
Abstract
Seizures are complex electrophysiological disturbances affecting one or more populations of brain neurons. Seizures following test article (TA) exposure pose significant challenges in drug development. This paper considers the diverse neurological manifestations, mechanisms, and functional and structural assessments needed to investigate TA-related seizure liabilities, with a particular focus on nonclinical species. Accurate discrimination of seizures from convulsions (irregular involuntary body and/or limb movements) and the nuanced presentation of different seizure types (partial vs. general) and phases (prodromal, ictal, and postictal) are essential for discerning their clinical implications. In nonclinical safety testing, the most direct evaluation method to confirm existence of seizures is electroencephalography (EEG) while clinical endpoints (e.g., functional observational batteries [FOB], comprehensive neurological examinations) and neuropathological findings (e.g., neuronal necrosis in tissue sections, raised biomarker levels in cerebrospinal fluid or serum) can indicate a seizure liability and provide additional guidance to identify the origin, frequency, and severity of seizures needed to align nonclinical effects with clinical relevance. In general, the regulatory perspective is that seizures identified in nonclinical species as well as potential risk management strategies (e.g., safety margin considerations, dosing paradigms, and clinical monitoring) translate effectively for purposes of clinical risk assessment.
The landscape of biomedical product development is continually evolving, marked by advancements in technology and a deeper understanding of potential adverse reactions. Despite significant efforts in nonclinical safety assessment, no current regulatory guidelines specifically address how to investigate the translational relevance (if any) of potential seizures associated with exposure to various test articles (TAs) including conventional drugs (small molecules) as well as biologics (nucleic acids and proteins), cell and gene therapies (including gene editing), medical devices, vaccines, and the like. With respect to drugs, the implementation of the International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) S7A guideline on safety pharmacology studies 1 has reduced post-market withdrawals due to belated recognition of seizurogenic potential. High levels of TA attrition also persist in premarket development of drugs targeting diseases of the central nervous system (CNS), thus underscoring the need for more reliable approaches to assessing seizure risk.
Seizure liability is difficult to investigate in the nonclinical setting. For example, a survey of drugs approved in Japan between 1999 to 2013 indicated that the majority of products with reported adverse drug reactions (ADRs) classified as seizures/convulsions in patients were not identified during nonclinical studies to have a seizure liability using conventional methods of evaluation (e.g., clinical observations, drug distribution, fluid biomarkers of organ damage, macroscopic and microscopic pathology findings). 2 Drug-induced seizures cut across diverse pharmacological classes and therapeutic indications and are not limited to agents designed to enter and act in the central nervous system (CNS); indeed, for a compilation of 266 TAs associated with seizure liability in nonclinical studies, approximately half of the seizurogenic agents were targeting non-CNS diseases. 3 Spontaneous seizures are reported in various species and need to be differentiated from drug-induced seizures, 4 adding complexity to the translation of nonclinical results to humans. This discordance between typical nonclinical and clinical outcomes underscores the need for a comprehensive evaluation of seizure risk during early stages of drug development. Regrettably, current industry practices for nonclinical assessment of seizure liability lack standardization, emphasizing the urgent need to bridge this knowledge gap.
Identification of seizure liabilities from nonclinical data to inform translation of risks to humans is an area of intense interest among biomedical product developers and regulators because of the safety risks posed to patients (and healthy volunteers) by brain dysfunction during clinical trials. This comprehensive review delves into major clinical manifestations, biological mechanisms, and preferred batteries of functional and structural assessments needed to evaluate TA-related seizure liabilities, with a particular focus on nonclinical safety studies for traditional drugs (small molecules). In particular, the content of this paper is based on a ½-day continuing education course entitled “Identifying and Understanding Seizure Liability in Pharmaceutical Development” that was first delivered in November, 2022 at the annual meeting of the American College of Toxicology (ACT) and then reprised in expanded form as a ½-day virtual continuing education course held in April, 2024 under the auspices of the Society of Toxicologic Pathology (STP). The goals of this paper are (1) to provide an understanding of seizure biology, including mechanisms and neurological manifestations, (2) to describe usual functional and structural findings in nonclinical studies indicating a possible seizure liability, (3) to understand the essential design parameters of a pivotal nonclincial study to assess seizure potential, and (4) to offer insights regarding selection and implementation of risk mitigation options to alleviate seizure liability in clinical trials. The information in this review seeks to craft an integrated model regarding how to detect and curtail the likelihood that novel TAs may pose a seizure risk to patients while defining the limits of existing state-of-the-art methods to inform drug discovery and development. Methods for predicting risk early in discovery for innovative product candidates with uncharacterized seizurogenic potential have been discussed previously. 82 While not considered further in this review, other biomedical product classes (biologics, cell and gene therapies, medical devices, vaccines, etc.) exhibiting a potential seizure liability may be assessed using the same principles and equivalent practices to those described here for small molecule drugs.
