
Abstract
Physiologically based pharmacokinetic (PBPK) modeling is increasingly used to anticipate, quantify, and strategically manage drug–drug (DDI) and herb–drug (HDI) interactions that can alter the exposure of chemotherapy agents together with co-administered phytochemicals or nutraceuticals. To evaluate current knowledge, we performed a comprehensive Google Scholar search (2003–2024) and selected studies that employed PBPK platforms, reported quantitative validation, and focused on chemotherapy-related interactions. From these reports, key modeling parameters, validation metrics, and clinically relevant outcomes were extracted, and then the information was synthesized to identify common trends. Collectively, the evidence indicates that unintended changes in drug exposure—most often mediated by CYP3A4 inhibition or induction—may modify efficacy, toxicity, and overall anticancer response; nevertheless, PBPK models reproduce these effects with high accuracy, and emerging AI-enhanced approaches promise even finer precision. Accordingly, our synthesis underscores how PBPK modeling can help clinicians forecast interaction risk, individualize dosing, and avert therapeutic failure, especially in polypharmacy settings. Integrating these models into routine oncology practice therefore offers a proactive path toward safer, more personalized chemotherapy and, ultimately, better patient outcomes within an increasingly complex therapeutic landscape.
Keywords
Introduction
Drug interactions occur when one substance affects the pharmacokinetic characteristics or pharmacological activity of another, potentially altering efficiency of drug or increasing the risk of toxicity, or even causing a loss of therapeutic effect overall, even though many of them are purposeful and are used to increase desired effects or decrease undesirable ones.1,2
Numerous studies have combined experimental data with PBPK models to elucidate the mechanisms of drug–drug and herb–drug interactions.3,4 Although these studies are very useful for examining particular drug interactions, a more holistic viewpoint is essential to bringing similar pathways together and creating thorough, trustworthy models.
Research on DDIs has concentrated on identifying significant interactions, classifying them, and building predictive models. 5 Key findings emphasize the importance of pharmacokinetic and pharmacodynamic mechanisms such as changes in drug metabolism or cumulative effects, in causing adverse interactions. Digoxin, NSAIDs, warfarin, and antidepressants are among the common medications linked to major DDIs.2,6
Unplanned DDIs are quite likely to emerge in oncology in which complex treatment strategies often involve chemotherapeutics, immunotherapeutics, targeted therapies, and supportive care medications. 1 This complex treatment landscape is further complicated by the increasing use of phytochemicals, herbal supplements, and nutraceuticals to promote tumor control, boost tolerance, or simply support quality of life, in addition to these pharmacological issues. 7 Numerous phytochemicals/herbal medications have antiproliferative properties and may alter drug transport and metabolism, offering both opportunities and risks for patient outcomes.
The majority of pharmacokinetic DDIs arise from alterations in absorption, distribution, metabolism, or excretion (ADME). 2 Primary contributors include transporters like P-gp, whose modulation (via induction or inhibition) can significantly impact plasma levels of medication and its safety/efficacy profile, and cytochrome P450 (CYP) enzymes, of which CYP3A4 is the primary contributor.1,2 For example, competition for tissue distribution sites or plasma protein binding may change distribution, whereas changes in gastrointestinal pH, motility, or transporter function may affect absorption. Drug exposure and half-life are also altered by changes in renal or biliary excretion, although metabolic interactions frequently rely on modulators of the CYP enzymes themselves.1,8
Examples of combination therapies include medications such as anti-inflammatory analgesics or phytochemicals or nutraceuticals that can change the metabolic pathways of concurrently delivered chemotherapeutic treatments. Dietary supplements and nutraceuticals such as St. John’s Wort (a CYP3A4 inducer) can quickly lower the plasma concentrations of various anticancer medications, jeopardizing their therapeutic effects.9-11 A further layer of uncertainty is added by polypharmacy, a condition that is prevalent, particularly with older patients and those with multiple illnesses. 5 Complex and frequently unpredictable interaction networks result from the interference of different anticancer agents working with various supportive therapy modalities and other additional medications, chemicals, or substances. 12
Most of the reviews in literature focus on the mechanisms of DDIs at various levels, including ADME to examine the clinical consequences of these interactions, ranging from enhanced toxicity to altered efficacy.1,13 As also highlighted in review studies14,15, the growing adoption of PBPK modeling, driven by advancements in computational tools and regulatory acceptance, underscores its potential to enhance drug safety and efficacy. These studies also identify challenges, including the need for more robust physiological data and the harmonization of in vitro experiments to improve predictive accuracy.
Drug interactions are especially critical in oncology due to the narrow therapeutic windows, multiple concurrent treatments, and potential for serious clinical consequences. This review discusses the pharmacokinetic principles underlying drug interactions in oncology, emphasizing PBPK modeling of drug–drug and herb–drug interactions. While recent work has often centered on specific cancer types, our primary goal is to synthesize these findings, evaluate the validity of mechanism-based models, and demonstrate how advanced PBPK tools can guide preventive strategies.
Methods
A literature search covering 2003–2024 was performed in Google Scholar with the keywords chemotherapy, PBPK modeling, drug interactions, and phytochemical interactions. Quantitative PBPK modeling studies on drug interactions using these platforms were included. The search was restricted to studies using a modeling software platform that also provided a quantitative model validation for chemotherapy drug interactions. For each eligible paper, modeling parameters, validation metrics, and clinical outcomes were abstracted to enable a structured synthesis and comparison. Figure 1 illustrates the common workflow observed across these studies, moving from experimental plasma concentration data through PBPK simulation to statistical validation. Taken together, the compiled results demonstrate the robustness and broad clinical applicability of PBPK models. General PBPK workflow for evaluating chemotherapy drug–drug and herb–drug interactions.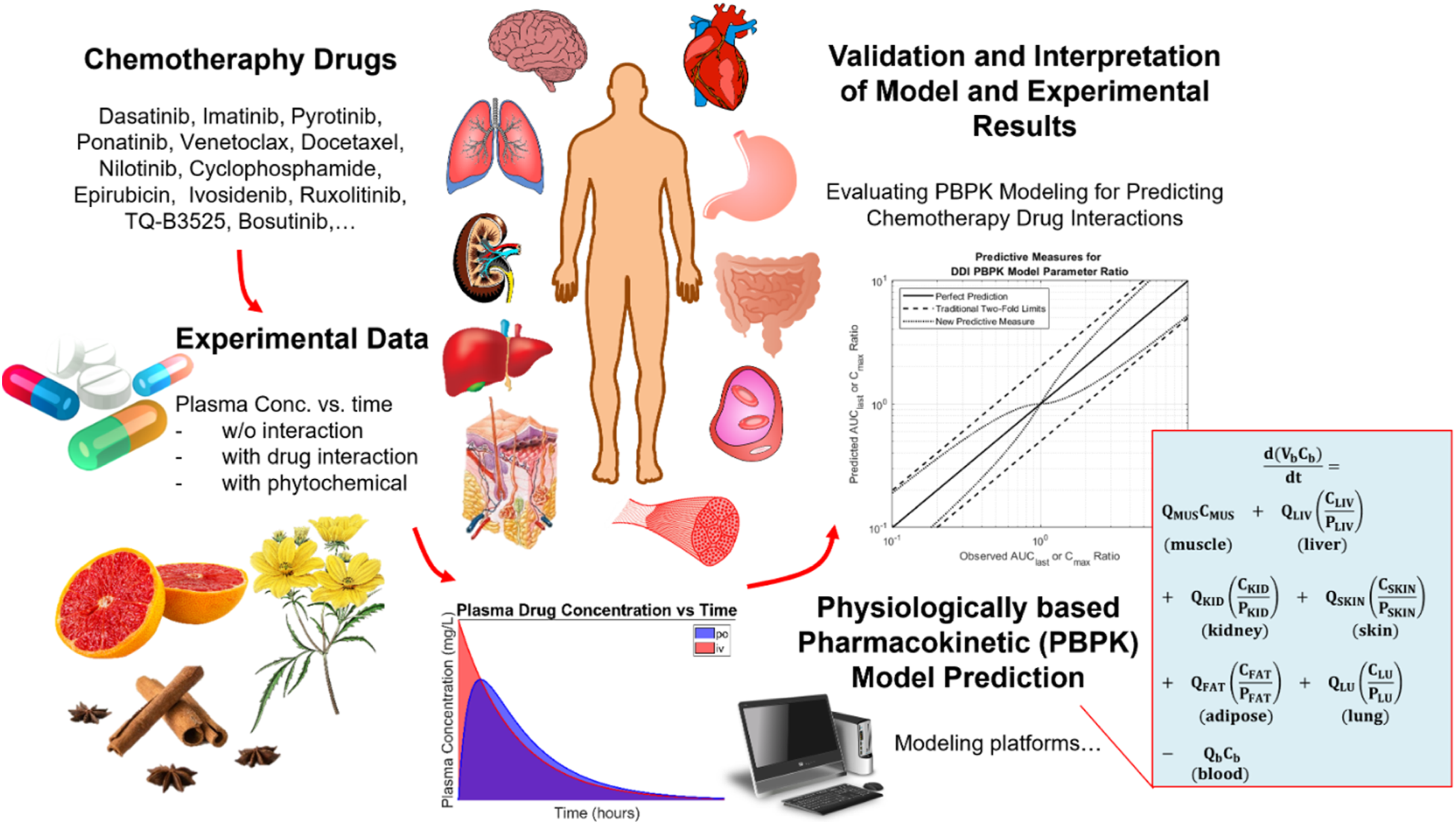
Drug–Drug Interactions in Oncology
Cancer patients frequently get novel treatments, which, while improving survival results, are susceptible to pharmacokinetic and pharmacodynamic interactions that might impair efficacy or increase toxicity. In those treatments, unknown DDIs present a critical challenge because of the complex pharmacological profiles of anticancer agents and their concurrent use with other medications. The increasing complexity of cancer treatments has therefore brought the management of DDIs to the forefront, significantly impacting therapeutic efficacy, patient safety, and treatment outcomes. 1
Common Mechanisms Underlying Pharmacokinetic DDIs.
