel exhaust (FDE) the exposed group compared to the non-exposed (NE) and clean air exposed (CAE) groups. This increased expression was diminished in the N-acetyl cysteine pre-treated FDE-exposed group (NAC + FDE). Scale bars in each image represent 100 μm. Graphs illustrating the mean ± SEM values of (A) alveolar septal thickness (μm), (B) alveolar cross-sectional area (μm2), and (C) TRPV1 fluorescence intensity. The mean alveolar septal thickness and mean TRPV1 fluorescence intensity were significantly higher in the filtered diesel exhaust (FDE)-exposed group compared to the non-exposed (NE) and clean air exposed (CAE) groups (P < 0.05, Dunnett’s test), as indicated by the asterisk (*) sign. The changes induced by FDE exposure were significantly reduced in the N-acetyl cysteine pre-treated FDE-exposed group (NAC + FDE) (P < 0.05, Dunnett’s test). Additionally, there was no significant difference in the mean alveolar area among the various groups of rats (P > 0.05, Dunnett’s test). (D) Heat map showing mean ± SEM values of the histopathological lesion scores across experimental groups (n = 6 per group), as per the INHAND (International Harmonization of Nomenclature and Diagnostic criteria for rat lung lesions) classification. Scores: 0 = absent, 1 = minimal, 2 = moderate, 3 = marked. (A)-(D) Photomicrographs of hematoxylin and eosin-stained lung sections show the histological changes associated with exposure to filtered diesel exhaust at higher magnification (×40). (A) Two pulmonary capillaries exhibit increased wall thickness and infiltration by lymphocytes and neutrophils. (B) Pulmonary capillary congestion is noted, along with extravasation of immune cell into the interstitial space. (C) Inflammation infiltration observed around a pulmonary arteriole. (D) Bronchial epithelial damage (red arrow) is accompanied by inflammatory infiltration around the bronchioles (black arrow). (E)-(F) Immunofluorescence images depict increased TRPV1 expression (red dots) around a congested pulmonary arteriole (E), and around a congested pulmonary capillary (yellow arrow) with surrounding inflammatory infiltration (F).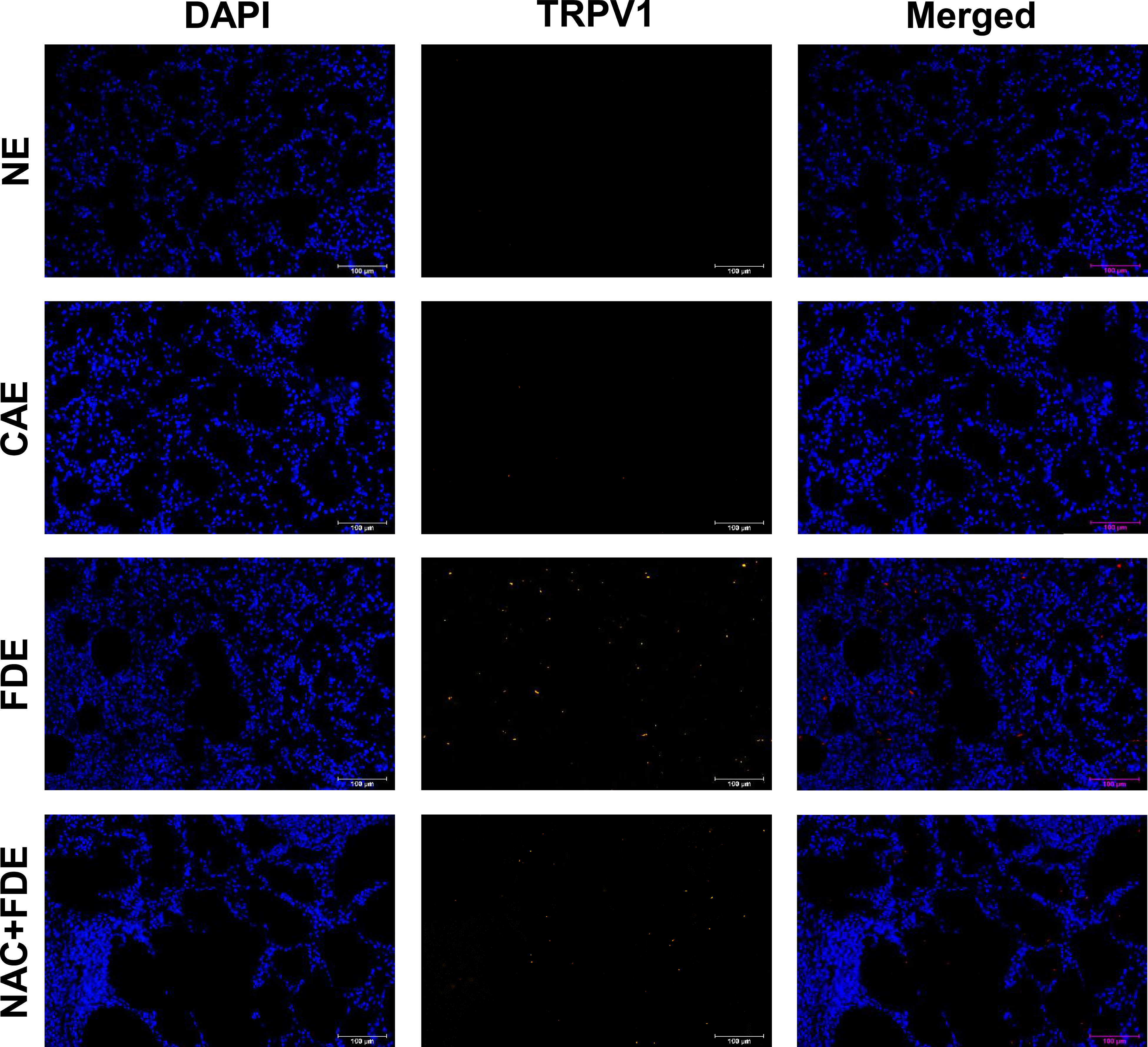