Basic Seizure Biology: Classifications and Mechanisms
Neurological signs observed in the course of clinical trials are considered to be adverse events (AEs), and with sufficient intensity are categorized as serious adverse events (SAEs). Because seizures occur commonly in the general population, 5 some seizures observed during clinical trials may be spontaneous events rather than sequelae induced by administration of a neuroactive TA. Therefore, identification of the mechanisms responsible for seizures and their relationship (if any) to TA exposure is a crucial consideration due to the inherent risk to patients associated with seizures.
Classification of Seizures
Several categories of in-life neurological events must be discriminated during the nonclinical and clinical evaluation of TAs for possible seizure liabilities (Figure 1). 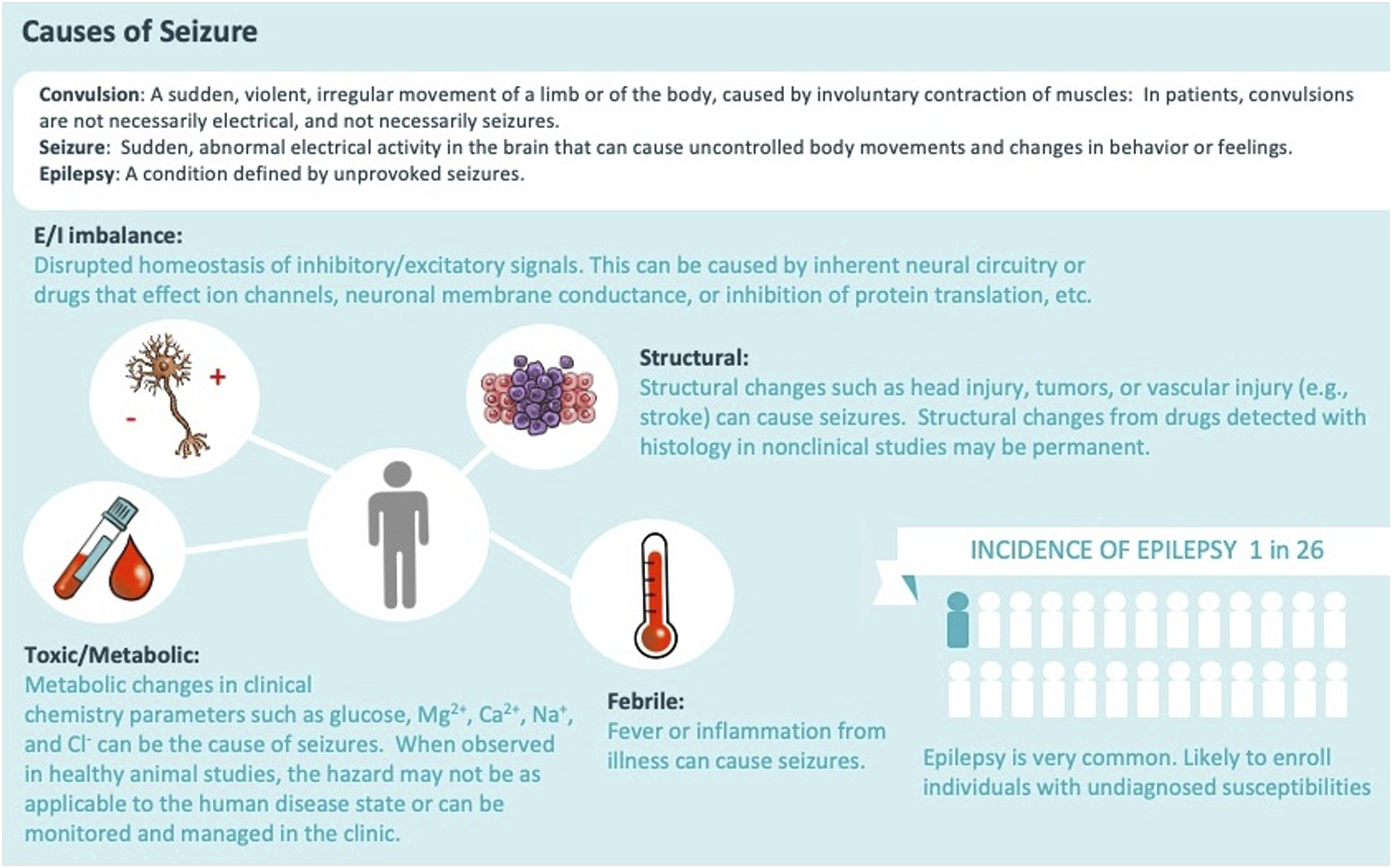
Seizure patterns depend on the nature of the brain injury (Figure 2).