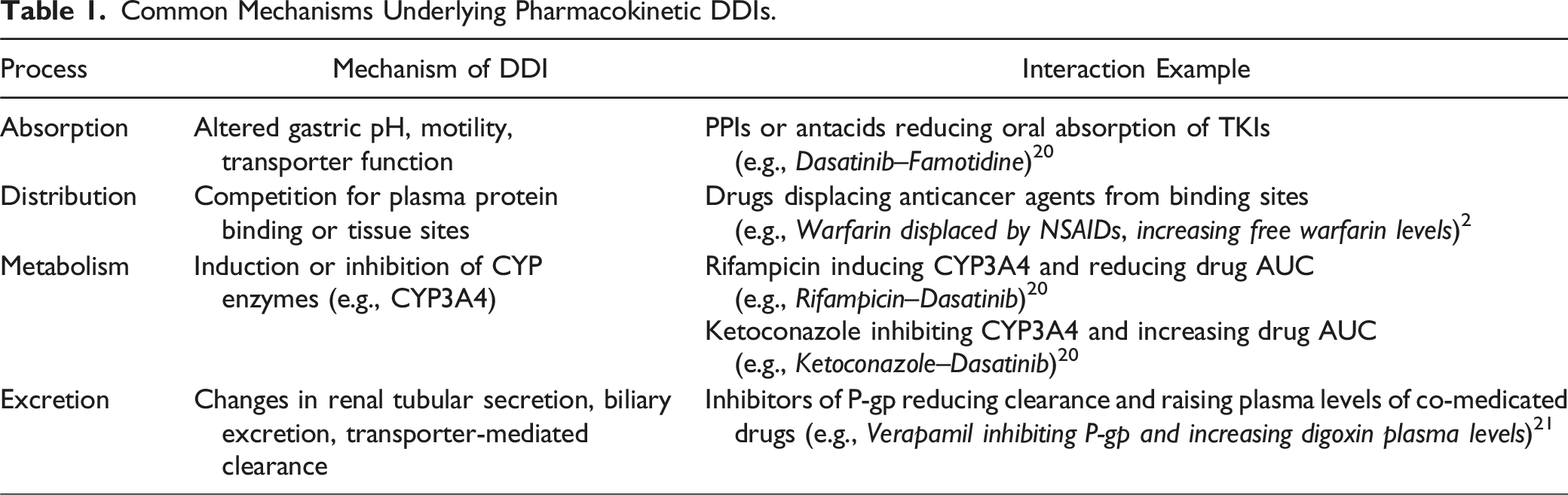
The complexity of DDIs during cancer treatment is underlined, especially when cytotoxic agents are being incorporated into targeted therapies. 17 The study specifically focuses on dose optimization methodologies developed under the FDA’s Project Optimus initiative that combines pharmacokinetic/pharmacodynamic modeling, quantitative systems pharmacology, and translational data in improving predictions related to clinical outcomes. It further highlights the collaboration with regulatory bodies and the use of iterative modeling techniques in the optimization of dosing strategies. In conclusion, tailored approaches are needed for the unique challenges each drug combination presents, as well as for the diverse patient populations. 17
The vulnerability of geriatric patients to DDIs is emphasized in consideration of physiological changes, polypharmacy, and related comorbidities. 18 During polymedication in geriatric oncology, there are increased chances of adverse drug reactions, thus needing geriatric assessment in order to guide proper dose adjustment in an effort to reduce toxicity. 18 A probabilistic model is developed for managing uncertain DDIs. 19 The model integrated the likelihood of an interaction with the interaction’s severity and avoidability to allow the ranking of patient safety with therapeutic efficacy. Pascal’s Uncertainty Principle underpinned the described approach to enable sound decision-making within clinical oncology. 19
Natural products also present some unique challenges regarding drug interactions. Phytochemicals in products such as grapefruit juice and St. John’s Wort may act as inhibitors or inducers of drug-metabolizing enzymes, which could result in subtherapeutic or toxic levels of anticancer agents.22,23 Hence, it emphasizes the importance of patient education and strict monitoring while using alternative medications alongside cancer treatments.
Physiologically Based Pharmacokinetic (PBPK) Modeling Strategies
Interest in PBPK modeling has recently increased, suggesting a closer link and predictive power with oncology drugs. The most interesting aspect of software such as PK-Sim and Simcyp is the ease with which in vitro and in vivo data integrate, providing simulations of the main pharmacokinetic processes of ADME through various physiological and pathophysiological states. 12 By modeling complex mechanisms such as enzyme inhibition, induction, competitive binding, and transporter modulation, it is possible to predict changes in drug exposure within clinical scenarios that have not been tested. This allows the clinician and researcher to anticipate potential DDIs, enlighten clinical trial design, and optimize therapeutic strategy.9,24-26
PBPK model development and validation typically follow four key stages: (1) data preparation, (2) model development, (3) calibration, and (4) validation (Figure 2).
27
The initial step often involves an extensive literature review to gather physicochemical properties and ADME parameters from clinical trials (e.g., concentration–time profiles, dosing regimens). Relevant data on interacting drugs, physiological parameters, and population demographics are then compiled, with the overall dataset being split into training and test sets. A stepwise strategy for PBPK model development.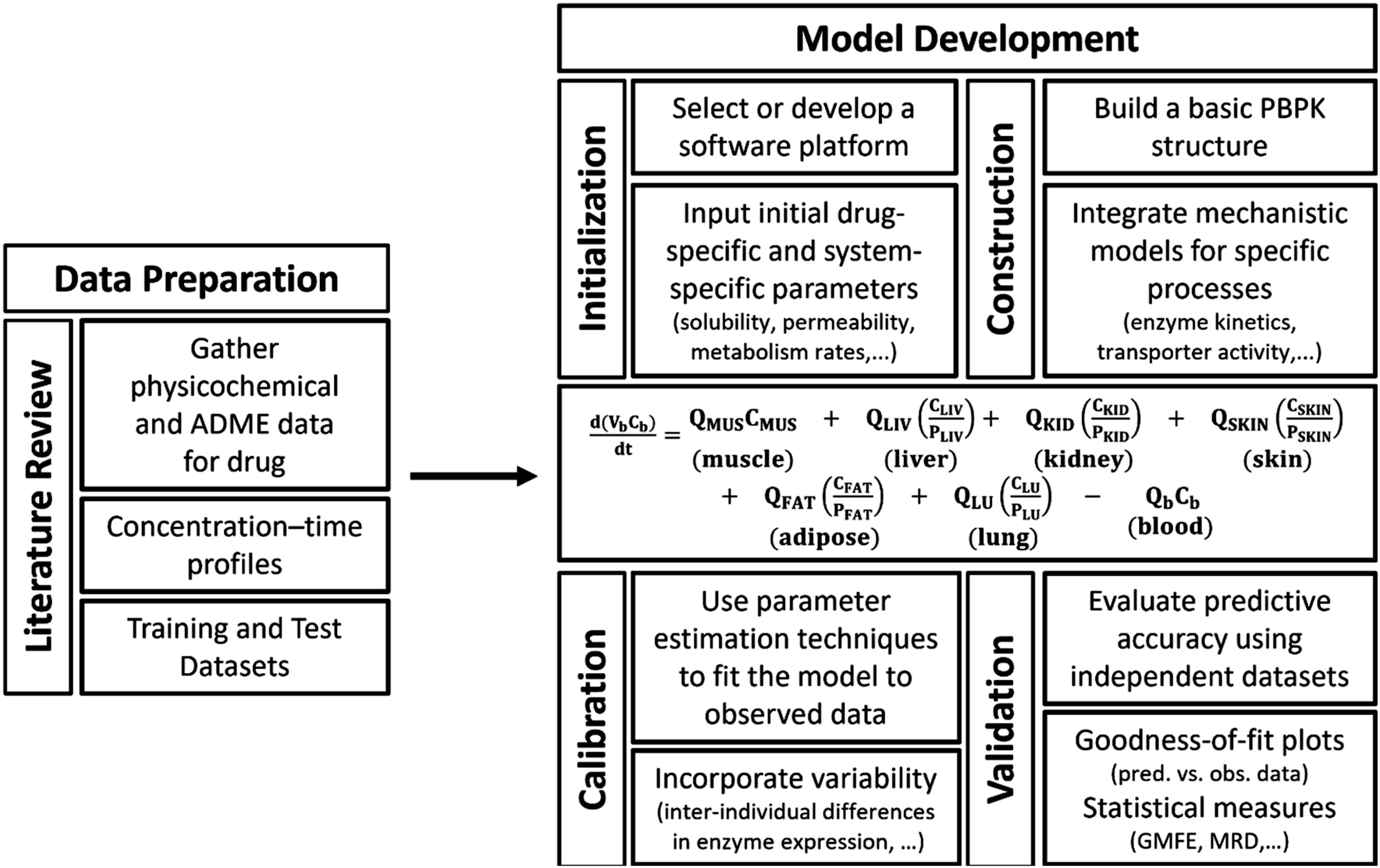
During the model development phase, a middle-out approach combining in vitro, in silico, and clinical data with parameter estimation is employed. Parameter estimation techniques (e.g., Levenberg–Marquardt algorithm) fit the model to training plasma profiles, while key metabolic constants (such as Michaelis–Menten parameters) are derived from the literature.