NAC Attenuated FDE Exposure-induced Alterations in Lung Histology
In the NAC pre-treated FDE-exposed group, the interstitial, capillary, and peri-capillary changes observed in the FDE-exposed group were significantly attenuated. The mean ± SEM alveolar septal thickness was significantly reduced in the NAC pre-treated FDE-exposed group (19.46 ± 2.03 μm), as compared with the FDE-exposed group of rats (Figures 2 and 4; P < 0.05, Dunnett’s test). Further, the mean ± SEM alveolar cross-sectional area was not significantly different in the NAC pre-treated FDE-exposed group (13145 ± 4957 μm2) as compared with the FDE-exposed group of rats (Figures 2 and 4; P > 0.05, Dunnett’s test). However, there were significant non-uniformities in the alveolar cross-sectional area in the NAC pre-treated FDE-exposed group, ranging from 1781 μm2 to 43630 μm2, unlike the other groups. The histopathological lesion scores (INHAND criteria) were significantly reduced in the NAC pre-treated FDE-exposed group in comparison with the FDE-exposed group (Figure 4D; P < 0.001, one-way ANOVA). The fluorescent microscopic images revealed that the mean ± SEM TRPV1 fluorescence intensity (pixels) was significantly reduced in the NAC pre-treated FDE-exposed group (0.709 ± 0.129), as compared with the FDE-exposed group of rats (Figures 3 and 4; P < 0.05, Dunnett’s test).
Discussion
Our study presents the histological changes in the lung sections associated with FDE exposure contrasting with the NE, CAE, and NAC pre-treated FDE-exposed groups of rats. The FDE-exposed group shows notable thickening of the alveolar septa with interstitial inflammatory infiltration, congestion of pulmonary capillary along with thickened walls, and surrounding inflammatory infiltration, in contrast to the normal pulmonary interstitium and capillaries observed in the NE and CAE groups. In the NAC pre-treated FDE-exposed group, the interstitial, capillary, and peri-capillary changes observed in the FDE-exposed group are significantly reduced.