6
For example, 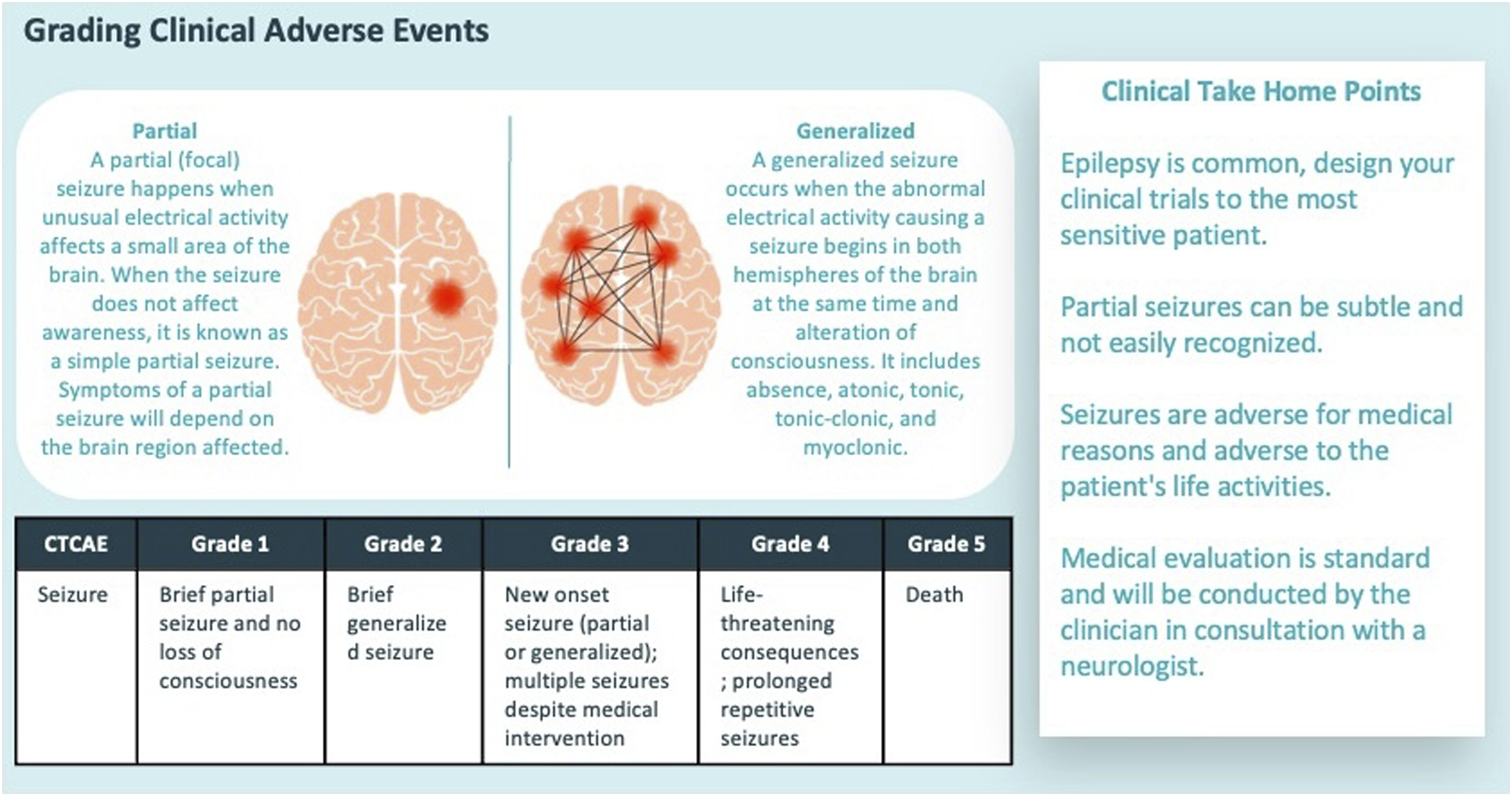
Some seizures exhibit a stereotyped progression with definable phases (Figure 3).12,13 Focal seizures often start with an 
Mechanisms of Seizures
Anything that disrupts normal brain function can cause seizures. The major causes differ in various patient populations (Figure 1). 9 In newborns and infants, common causes include birth trauma, congenital (i.e., present at birth) problems, fever (typically related to infection), genetic disorders, or systemic metabolic imbalances. In children, adolescents, and young adults, seizures are most commonly related to alcohol or illicit drug use, brain injury (often a consequence of head trauma), genetic conditions, infection, or late-manifesting congenital defects. In mature and elderly adults, seizures usually result from alcohol or illicit drug use or withdrawal, brain neoplasms (primary or metastatic), neurological conditions (e.g., dementia, multiple sclerosis, stroke), and some prescribed medications. Similar causes are posited for seizure induction in animals except that voluntary self-exposure to illicit drugs or prescribed medications is not a consideration in nonclinical species.