20
Predicted plasma concentration–time profiles are compared to observed values, and performance is evaluated using metrics like mean relative deviations (MRDs) (equation (1)) and geometric mean fold errors (GMFEs) (equation (2)).
28
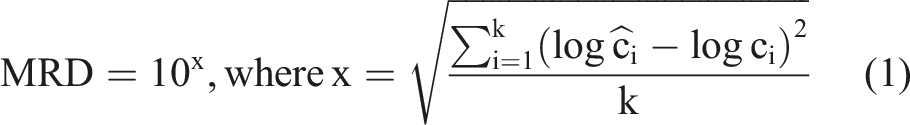
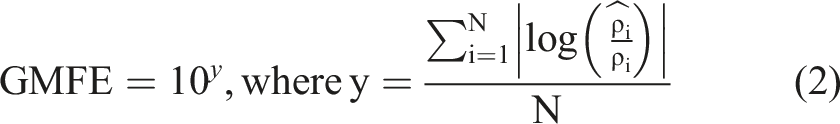
Changes in parameters like Cmax and AUC are frequently evaluated by comparing values measured under DDI conditions to results obtained without DDI (equation (3)). This approach applies to both model predictions and clinical observations. A prediction/observation ratio is often used to assess model validity. Prediction success is commonly measured using the two-fold criterion, a widely used metric in pharmacokinetics and drug–drug interaction (DDI) studies to evaluate prediction accuracy.
29
According to this criterion, a prediction is considered successful if the predicted value (e.g., the area under the concentration–time curve, or AUC) falls within a range that is double or half the observed experimental value.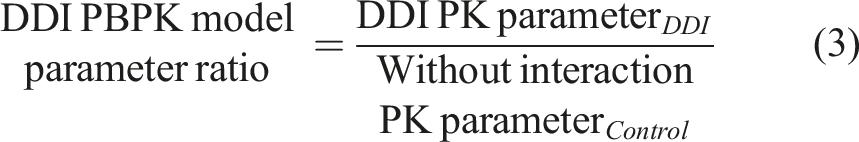
For example, if the observed AUC ratio in the presence of a drug interaction is 1.0, the two-fold range would be between 0.5 and 2.0. This method provides a broad tolerance range for predictions, making it useful for addressing variability in experimental data. However, the two-fold range can sometimes create false impressions of prediction success, particularly when the observed AUC ratio is close to 1 (indicating no interaction). To address this limitation, a new predictive approach is proposed, as illustrated in Figure 3, to refine the success criteria and improve prediction accuracy.
29
Schematic representation of the limits for different predictive methods, comparing the traditional two-fold measure (dashed lines) with the proposed new measure (dotted lines) by Guest et al.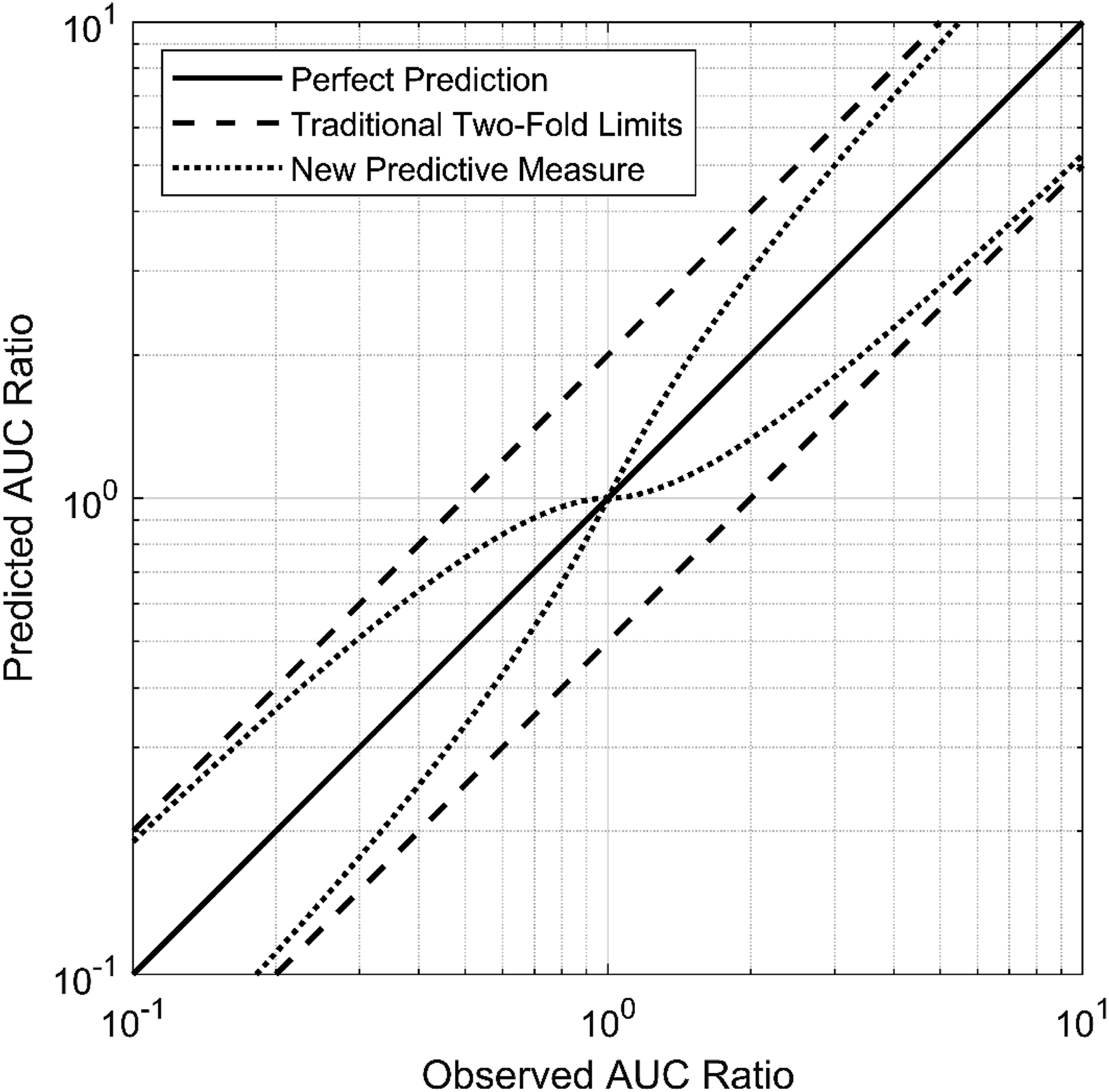
It is challenging to apply physiologically based pharmacokinetic modeling to herbal drugs because the PBPK approach necessitates the physicochemical properties of these phytochemical/food component(s) causing the herb–drug interaction. Moreover, the uniformity of content of these components within and across marketed products and knowledge of the systemic bioavailability and human PK of these component(s) are usually missing.29-35
Modeling Cancer Drug–Drug Interactions Using PBPK Approaches
The targeted and diverse mechanisms of therapies across different cancer types (chronic myeloid leukemia (CML), chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), acute myeloid leukemia (AML), myelofibrosis (bone marrow cancer), follicular lymphoma, HER2-positive metastatic breast cancer, lung, prostate, stomach, ovarian, blood, and head/neck cancers) are investigated in terms of drug–drug and drug–nutraceutical interactions.