Ambient air pollution is linked to a rise in the incidence of cardiorespiratory diseases. 18 The associations are stronger even for short-term exposure to either particulate matter or gaseous pollutants. 19 Gaseous pollutants are particularly concerning as they can penetrate deeply into the respiratory system and cross into the bloodstream, causing harmful effects not only to the respiratory and but also to the cardiovascular systems. 20 Diesel engine emissions are principally rich with ultrafine particulates and gaseous oxides, which significantly lead to health impacts in metropolitan and industrialized areas. 21 The negative effects of gaseous pollutants from diesel exhaust on blood pressure, coronary vascular function, atherosclerosis, and clotting processes have been documented, leading to a higher incidence of adverse cardiorespiratory events. 22 The precise mechanistic basis of the heightened risk of cardiovascular and respiratory diseases with exposure to diesel exhaust pollutants remains unclear. One possibility is that systemic inflammation, which follows pulmonary inflammation, may facilitate the transfer of harmful substances from the lungs to the vascular system. Alternatively, inhalation of gaseous pollutants could affect distant organs through neural and humoral pathways. 23 Additionally, sensory feedback from the lungs can also modulate autonomic outflow from the central nervous system,7,24 and multiple studies have shown that pollutants can modulate these sensory pathways6,25-28 and produce alterations to lung morphology and histology.9,10
In the present study, the FDE-exposed group shows notable thickening of the alveolar septa with interstitial inflammatory infiltration, congestion of pulmonary capillary along with thickened walls, and surrounding inflammatory infiltration, contrary to the normal pulmonary interstitium and capillaries observed in the NE and CAE groups. We further quantified these changes using INHAND-based lesion scoring, which allowed objective comparison across groups and provided graphical representation of lung injury severity. Studies elsewhere involving particulate matter from diesel exhaust for prolonged exposure periods have reported that exposure to traffic-derived air pollutants produced similar patterns of lung damage manifesting as diffuse alveolar damage, vascular congestion, interstitial thickening, fibrosis, and inflammatory infiltration.9,10 However, in this study we demonstrate that subacute exposure to the particulate-free gaseous fraction of diesel engine exhaust, in the absence of particulate matter, produces significant alterations in the lung histo-morphology, which could be attributed to the pathogenesis of respiratory ailments. This distinction is critical, as most prior studies evaluated particulate-containing exhaust, whereas we used a real-world urban exposure proxy filtered to exclude particulate matter. Our findings reveal that even the gaseous phase alone can produce TRPV1-mediated histological injury. A developing hypothesis regarding the hostile cardiorespiratory consequences of various components of diesel engine exhaust focuses on the activation of TRPV1 ion channels. These channels, which are inconsistently present in the pulmonary tract, act as environmental sensors, reacting to certain nociceptive chemicals, like endogenous mediators of inflammation and exogenous irritant substances including ambient air pollutants.29-31 Research has shown that the activation of TRP channels is associated with cardiorespiratory effects linked to diesel exhaust exposure in rats, along with the emergence of asthma-like symptoms in mice. 32 Mechanistically, the TRP channel activation in pulmonary C-fibers can trigger neurogenic inflammation and influence the expression of pro-inflammatory cytokines.11,12 Previous studies on particulate matter have highlighted the said involvement of TRPV1 receptors, pulmonary C-fiber afferents, and neurogenic inflammation in the vicinity13,14; however, the effects of the particulate-free gaseous fraction remained largely underexplored.
In the present study, involving short-term subacute exposure to particulate-free filtered diesel exhaust, the FDE-exposed lung sections demonstrate an increased expression of TRPV1 compared to the NE and CAE groups, and more specifically, the increased TRPV1 expression was majorly localized near congested pulmonary vessels and along thickened alveolar septa with inflammatory cell infiltration. There are numerous diverse sensory nerve subtypes located in the lung; some of which are mechanically sensitive, and others are chemo sensitive, namely, C-fibers and Aδ-fibers. TRP channels situated on the sensory terminals are triggered by a wide diversity of mechanical and chemical stimuli to produce reflexes, failure of which leads to respiratory symptoms. 6 After activation, TRP channels permit calcium influx into the cell, causing consequent depolarization of membrane and in due course generation of an action potential that spreads along the vagus nerve to the ponto-medullary centers of the brain, which modulate the cardiorespiratory parameters in a reflex fashion. In our recent study, we have demonstrated that FDE exposure, the capsaicin-induced protective nociceptive cardiopulmonary reflex responses in rats, in comparison with the NE group of rats. Additionally, the FDE-induced reduction of capsaicin-evoked cardiorespiratory responses are minimalized in the NAC-treated FDE rats, which suggested that gaseous chemicals from diesel engine exhaust attenuate capsaicin-induced protective cardiorespiratory reflex responses, through conceivable participation of oxidant stress mechanisms. 6 Several pharmacological agents with antioxidant properties, such NAC, have demonstrated the ability to reduce the making of reactive oxygen species, and alleviate damages caused by inflammatory cells to pulmonary tissue in rat acute lung injury models, caused by toxins or chemicals. NAC not only promotes the synthesis of glutathione but also possesses intrinsic antioxidant characteristics, allowing it to neutralize various reactive oxygen species.