The potential for a neuroactive TA or metabolite to incite a seizurogenic effect is tied to its ability to access the brain. Entry of a blood-borne molecule into the brain depends on traits of both the TA and the blood–brain barrier (BBB). 3 Molecule-related properties include electrical charge, hydrogen-binding capacity, lipid solubility, and molecular size. 14 Modifications to TAs may be engineered to facilitate penetration of the BBB. 15 Given the absence of paracellular and transcellular channels in the BBB, circulating molecules generally reach the brain interstitial fluid and parenchymal cells via either lipid-mediated free diffusion (a slower process) or a transport system. Most non-polar lipid-soluble molecules <500 Da in mass (e.g., ethanol, oxygen) readily cross the BBB by passive diffusion through endothelial cells (which requires crossing the lipid-rich luminal and abluminal plasma membranes) or via transcellular channels between endothelial cells (which necessitates transit through cell adhesion complexes formed by adherens junctions, gap junctions, and tight junctions). Charged, hydrophilic, and/or large entities typically require a molecule-specific mechanism (e.g., active, carrier-mediated, or receptor-mediated transport) to penetrate the BBB. The BBB is largely intact during health. In contrast, the BBB structure may be disrupted by many factors including properties of the TAs (e.g., lipolytic or surfactant activity) or existing damage to the brain parenchyma (e.g., epilepsy, hypertension, and neurodegenerative diseases). Notably, BBB malfunctioning may result in higher-than-expected brain exposure.
Several brain neuron populations have been closely linked to seizure induction, including neuroactive TAs.
16
Historically, affected cells have been considered to be either excitatory neurons using the neurotransmitter glutamate or inhibitory neurons using the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). The
Recently, the
Clinical Evaluation of Seizures
Seizure patients undergo a battery of medical tests to define the cause(s) and mechanism(s) of their condition as well as to chart a course of therapy. The same tests are used for both new-onset seizures and individuals with established epilepsy. This section describes test options in the context of patients who exhibit seizures during clinical trials of neuroactive TAs.
Implications of Seizures during Clinical Trials
For the most part, epilepsy is an exclusion criterion for clinical trials involving neuro-targeting TAs. The clear exceptions are clinical trials for anti-seizure medications (ASMs). In general, patients with epilepsy must have refractory seizures that are unable to be adequately controlled after multiple trials of currently approved ASMs in order to take part in clinical trials of new ASM candidates. Thus, epileptic patients participating in clinical trials for ASMs suffer more than one seizure each week with prominent motor signs. As such, these patients are not evaluated medically if they merely exhibit their usual numbers of seizures in the course of the trial.
In contrast, if a subject without a history of epilepsy has a seizure during a clinical trial, they are evaluated and treated similarly to any other patient with a first-time seizure. Because epilepsy is so common in the general population, a seizure that occurs during a clinical trial may represent new-onset epilepsy rather than exposure to the neuroactive TA. Therefore, standard principles of medical evaluation will apply in order to distinguish TA-induced and spontaneous (background) epilepsy. The conventional menu of methods used to evaluate epilepsy in patients consists of a neuro-imaging procedure, an EEG evaluation, and (where warranted) assessments of potentially seizurogenic agents including endogenous metabolites (at abnormal levels) and accumulation of neurotoxic TAs and their principal neuroactive metabolites.9,11
Clinical Methods for Evaluating Seizures
Initial evaluation for a first-time seizure event includes collecting the patient’s history and physical examination. The medical history should target signs and symptoms that indicate the potential for epilepsy. Major clues in the history include prior symptoms of sub-acute seizures, a family history of seizures, and toxic/metabolic conditions. The physical exam involves both a comprehensive neurological exam for subtle signs of lateralization (i.e., signs indicating that the seizure originates on one side of the brain) and an assessment of trauma resulting from seizure episodes. These two evaluations are complimented by laboratory tests including a complete serum chemistry (i.e., metabolic) panel, a toxicology screen (if deemed relevant), and if feasible analysis of blood concentrations for the TA being tested in the clinical trial. The history, physical exam, and initial serum chemistry panel are obtained in the Emergency Department (ED) while a more detailed history, the comprehensive neurological exam, and expanded laboratory testing are undertaken by the Neurology Department (ND).