Dasatinib
A PBPK model of dasatinib, which exhibits high dependence on CYP3A4-mediated metabolism and pH-dependent solubility, was developed and implemented in PK-Sim and MoBi modeling platforms based on experimental data from various clinical studies using different dosages and dosing intervals. 20 The model includes three elimination processes: (i) renal excretion, which accounts for passive glomerular filtration, (ii) metabolic CYP3A4-dependent elimination, which follows Michaelis–Menten kinetics, and (iii) a nonspecific hepatic clearance component, which accounts for metabolic pathways other than CYP3A4.
Summary of DDI Studies.
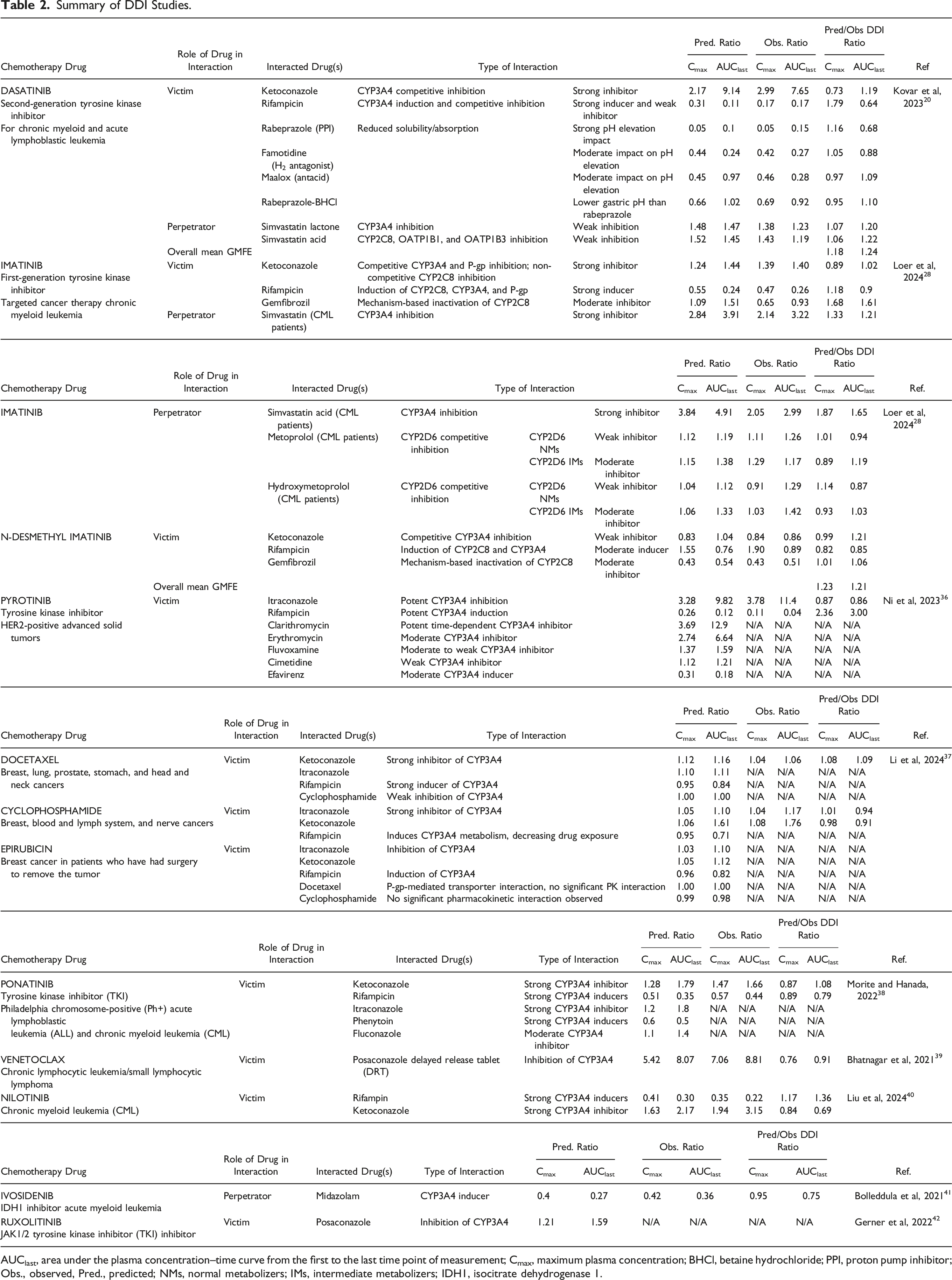
AUClast, area under the plasma concentration–time curve from the first to the last time point of measurement; Cmax, maximum plasma concentration; BHCl, betaine hydrochloride; PPI, proton pump inhibitor; Obs., observed, Pred., predicted; NMs, normal metabolizers; IMs, intermediate metabolizers; IDH1, isocitrate dehydrogenase 1.
Ketoconazole and itraconazole are potent CYP3A4 inhibitors that greatly raise dasatinib’s AUC and Cmax, whereas rifampicin is a powerful CYP3A4 inducer that decreases dasatinib exposure. On the other hand, increasing stomach pH inhibits dasatinib absorption by around 90%. Thus, it emphasizes the importance of time-dependent medication administration, drug-avoidance measures, and switching techniques.
To forecast probable interactions, simulations of various scenarios were run using obtained PBPK model. Even more complex multi-agent combinations, such as a moderate inhibitor erythromycin and a high inducer carbamazepine, demonstrated balanced effects that reduced the size of the AUC drop, reflecting the increased complexity of induction versus inhibition in the combination. 20 This finding emphasizes the complexities of managing DDIs with dasatinib and the need for PBPK in guiding dosage adjustment.
Imatinib
Similarly, a physiologically based pharmacokinetic model is developed for imatinib and its main metabolite, N-desmethyl imatinib, while considering the complex metabolic processes of CYP3A4, CYP2C8, and P-glycoprotein in transport. 28 This model accurately predicts the behavior of imatinib not only as a victim, where its exposure was considerably altered by inducers, with a 74% decrease in AUC following co-administration of rifampicin, but also as a perpetrator, affecting the clearance of co-administered drugs such as simvastatin and metoprolol due to inhibition of CYP3A4 and CYP2D6 as given in Table 2. The drug–drug gene interaction (DDGI) studies are also included for metaprolol and hydroxymetaprolol cases by considering CYP2D6 normal and intermediate metabolizer patients. Over 90% of its predictions were within a two-fold margin of error and hence more reliable and clinically significant. However, it still acknowledged a few limitations, like not considering data on genetic polymorphisms. 28
Pyrotinib
PBPK modeling approach is applied to assess CYP3A4-mediated DDIs affecting pyrotinib, another TKI used in HER2-positive breast cancer. 36 As shown in Table 2, strong inhibitors, such as itraconazole, approximately 10-fold increased pyrotinib AUC, whereas strong inducers, such as rifampicin, comparably decreased AUC. Moderate and weak CYP3A4 inducers/modulators demonstrated graded effects on exposure. Considering these pronounced changes, the authors recommend a general precaution and avoidance of co-administration of pyrotinib with strong and moderate CYP3A4 modulators. Poor inhibitors, however, pose an insignificant threat and can be given together with less concern. These results highlight even more the utility of PBPK modeling in defining safe and efficacious dosing regimens, informing the development of clinical study designs, and minimizing the need for extensive empirical testing in complex polypharmacy scenarios. 36 The high predictive performance of the model (GMFE <2.0) enhances its usefulness in informing clinical decisions.