In recent times, data has suggested bi-directional relationship between neurogenic and immunogenic mechanisms in pulmonary inflammation. In addition to earlier reports regarding the presence of TRPV1 on respiratory afferent neurons, bronchial smooth muscle cells, and epithelial cells in the lung and respiratory dendritic cells, current discoveries have directed that there is the expression of TRPV1 on T cells, which adjusts the inflammatory capabilities and activation of leucocytes. Results from the current study demonstrate that the interstitial, capillary, and peri-capillary changes observed in the FDE-exposed group are significantly reduced in the NAC pre-treated FDE-exposed group. Further, NAC pre-treatment also attenuates the increased TRPV1 expression observed in the FDE-exposed group. Thus, the FDE exposure-induced increased TRPV1 expression could possibly mediate the pathogenesis of increased pulmonary and cardiovascular diseases associated with diesel exhaust air pollutants, by mechanisms possibly involving oxidative stress mechanisms. On the whole, this study investigates the respiratory effects of FDE exposure rather than to elucidate molecular mechanisms of TRPV1 receptor activation. While mechanisms were not directly studied, the results provide foundational data for future research exploring pathways underlying these effects. The study also adds value by examining the impact of FDE exposure, which remains underreported compared to unfiltered exhaust, especially in the context of urban air pollution and lung health.
While our study provides valuable insights into the histological and molecular changes in lung tissues following FDE exposure and the protective effects of NAC, we acknowledge certain limitations. First, the study was conducted on a rodent model, which, despite its translational value, may not fully replicate the complex physiological responses observed in humans. Second, the focus on short-term subacute exposure to the gaseous fraction of diesel exhaust does not account for the potential cumulative or long-term effects of chronic exposure, which may involve additional pathological mechanisms. Third, while TRPV1 expression was examined in lung tissue, the study did not explore its downstream signaling pathways or interactions with other TRP channels that may contribute to respiratory dysfunction. Another limitation is the lack of chemical speciation of volatile organic compounds by employing gas chromatography or Fourier Transform infrared spectroscopy analysis, which could help isolate the key toxic components. Future studies incorporating chronic exposure models, simulating more human-relevant exposure conditions, and a broader investigation into TRP channel-mediated inflammatory pathways will be essential for further elucidating the precise mechanistic role of TRPV1 in diesel exhaust-induced pulmonary pathology. In addition, comprehensive biochemical quantification of oxidative stress markers, cytokine profiling, functional respiratory measurements, inclusion of additional timepoints or exposure doses, comparative analysis with whole diesel exhaust, and advanced validation through TRPV1 antagonism or genetic models were beyond the scope of the current work. Further studies are necessitated to systematically address these aspects to establish dose dependence, temporal progression, and mechanistic causality in greater depth.
Conclusions
In summary, exposure to filtered diesel exhaust (FDE) elicits significant alterations in lung architecture, characterized by pronounced thickening of the alveolar septa, interstitial inflammatory infiltration, and congestion of the pulmonary capillaries, contrasting sharply with the normal lung interstitium and capillaries seen in the non-exposed and clean air exposed groups. In the N-acetyl cysteine (NAC) pre-treated FDE-exposed group, the interstitial, capillary, and peri-capillary changes observed in the FDE-exposed group are significantly reduced. Additionally, we observed an upregulation of TRPV1 expression in the FDE-exposed lung tissues compared to the NE and CAE groups, which was markedly reduced following NAC treatment. This increased TRPV1 expression was primarily localized around congested pulmonary vessels and along the thickened alveolar septa with infiltrating inflammatory cells. These results highlight a potential mechanistic relationship between TRPV1 receptor expression and oxidative stress pathways in the lung damage induced by FDE exposure, underscoring the therapeutic promise of NAC in countering these effects.
Footnotes
Author Contributions
Ravindran Revand: Conceptualization, data curation, formal analysis, investigation, methodology, and writing—original draft. Aditya Dontham: Investigation. Swarnabha Sarkar: Investigation. Abhishek Kandpal: Validation. Debabrata Dasgupta, Bahni Ray, and Mayank Kumar: Conceptualization, methodology, and validation. Asmita Patil: Conceptualization, supervision, validation, and writing—reviewing and editing.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
This study received approval from the Institutional Animal Ethics Committee at the All India Institute of Medical Sciences in New Delhi, India (Letter No. 391/IAEC-1/2022-17.03.2023). All animal procedures obeyed to the guiding principle laid down by the US National Institute of Health’s “Guide for the Care and Use of Laboratory Animals” and reported in line with ARRIVE guidelines.
Data Availability Statement
All data generated or analyzed during this study are included in this published article at relevant places. Data from the study will be made available by the corresponding author upon reasonable request.