Magnetic resonance imaging (MRI) is the neuro-imaging gold standard for finding structural abnormalities that may underlie seizures. Different imaging conditions and image resolutions may be employed to highlight distinct brain structures.9,11 Depending on the level of concern for malignancy, the MRI may be performed with a contrast agent. Computed tomography (CT) may also be used and is particularly helpful for evaluating the extent of acute trauma (e.g., fractures leading to superficial brain damage, hemorrhage in the brain meninges or parenchyma). A CT scan is faster and so is often the diagnostic imaging modality of choice in the ED while slower high-resolution MRI scans are typically ordered by the ND.
An EEG study may be performed after a first-time seizure. For toxic/metabolic exposures, EEG abnormalities indicative of encephalopathy (i.e., diffuse slowing of brain electrical activity) should return to baseline after the resolution of the medical condition. In patients with epilepsy, regression of electrical abnormalities may be delayed or incomplete. A standard EEG study may exhibit wave patterns pointing to epilepsy even when the patient is not actively seizing, such as focal epileptiform abnormalities (e.g., sharp, slow, or spike waves characteristic of IED); such features pre-dispose the patient to unprovoked recurrent seizures in the future. Certain structural anomalies that disrupt one or more brain regions (e.g., developmental malformations, infarcts, masses [abscesses, neoplasms, parasitic cysts]) also may cause focal areas of “slowing” in the EEG wave pattern.
In general, neuropathological evaluation in patients with seizures is focused on assessment of fluid biomarkers rather than microscopic examination of tissue sections. For biomarker analysis, the usual samples are blood and/or cerebrospinal fluid (CSF). Neuron markers of interest include proteins, such as neurofilament light chain (NfL), S100β, and tau; enzymes, like neuron-specific enolase (NSE); and various microRNAs (miR). The presence of these molecules is indicative of neuronal injury (non-lethal or lethal) leading to plasmalemma damage and release of intracellular constituents. Alternatively, high fluid levels of a glial component like glial fibrillary acidic protein (GFAP, a biomarker for astrocytes) is often indicative of a secondary glial reaction to primary parenchymal injury. These fluid biomarkers may be elevated in many neurological conditions and therefore are not specific for seizure disorders.20–24 Diagnostic and/or excisional biopsy specimens may be acquired by conventional neurosurgical techniques in certain cases of epilepsy. Diagnostic biopsies are a relatively non-invasive approach used to investigate cause(s) and mechanism(s) in patients (usually pediatric) with severe, often intractable seizures. In such instances, one or a few small tissue cores are acquired from the brain area—usually the cerebral cortex—thought to represent the seizurogenic nidus (as defined by prior EEG, neuro-imaging, and/or neurological studies). In contrast, excisional biopsies involve more aggressive tissue removal in situations where a clear underlying structural lesion (e.g., neoplasm) has been identified, and must be extirpated as part of the therapeutic course. Macroscopic findings may occur in patients with chronic epilepsy, including attenuated cerebellar folia (termed “cerebellar atrophy” or “cerebellar degeneration”) and/or hippocampus (termed “hippocampal sclerosis”).25-27 These gross changes result from regional neuron loss with subsequent astrocytic and microglial proliferation, and they may be visible as well during life by MRI.28,29
Nonclinical Evaluation of Seizure Liability during Pharmaceutical Development
Pharmaceuticals vary in their capacity to induce seizures. Certain molecular attributes raise the likelihood that a TA will carry a seizure liability, and that liability may be explored during the discovery phase of drug development. This section provides points to consider in designing a nonclinical safety strategy to develop a TA with either a proven or biologically plausible ability to induce seizures. In particular, TAs with seizurogenic potential are expected to cross the BBB, interact with cellular targets known to disrupt brain electrophysiological processes, and/or belong to a product class with known seizure liability.