Neoadjuvant Chemotherapies: Docetaxel, Cyclophosphamide, and Epirubicin
By concentrating on three common neoadjuvant chemotherapeutic agents, docetaxel, cyclophosphamide, and epirubicin, a study on the utility of PBPK modeling in DDI prediction is conducted. 37 Although CYP3A4 and P-pg activity affected all three of these medications, it is more likely that co-administration of CYP3A4 inducers or inhibitors with docetaxel and cyclophosphamide, but not epirubicin, will produce clinically noticeable changes based on systemic exposures. Predicted values of AUC and Cmax changes are very close to observed values (Table 2). Strong inhibitors like ketoconazole and itraconazole intensely increased AUC and thus needed lower doses whereas inducers like rifampicin decreased AUC, which may affect the drug’s efficacy, and thus higher doses may be necessary. 37 Epirubicin was the least affected among all these agents; it showed the least variation in exposure, and thus, this drug may be of lesser concern when DDI is considered for this class of drugs. Based on the predicted PBPK model results, docetaxel and cyclophosphamide should be carefully evaluated and their dosages adjusted when taken with strong CYP3A4 inhibitors or modulators, with some assurance that epirubicin will be only little impacted. Once more, this provides an excellent example of how population pharmacokinetic modeling can be used to forecast, measure, and control drug–drug interactions in complex oncologic regimens. 37
Ponatinib
Ponatinib is a tyrosine kinase inhibitor (TKI) used in the treatment of Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL) and chronic myeloid leukemia (CML).38,43 PBPK modeling techniques are employed for predicting possible DDIs with ponatinib (Table 2). Because ponatinib is primarily metabolized via that pathway and exhibits very low renal clearance, the scientists focused on the measurement of the effects of co-administered medications that inhibit or increase CYP3A4. 26 According to their validated model, which has given very good agreement with observed clinical data, strong CYP3A4 inducers, such as rifampicin, greatly reduce ponatinib levels, potentially undermining therapeutic efficacy, while strong CYP3A4 inhibitors, such as ketoconazole, may greatly increase ponatinib exposure, increasing the risk of toxicity. 38 Changes with both moderate inhibitors and weak inducers were modest, pointing at safer profiles upon co-administration. 38
Venetoclax with Posaconazole
Posaconazole, a powerful CYP3A4 inhibitor, when dosed in a delayed release tablet (DRT) manner of 300 mg in combination with venetoclax, led to an approximately 8.8- and 7.1-fold increase in venetoclax AUC and Cmax, respectively, and the safe dose was reduced from 400 mg to 70 mg daily for safety reasons. 39 Treatment with high-dose posaconazole (500 mg DRT) resulted in a 12% increase in venetoclax exposure, well within a concentration range tolerated for safety. 39 All of these interactions were predicted when applying the validated PBPK model which further supports its use in guiding venetoclax dose adjustment to ensure safety for individual patients and to limit the number of clinically relevant studies performed. These results emphasize the need for personalized dosing approaches to mitigate CYP3A4-dependent drug–drug interactions in clinic settings. 39
Nilotinib and Special Populations
The physiologically based pharmacokinetic model of the drug nilotinib in CML is established and then improved to describe its pharmacokinetic and distribution pattern in functional spaces that comprise drug–drug interaction and select subpopulations such as children, pregnant woman, and lactating woman. 40 The AUC and Cmax of nilotinib increased 3.15-fold and 1.94-fold, respectively, when it was co-administered with the potently CYP3A4 inhibitor ketoconazole making dose reductions necessary when strong CYP3A4 inhibitors are co-administered to avoid toxicity. Conversely, rifampin, an inducer of CYP3A4, was reported to reduce the AUC of nilotinib by as much as 78%, potentially leading to subtherapeutic levels; concomitant use should be avoided or dosage adjusted upwards. 40
The PBPK model was also used to explore other pharmacokinetic parameters that may be of special interest to certain populations. Considering the slower maturation trend of CYP1A2 in pediatric subjects and the impact of pregnancy on enzyme activity, these factors all play a dramatic impact on nilotinib’s PK along with a requirement for personalized dosing strategies. 40 Findings underscore the utility of PBPK modeling in forecasting pharmacokinetic changes, optimizing nilotinib therapy, and confirming safety and efficacy in challenging clinical scenarios. 40 This study further reinforces a growing role of PBPK modeling in clinical precision oncology, acting to facilitate the ability to navigate drug–drug interactions and population-specific issue. 40
Ivosidenib (IDH1 Inhibitor)
The pharmacokinetics and drug–drug interactions of ivosidenib (AG-120), a novel inhibitor of isocitrate dehydrogenase 1 (IDH1)—a cytosolic enzyme of the tricarboxylic-acid cycle whose gain-of-function mutations drive the oncogenic accumulation of 2-hydroxyglutarate in acute myeloid leukemia (AML)—are studied using the Simcyp platform. 41 Within a physiologically based pharmacokinetic modeling approach, the perpetrator potential of ivosidenib with respect to CYP-mediated and transporter-mediated DDIs is described, capturing inductive and inhibitive mechanisms on key PK parameters such as hepatic clearance, absorption, AUC, Cmax, and elimination. The study shows a highly significant induction of CYP3A4, increasing hepatic clearance as high as 371% with moderate induction of CYP2B6, CYP2C8, and CYP2C9. Also, inhibition of transporters, such as P-glycoprotein, OATP1B1/1B3, and OAT3, is observed among others. 41 There is no significant affect observed on the substrates of CYP2C9-like warfarin and substrates of P-gp-like digoxin. 41 Thus, it allows for appropriate predictions of drug–drug interactions and sets requirements for labeling. This research underlines how dosing adjustment of concomitant drugs can play an important role in patients being treated with ivosidenib to effect therapy with reduced possible clinical interactions.
Ruxolitinib and Disease-Specific Factors
PBPK modeling is performed for the estimation of DDI for ruxolitinib (RUX), a JAK1/2 inhibitor, co-administered with posaconazole (POS), a potent CYP3A4 inhibitor. 42 Since RUX is predominantly metabolized by major metabolic pathways via CYP3A4 and CYP2C9, it was highly susceptible to interact with substantial CYP3A4 inhibitors like POS. 42 The current investigation utilized clinical data from patients with graft versus host disease (GvHD) to simulate and validate plasma concentration profiles in predicting the change in exposure upon co-administration. Based on the developed PBPK model, POS increased the AUC of RUX by 59%, the Cmax by 20.5%, and placed emphasis on dose adjustment for GvHD patients in an attempt to avoid overexposure. GvHD reduces hepatic clearance and triggers changes in the pharmacokinetics of drugs that raise the risk of overexposure to RUX. This effect is potentiated by the receipt of other concomitant CYP inhibitors like atorvastatin and amiodarone, making an overall review of the medication important. The developed PBPK model takes into consideration both pharmacokinetic parameters along with disease-specific factors, providing actionable insights to optimize RUX therapy in GvHD patients receiving POS, thereby ensuring safety and efficacy. 42
TQ-B3525 and Multiple Metabolites
A physiologically based pharmacokinetic model is described for TQ-B3525, which is a dual inhibitor of PI3Kα and δ, and its metabolites M3 and M8-3, that would be used to study the emerging pharmacokinetic profile after the co-administration of modulators of CYP3A4 and P-gp. CYP3A4 and FMO3 are major enzymes participating in TQ-B3525 metabolic clearance, and TQ-B3525 is a substrate of P-gp, significantly affected by both its inhibitors and inducers. 44 Co-administration with rifampicin, the potent inducer of the cytochrome P450 isoenzyme 3A4, resulted in a pronounced decrease of the AUC of TQ-B3525 by 46% and reached only 76.1% of the baseline value in Cmax. This interaction significantly reduced the systemic exposure of TQ-B3525. In contrast, M3 and M8-3 experienced significant elevations as a result of the increased metabolism and distribution change. In contrast, concomitant administration of itraconazole, a strong CYP3A4/P-gp inhibitor, significantly increased the AUC (204%) and Cmax (131%) of TQ-B3525, and inhibited its formation of M3 and M8-3 (73–80%), indicative of a reduced elimination and increased bioavailability of the parent compound. PBPK results show that modulators of CYP3A4 and P-gp significantly impact exposure to TQ-B3525 and metabolite profiling. These agents significantly modify the pharmacokinetic landscape for TQ-B3525 by accelerating (or delaying) hepatic clearance and altering bioavailability. 44
Apatinib
Apatinib is a tyrosine kinase inhibitor used in cancer treatment. 45 To forecast the pharmacokinetics of apatinib in both healthy individuals and cancer patients, as well as to evaluate possible DDIs and drug–disease interactions (DDZIs), Liu et al conducted a PBPK modeling study by Simcyp Simulator and incorporating experimental data and settings from the literature. 45 Simulations showed that moderate CYP3A4 inducers significantly reduced apatinib exposure, while moderate CYP3A4 inhibitors increased it by 2–4 times (AUC). The exposure to apatinib increased 1.25–2 times when strong CYP2D6 inhibitors were used. For patients with drug–disease interactions, such as hepatic impairment, apatinib’s AUC showed a 2.25-fold increase in Child–Pugh B and a 3.04-fold increase in Child–Pugh C, accompanied by a slight reduction in peak plasma concentration (Cmax). 45 The PBPK model effectively reduced the reliance on resource-intensive in vivo studies by delivering reliable in silico predictions. This approach is especially beneficial for anticancer treatments, where ensuring patient safety is critical and clinical trial populations are often limited.
Another paper on apatinib explores the evaluation of its interactions with the calcium channel blocker nifedipine using PBPK models. 46 Given that the CYP3A4 enzyme metabolizes both medications, apatinib co-administration considerably raises nifedipine’s Cmax and AUC, suggesting extended systemic exposure. 46 The absorption rate does not alter much, as evidenced by the Tmax (time to achieve Cmax), which is almost unchanged. Other distribution characteristics, including the volume of distribution (Vd), did not show any significant variations. By blocking CYP3A4-mediated nifedipine metabolism, apatinib raises systemic exposure. This DDI may increase the chance of nifedipine-related side effects, like toxicity or hypotension. 46
Paclitaxel, Zanubrutinib, Oprozomib, and Fedratinib
One cytotoxic drug that is authorized for the treatment of several cancer types is paclitaxel which has many side effects, including neutropenia (low white blood cell count). 47 In order to forecast the incidence of neutropenia in cancer patients receiving paclitaxel treatment, a physiologically based pharmacokinetic–pharmacodynamic (PBPK-PD) model is constructed, particularly taking possible DDIs into account and integrating the metabolic routes via the CYP3A4 and CYP2C8 enzymes. 47 Simulations indicated that the combination of paclitaxel with potent CYP3A4 inhibitors might increase the incidence of neutropenia. 47 When paclitaxel is co-administered with potent CYP3A4 or CYP2C8 inhibitor ketoconazole, the Cmax increases up to two times, as a result of decreased metabolism. Cmax is lowered by CYP3A4 inducer rifampin. 47 Due to decreased clearance when CYP enzymes are inhibited, AUC dramatically rises (by two to three times in the case of CYP3A4 inhibitor ketoconazole and to a lesser amount with CYP2C8 inhibitor gemfibrozil). The toxicity of paclitaxel, particularly the possibility of severe neutropenia, is exacerbated by elevated Cmax. 47 When CYP inducers, like rifampin, are administered concurrently, the AUC is reduced by 50–80%, resulting in subtherapeutic levels. CYP inducers improve clearance, which speeds up the body’s removal of the substance. Changes in clearance affect toxicity as well as efficacy. Because of their slower metabolism and clearance, inhibitors extend the half-life of paclitaxel. On the other hand, inducers shorten it. Extended drug exposure due to a prolonged half-life may result in cumulative toxicity. Since Vd mostly relies on tissue binding and distribution characteristics rather than metabolic pathways, it is less impacted by DDIs. 47 The dosage of paclitaxel may need to be increased with inducers to maintain therapeutic efficacy or decrease d with CYP inhibitors to avoid toxicity. 47
The tyrosine kinase inhibitor zanubrutinib is used in the treatment of B-cell malignancies. 48 The primary enzyme responsible for the metabolism of zanubrutinib is cytochrome P450 3A (CYP3A). Based on its interactions with cytochrome P450 (CYP) enzymes, specifically CYP3A, CYP2C9, and CYP2C19, zanubrutinib exhibits significant changes in important pharmacokinetic (PK) parameters when used in combination with other medications. 48 Optimizing therapeutic approaches requires an understanding of its dual roles as a victim and a perpetrator in DDIs. Physicochemical characteristics, in vitro data, and clinical pharmacokinetic data from patients with B-cell malignancies and healthy volunteers are used to build the PBPK model for zanubrutinib. 48 Strong CYP3A inhibitor (itraconazole), as well as mild inhibitors (erythromycin, diltiazem), raises Cmax if zanubrutinib is a victim (affected by CYP modulators) because they decrease first-pass metabolism and slow systemic clearance. The impairment of metabolism results in a considerable increase in AUC (2–4 times). Because CYP3A-mediated elimination is hindered, clearance is drastically reduced. The longer the drug is in the systemic circulation, the longer its half-life (t1/2) is. Clinically, these could raise the chance of zanubrutinib-related side events such hematologic toxicity, tiredness, or diarrhea. On the other side, CYP3A inducer rifampin raises metabolism, which lowers Cmax. Subtherapeutic exposure results from a large (up to 80%) reduction in AUC. 48 As zanubrutinib is digested more quickly and clearance increases, 48 t1/2 is shortened, indicating a quicker rate of drug removal. Zanubrutinib’s effectiveness is decreased in these circumstances; a dose increase or other treatment may be required. 48
The oral proteasome inhibitor oprozomib was created to treat advanced cancers, such as multiple myeloma, among other cancers. 49 The Jak2 inhibitor fedratinib is used in the treatment of myelofibrosis, a kind of blood cancer. 50 Oprozomib and fedratinib’s drug–drug interaction risks are evaluated using physiologically based pharmacokinetic (PBPK) modeling by Simcyp population-based simulator (Simcyp, Sheffield, UK), with an emphasis on their effects on cytochrome P450 enzymes, to guarantee safe use in patients receiving cancer therapy.49,50 The pharmacokinetic (PK) parameters of fedratinib and oprozomib were simulated under various drug–drug interaction scenarios in the PBPK modeling experiments. When oprozomib and fedratinib are taken with potent CYP3A4 inhibitors (midazolam and ketoconazole, respectively), key PK outcomes include elevated AUC and Cmax, indicating enhanced systemic exposure. Additionally, when co-administered with CYP3A4 inducers like rifampin, simulations revealed a lower AUC.49,50 The fedratinib model also evaluated the effect of CYP2D6 inhibition. These findings inform dose optimization for minimizing toxicity and ensuring efficacy in cancer patients.
Performance of PBPK Modeling in Chemotherapy DDIs
Drug-interaction modeling is only as useful as the criteria against which its predictions are judged. The acceptance limits presented in Figure 3—the traditional two-fold window and the concentration-dependent limits proposed by Guest et al 29 —are used as quantitative yardsticks for deciding whether a simulated change in exposure (e.g., Cmax or AUClast) lies “close enough” to its clinical counterpart for dosing, labeling, or further study. Because chemotherapy regimens are characterized by narrow therapeutic indices and complex polypharmacy, both sets of limits were applied in the present work to provide a balanced assessment of model performance: the two-fold range is regarded as representing the regulatory standard for PBPK verification, whereas the tighter Guest boundaries are intended to reduce false indications of success when the observed DDI ratio is near unity. 29
Predicted-to-observed ratios for Cmax (panel a) and AUClast (panel b) obtained from all chemotherapy DDI studies in Table 2 are presented in Figure 4. Predicted versus observed DDI Cmax (a) and DDI AUClast (b) ratios of given chemotherapy drugs in DDI studies in Table 2.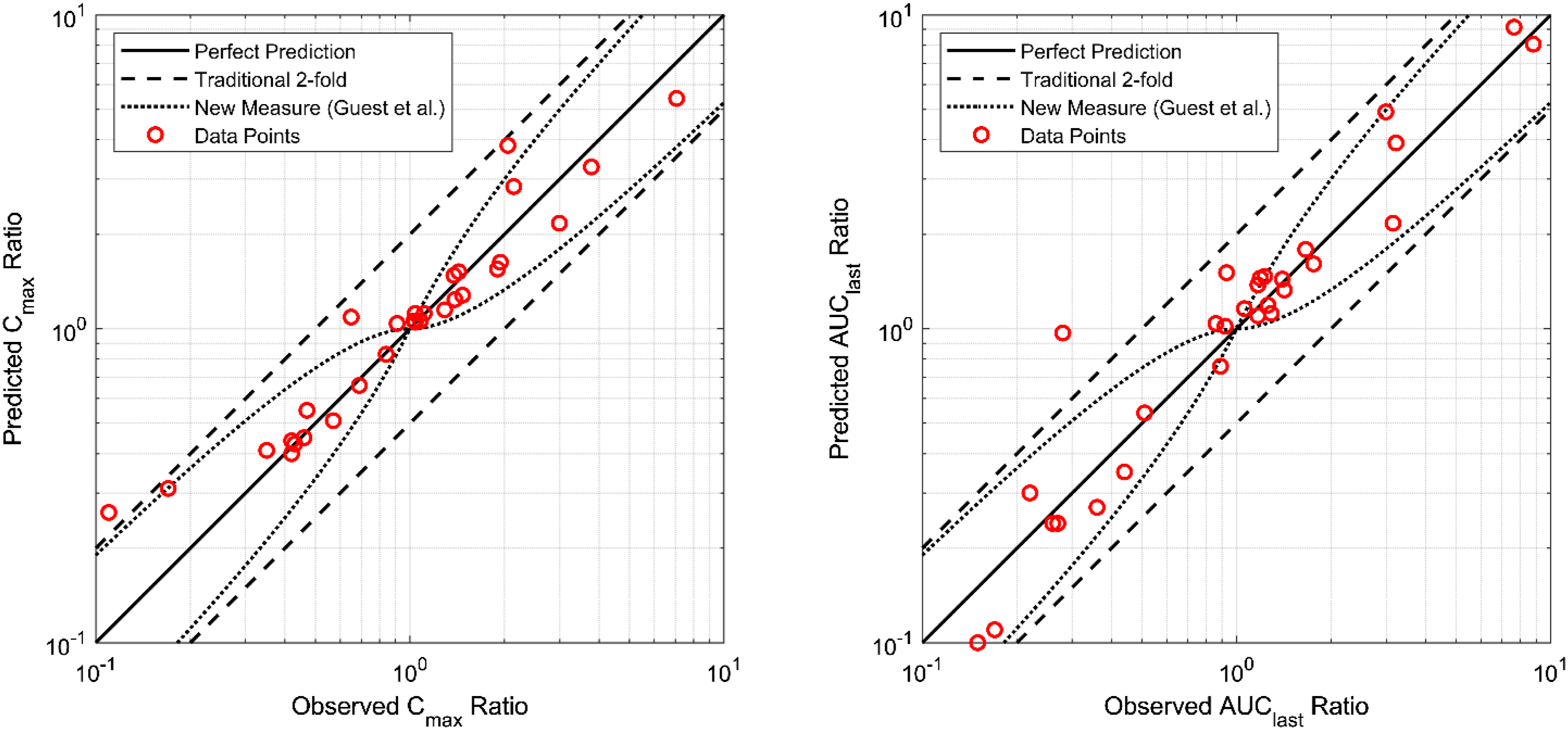
Close alignment with the identity line is evident for most simulations, and interaction magnitudes between 0.1 and 10 are spanned. Specifically, 79 % (n = 34) of the Cmax ratios and 86 % (n = 35) of the AUClast ratios fall within the conventional two-fold acceptance limits, whereas 54% and 61%, respectively, fall within the stricter Guest boundaries. 29 Deviations beyond two-fold are symmetrically distributed above and below unity, so no systematic prediction bias is apparent. Accordingly, the PBPK framework is judged to capture both peak- and exposure-based interaction magnitudes across a broad spectrum of oncology DDIs and is considered suitable for prospective clinical DDI risk assessment.