Program toxicologists can preemptively modify product development programs if a seizure liability is suspected or detected prior to initiation of Good Laboratory Practice (GLP)-compliant nonclinical toxicology studies. For example, further in vitro screening may be undertaken prior to launching additional in vivo studies to de-risk TAs if a seizure liability is suspected based on the agent’s molecular structure, mechanism of action (MoA), or chemical class effect. Similarly, electrophysiological studies (e.g., micro-electrode array recordings of neurons in brain slices or animals) may be performed to screen or characterize one or a series of high-risk molecules for any seizurogenic potential; in particular, agents may be rank-ordered to identify less risky candidates or define chemical moieties suitable for re-engineering. Such ancillary screening assays are de-risking strategies that may improve the likelihood of success during in vivo testing. However, at present no in vitro assays are available to de-risk TAs once a seizure liability has been observed in whole animal models. 30
Nonclinical Approach to Hazard Identification and Risk Characterization for Seizure Liability
Hazard identification approaches as applied to detecting potential seizure liability may be approached using the standard nonclinical safety strategy described in ICH M3(R2)
31
(Figure 4). Across the distinct types of in vivo studies conducted to support the clinical development of drug candidates, any nonclinical data may suggest potential seizure liability, including pharmacology and toxicology studies. However, as in the clinical setting, seizures in animals can be definitively confirmed only by detecting altered brain electrical activity (EEG); such electrophysiological assays are not standard practice in conventional safety pharmacology and general toxicology studies conducted for drug development. Therefore, in a practical sense identification of seizure liability during conventional nonclinical safety programs is restricted to certain in-life signs indicating altered neurological function (e.g., ataxia, convulsions, muscle tremors or twitching) and/or post mortem demonstration of neuron death in brain domains with a known vulnerability to seizures. Here, we discuss what types of nonclinical data may indicate a seizure liability.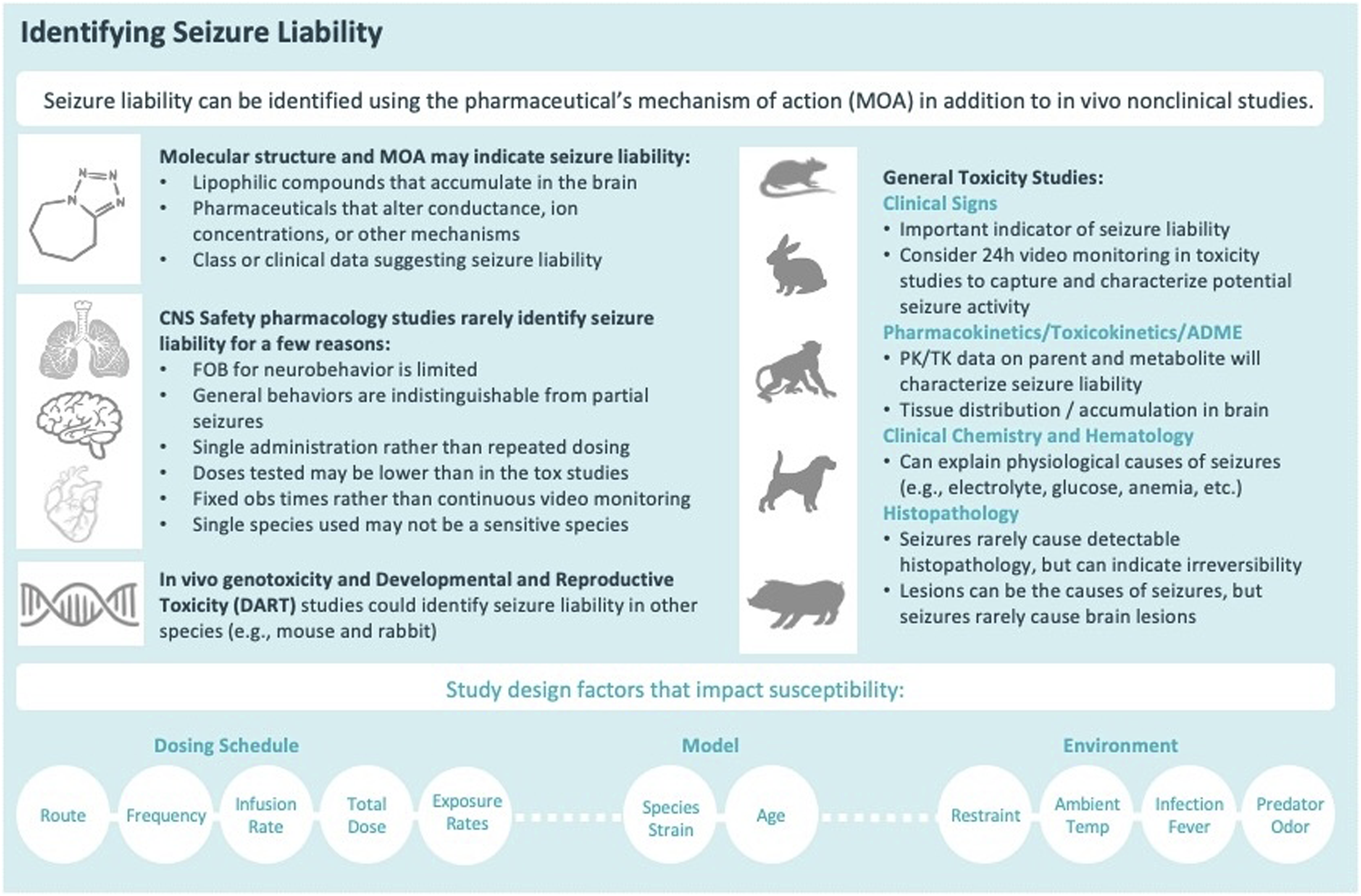
As part of a routine nonclinical drug development program, seizure liability as a hazard can be identified based on data from various types of in vitro and in vivo studies. Relevant in vitro data may be obtained from discovery pharmacology or safety pharmacology studies designed to characterize the activity of TAs at intended and unintended targets. Recently, attention has been placed on in vitro and ex vivo assessment of seizure liability, including multi-electrode arrays (MEA) on ex vivo hippocampal slices, human induced pluripotent stem cell (hiPSC)-derived neuronal cultures, larval zebrafish