PBPK-Based Evaluation of Herb–Drug and Food–Drug Interactions in Oncology
Phytochemicals and herbal products have gained increasing popularity in oncology practice for their perceived health benefits and antiproliferative properties. Unfortunately, their abilities to modulate critical drug-metabolizing enzymes and transporters add yet another layer of complexity to cancer pharmacotherapy. Concomitant administration of herbal medications (e.g., St. John’s Wort, Curcuma longa, Schisandra species) with anticancer agents can precipitate clinically relevant herb–drug interactions. These HDIs share mechanistic drivers with classical drug–drug interactions—notably CYP3A4 modulation and P-gp transport—yet are complicated by variable phytochemical content and sparse pharmacokinetic data.23,51 PBPK modeling provides a mechanism-based quantitative methodology to understand how the agents influence the disposition of anticancer therapies and allows forecasting of clinically significant herb–drug and food–drug interactions without exhaustive empiricism. 31
Schisandra Lignans and Targeted Therapy (Tyrosine Kinase Inhibitors)
Pharmacokinetic Effects of Various Phytochemicals/Herbal Medications on Chemotherapy Drugs.
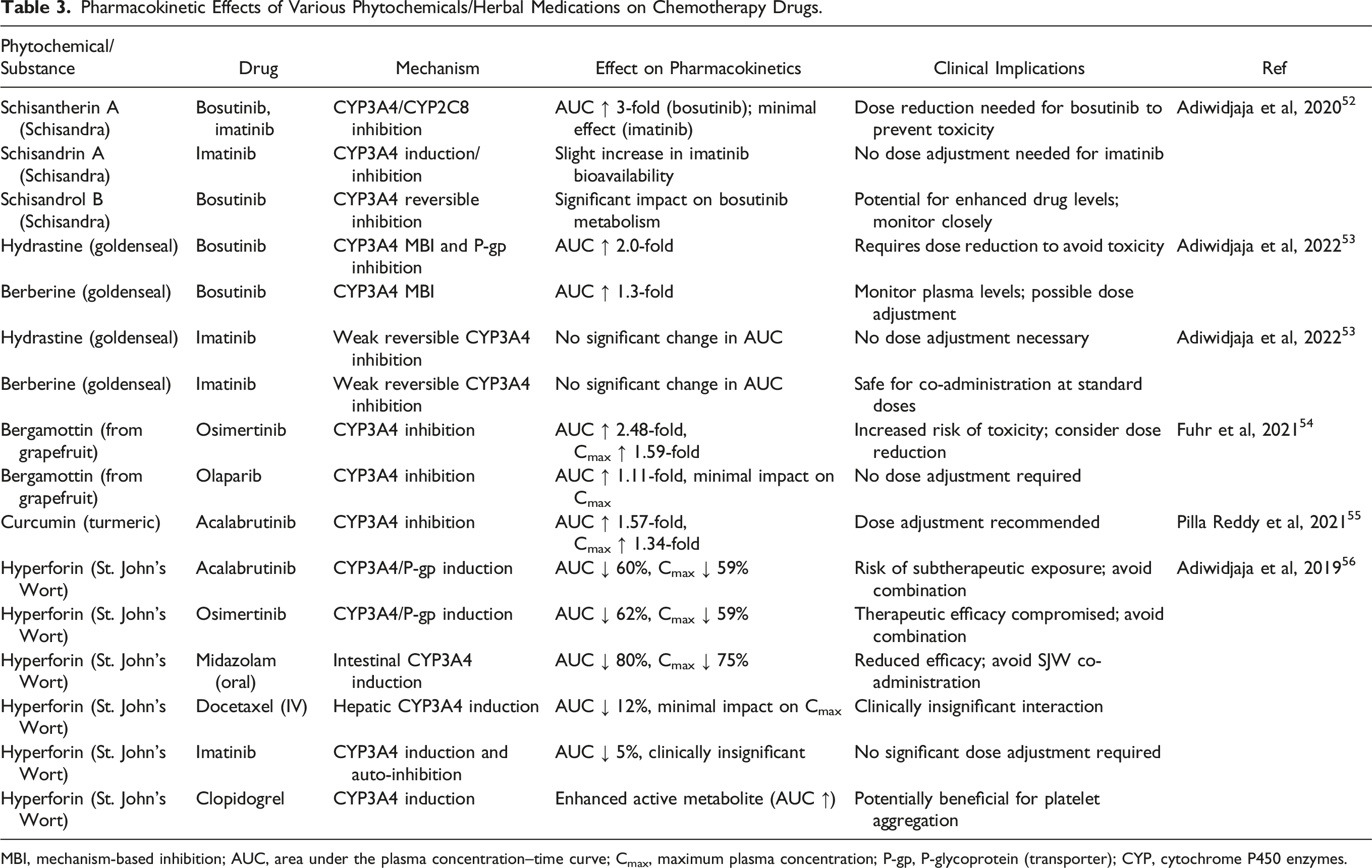
MBI, mechanism-based inhibition; AUC, area under the plasma concentration–time curve; Cmax, maximum plasma concentration; P-gp, P-glycoprotein (transporter); CYP, cytochrome P450 enzymes.
A three-fold increase of the AUC of bosutinib is observed (Table 3) when the extract was co-administered because of the dual effect involving the reversible inhibition and mechanism-based inactivation of the CYP3A enzymes. 52 However, the lignans only slightly altered the pharmacokinetics of imatinib at its steady state, with AUCs only 5% higher. Schisandra lignans may act both as inhibitors and inducers of CYP3A enzymes, depending on the type of interaction mechanism. Such duality is of importance in the context of cancer therapy, since TKIs, such as bosutinib, are highly sensitive to CYP3A modulation. 52 Schisandra might increase bosutinib exposure to potentially toxic levels and thus may require bosutinib dose reductions. On the other hand, the negligible effect on imatinib would indicate selective herbal medicine interactions with drugs driven by metabolic pathways. 52
This study demonstrates that PBPK modeling has the potential to foresee clinically relevant herb–drug interactions of Schisandra sphenanthera when combined with cancer therapeutics. Besides being a very encouraging hepatoprotective agent, its potent change in bosutinib pharmacokinetics may lead to adverse effects. Thus, combination therapy of Schisandra with CYP3A4-metabolized cancer drugs requires dose adjustment with careful monitoring; however, the drugs like imatinib are not affected significantly.
Goldenseal Alkaloids and Targeted Therapy (Tyrosine Kinase Inhibitors)
Berberine, hydrastine, palmatine, and smaller levels of canadine and hydrastinine are among the alkaloids found in the dried root or rhizome of goldenseal (Hydrastis canadensis L.). Goldenseal-based preparations have been used to treat conditions affecting the skin and eyes. PBPK models of two key goldenseal alkaloids, hydrastine and berberine, were created with great accuracy to predict the degree of CYP3A- and ABCB1-mediated drug interactions with goldenseal extracts and high-dose berberine.24,25,53 Clinically relevant dosages of goldenseal extract and berberine supplements were anticipated to have no effect on imatinib pharmacokinetics while increasing systemic exposure to bosutinib by up to 2.0 and 1.3 times, respectively. 53 The interactions were attributed to a reduction in hepatic clearance and alterations in transporter-mediated drug efflux. However, imatinib’s exposure was unaffected due to its lower dependency on CYP3A4 and auto-inhibition of its own metabolism. Bosutinib dose reductions may be required to maintain exposure within the defined therapeutic range, although more clinical trials to confirm these interactions and their consequences for clinical outcomes are warranted. 53
PBPK model results underline the clinical importance of goldenseal alkaloids in altering tyrosine kinase inhibitor pharmacokinetics, especially for CYP3A4-dependent drugs such as bosutinib. 53 These results signify that dose adjustments of bosutinib are essential when combined with goldenseal products to evade its adverse effects. For imatinib, the absence of significant interactions seems to present a relatively safer profile for concurrent use with goldenseal extracts. Therefore, this study represents an important way to understand how herb–drug interactions can be managed in cancer therapy using PBPK modeling.
Food-Derived Phytochemicals: Bergamottin, Curcumin, and Hyperforin
PBPK modeling is used to investigate the interactions of common phytochemicals derived from food with other oncology drugs. These included bergamottin, curcumin, and hyperforin (found in grapefruit juice, turmeric, and St. John’s Wort, respectively) acting on anticancer drugs.9,55 These agents act by inhibiting hepatic drug-metabolizing enzymes and transporters such as P-gp and BCRP (Table 3).
According to the results of PBPK models, bergamottin increases the AUC of osimertinib (used to treat non-small cell lung cancer (NSCLC)) approximately 2.5-fold, and curcumin increases acalabrutinib (used to treat mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), and small lymphocytic lymphoma (SLL)) AUC by 1.57-fold. 55 On the other hand, the induction of both CYP3A4 and P-gp by hyperforin lowers the acalabrutinib and osimertinib AUC by 60%–62% which can lead to subtherapeutic drug levels.
St. John’s Wort (SJW) interactions with pharmacological drugs are investigated via its main ingredient, hyperforin, using physiologically based pharmacokinetic modeling and in silico analysis. 35 Researchers demonstrated that hyperforin induces intestinal CYP enzymes such as CYP3A4, CYP2C9, and CYP2C19, as well as ABCB1 transporters (Table 3). This method significantly decreases bioavailability and, as a result, the therapeutic efficacy of any given medicines metabolized by these pathways. This was shown to be effective against a variety of CYP substrates, including imatinib, midazolam, and docetaxel that are used to treat certain types of cancers (breast, lung, prostate, stomach, head, and neck). Furthermore, with ratios of 15.5 to 1.1, respectively, it demonstrated a considerable preference for intestinal induction over hepatic induction. Imatinib, a CYP3A substrate, had a lower impact due to its auto-inhibitory action, resulting in a clinically insignificant steady-state effect. 56
Concerns about possible interactions between SJW and cancer treatments that rely on CYP3A4 metabolism or P-gp transport are brought to light by literature studies. However, with prodrugs such as clopidogrel, increased metabolic activity may result in a superior therapeutic response, even if such pathway activation restricts drug intake, as demonstrated with midazolam and docetaxel. 56
Artificial Intelligence (AI) and Drug–Drug Interactions
The fusion of artificial intelligence (AI) with physiologically based pharmacokinetic (PBPK) modeling is rapidly redefining how oncology teams anticipate, quantify, and ultimately manage DDIs. Classic PBPK workflows—built on in vitro and in vivo parameter estimation—are now being augmented by machine-learning (ML) algorithms that uncover latent relationships, tighten parameter uncertainty, and drive more reliable forward simulations. 57 Comparative work by Gill et al shows that combining ML with PBPK or population-PK (PopPK) frameworks improves both interpretability and clinical utility, while Chou and Lin demonstrate how ML aids feature selection, parameter fitting, and virtual-population generation, thereby reducing reliance on labor-intensive experimental datasets.57-60
Across representative studies, ML models built with random forests, support-vector machines, or deep neural networks routinely predict key ADME parameters with high fidelity (R2 >0.8, RMSE <0.5) and, once embedded in PBPK simulators, yield exposure metrics (AUC, Cmax, Vd) that match—or surpass—traditional mechanistic efforts. 59 Particularly noteworthy is the Neural-ODE architecture, which can learn irregular time-series PK trajectories directly from data, opening a path toward fully data-driven PBPK simulations that require minimal prior structural assumptions. 59
A prominent application is ML-assisted PBPK hybridization for whole-body human PK prediction. Independent investigations by research groups confirm that such hybrids accurately reproduce absorption, distribution, metabolism, and excretion across diverse chemotypes and dosing routes, outperforming standalone statistical or mechanistic models in both scalability and adaptability.61-63
Complementing these technical advances, a wider AI-DDI field has recently been catalogued. 64 After quantifying the clinical burden of interaction-related harm, the researchers mapped the data landscape—DrugBank, ChEMBL, PubChem, TWOSIDES, DDInter, and others—that now feeds multimodal features (structures, targets, pathways, side effects, real-world reports) into AI pipelines. 64 Methods fall into three tiers: (i) undirected (“interact or not”) models that rely on similarity scores or matrix propagation; (ii) event-level systems (e.g., DeepDDI, MDF-SA-DDI) that use transformers or GNNs to label tens of PK/PD outcomes; and (iii) asymmetric approaches (e.g., DGAT-DDI) that capture perpetrator to victim directionality. 64 On public benchmarks, the best binary tools exceed AUC 0.90, and leading multiclass models approach F ≈ 0.8 despite much larger label spaces. 65
Although momentum is strong, several obstacles must be overcome before AI-DDI tools can be deployed with confidence at the bedside. First, most deep models lack interpretability, preventing clinicians from understanding which molecular or clinical features drive an interaction alert. Second, the data are exceedingly imbalanced: confirmed DDIs represent a tiny fraction of all possible drug pairs, and many presumed “negatives” may simply be unreported positives, biasing model training. Third, no harmonized, gold-standard corpus exists—research groups curate different slices of DrugBank, FAERS, and other sources—so results are difficult to compare or validate. Finally, current algorithms incorporate little patient-specific context such as genotype, organ function, comorbidities or dosing schedules, even though these factors critically influence interaction risk. Addressing these four gaps is essential for translating AI-driven insights into reliable, personalized pharmacotherapy. 66
Future Perspectives: Potential for Personalized Medicine
DDIs that occur during cancer treatment remain a significant challenge, particularly as treatment protocols increasingly integrate multiple oncologic agents, adjuvant medications, and various phytochemical or nutritional supplements. In fact, PBPK modeling has demonstrated remarkable utility across a broad range of clinical scenarios—from tyrosine kinase inhibitors and conventional chemotherapeutics to novel targeted therapies—by predicting the magnitude and directionality (induction vs inhibition) of potential interactions. Such predictions, derived by combining in vitro and in vivo data, enable evidence-based guidance on avoiding potent enzyme inducers or inhibitors, timing administration around gastric pH modifiers, and adjusting dosages when herb–drug or food–drug interactions threaten safety or efficacy (Table 3).
PBPK modeling can be used to anticipate and address issues that reduce therapy effectiveness. For example, when cancer cells or the surrounding tumor microenvironment adapt and avoid immunosurveillance through mechanisms that reduce the therapeutic impact of PD-L1 inhibition, PD-L1 treatment resistance develops. PBPK-guided dose modifications may lessen specific resistance patterns associated with insufficient drug exposure in the context of PD-L1 inhibitors. However, complex immunobiological pathways that necessitate a wider range of approaches—from combination regimens to biomarker-based treatment optimization—are frequently involved in immune checkpoint therapy resistance. As a result, whereas PBPK-driven dose modifications can be very helpful in treating some aspects of PD-L1 inhibitor resistance, coordinated, mechanism-based treatment approaches yield the best outcomes.
Beyond the traditional applications, PBPK models also have the potential to reveal rare or underreported interactions caused by newly approved drugs, natural supplements, and off-label therapy, thus helping clinicians avoid such unanticipated and potentially lethal risks. Moreover, when combined with AI and machine learning, models can identify hidden patterns in pharmacokinetic data because its predictability increases. The synergy of PBPK modeling and advanced analytics with proactive clinical planning might well help shift treatment strategies from a reactive problem-solving approach to a more preventive, individualized one.
PBPK modeling has the potential to become a key component of precision oncology in the future. PBPK-informed decisions can improve clinical outcomes, optimize therapeutic efficacy, and strengthen patient safety by empowering clinicians to make data-driven changes instead of depending on indirect or sparse evidence. This ensures that all possible advantages are realized in the continuous endeavor to improve cancer care.
Conclusion
The possibility of pharmacokinetic and pharmacodynamic interactions is growing as polypharmacy and new therapeutic drugs are used more frequently. These interactions have the potential to worsen toxicity, impair patient outcomes, and have a substantial effect on therapeutic efficacy. Therefore, improving cancer therapy requires a thorough understanding of DDIs. To reduce hazards and improve therapeutic success, future research should concentrate on developing predictive tools, improving medication monitoring procedures, and incorporating personalized medicine approaches. To reduce the negative effects of DDIs and enhance the safety and effectiveness of cancer treatment, cooperation between pharmacologists, researchers, and clinicians is essential.
Footnotes
Author Contributions
E.E.T.: Conceptualization and design, Data acquisition, analysis, interpretation, Writing - drafting, Final approval, Accountability for integrity and accuracy; K.O.U.: Conceptualization and design, Data acquisition, analysis, interpretation, Writing - review and editing, Final approval, Accountability for integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors gratefully acknowledge the support from the Scientific Research Projects (BAP) Coordination Unit (Project No 20161).