locomotor assays, and ion channel panels.30,32 However, these methods are still limited to predict some drug-induced seizures that arise from stress or inflammation. While such methods may be helpful for early discovery screening, they are not sufficient to derisk a seizure liability identified in vivo. 30 Prior to first-in-human (FIH) clinical trials, Investigational New Drug (IND)-enabling in vivo data that suggest a potential seizure liability are generally limited to in-life clinical signs in safety pharmacology and short-term (up to 13 weeks) repeat-dose general toxicology studies as EEG is not a standard assay performed routinely during nonclinical drug development. Clinical signs suggestive of a seizure may occur at any in vivo stage of drug development including specialized experiments like genetic toxicology, developmental and reproductive toxicology (DART), and carcinogenicity studies.
For risk characterization, nonclinical assessment of TAs for a potential seizure liability may be characterized in various ways to ensure that an accurate and detailed narrative is developed for corporate managers, regulatory agencies, and others who make key product development and safety decisions. Ideally, this narrative should establish that the hazard is related to TA exposure; define its relationship to the TA (e.g., dependence on the TA dose, frequency, repeatability, and timing of TA administration); outline the features of the neurological dysfunction (e.g., the pattern and persistence of in-life clinical signs); and illuminate any correlation of seizures to other toxicities. In particular, comprehensive descriptions of neurological signs and the TA dose-response should be presented with other information on brain function such as clinical pathology data (e.g., fluid-based biomarkers of neural cell injury) and electrophysiological (EEG) data when available. In general, routine anatomic pathology data reporting structural lesions in various brain regions is less informative when characterizing seizure causes and mechanisms in the nonclinical setting. 33 Pharmacokinetic (PK) data matching blood (or less often CSF) concentrations of neuroactive TA and any principal metabolites also provide useful context for understanding seizure liability.
Nonclinical Methods for Evaluating Seizure Liability
As noted above, a plethora of data will be generated for routine nonclinical pharmacology and toxicology studies that, when integrated, will provide a reasonable understanding of the extent and possible severity of any potential seizure liability posed by exposure to a novel TA. This section provides a brief but detailed explanation of the main data types that are collected in various nonclinical studies and their utility in translating animal-derived data for assessing the potential risk of TA-related seizures when patients are to be exposed to new drugs candidates.
In-life Evaluation in Assessing Seizure Liability
In conventional nonclinical studies, the most concerning in-life clinical signs that suggest a potential seizure liability are overt convulsions and 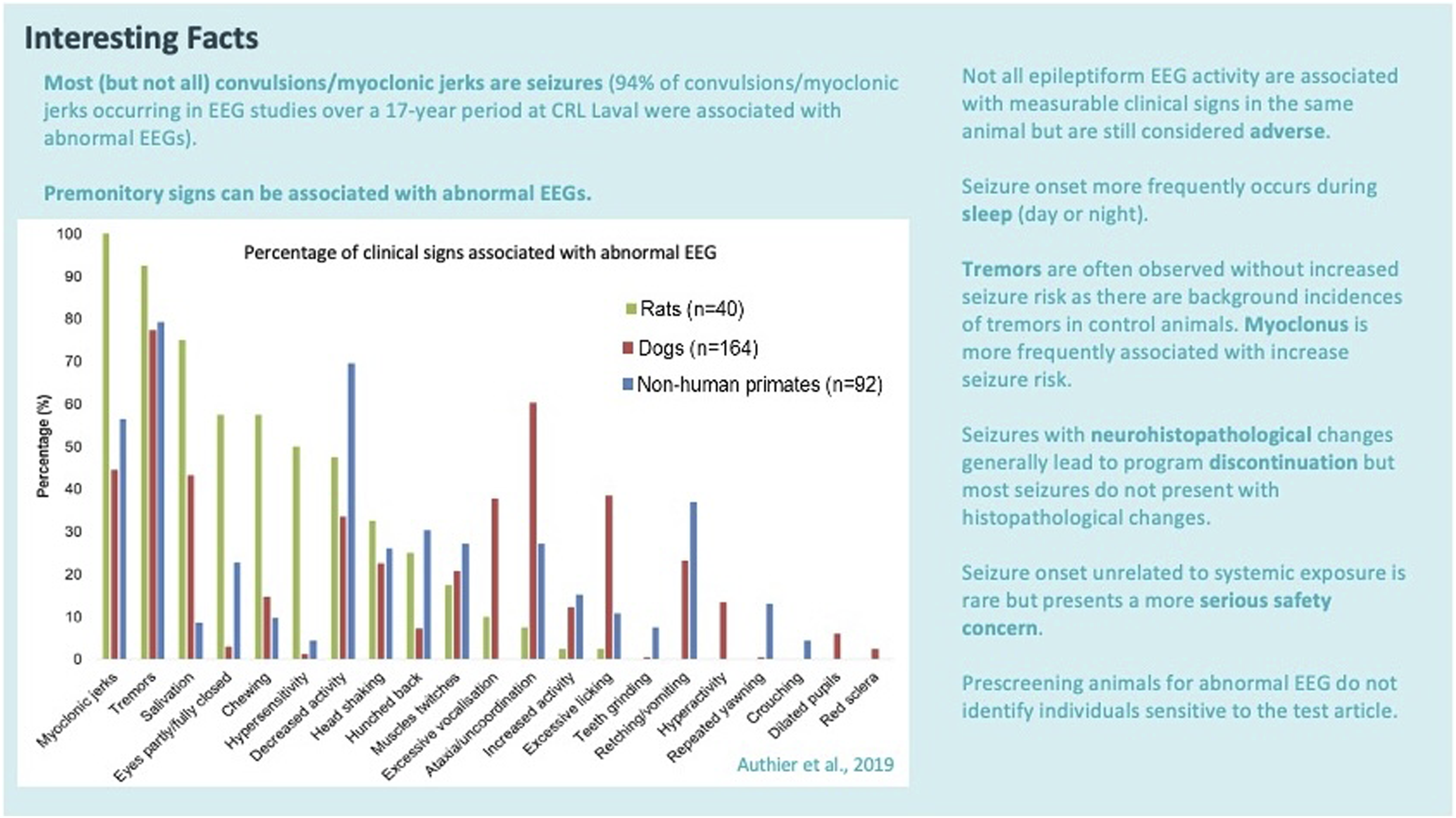
Accordingly, nonclinical drug development programs faced with a potential seizure liability generally may require additional investigation to characterize clinical signs that are accompanied by corresponding EEG abnormalities. Such information is essential when translating animal data to assess whether patients might be at risk for TA-related seizures. Neuro-imaging methods (e.g., micro-CT, micro-MRI) are technically feasible in conventional test animals 34 but are rarely used in nonclinical pharmacology and toxicology studies due to their cost (in terms of labor, money, and time) and low throughput. Similarly, EEG procedures are readily adapted for use in animals, but this technology is not used as a first-line screen in nonclinical studies due to the cost and low throughput. Nonetheless, an EEG analysis that specifically measures brain electrical activity is the sine qua non for demonstrating the absence of a seizure liability when the pattern of clinical signs includes convulsions, myoclonus, and/or a collection of less specific functional alterations. For this reason, details regarding design elements used in nonclinical EEG studies are described in later sections.
Clinical signs related to seizures are often intermittent, and therefore they may not be seen at the once- or twice-daily observation periods used for standard nonclinical toxicology studies. For this reason, 24 h video-monitoring may be important to document findings during times when technical staff are absent, thereby providing confidence that the full spectrum of clinical signs indicative of a possible seizure liability have been captured. For example, when used during dose range-finding (i.e., high-dose and short-term) studies, 24 h video-monitoring can inform the selection of observation times in the pivotal studies and may influence species selection. Video-monitoring may be employed in combination with EEG analysis to create a scientifically based justification that any clinical signs are not associated with seizures.
Clinical signs are often challenging to describe in a nonclinical toxicology study report, but they often provide the bulk of the evidence for a potential seizure liability. Effective documentation will offer detailed descriptions of the clinical signs consistent with seizure activity (Figure 6). Written descriptions of convulsions and other relevant prodromal signs should document the nature of the event (e.g., affected body part, degree of incapacitation, progression, and severity) as well as the temporal pattern (e.g., initial [onset] and ending [offset] times, rate of repetition, and the relation to any apparent stimulus). Onset is often related to the time after dosing while offset is indicative of the event duration. In repeat-dose studies, clinical signs indicative of seizures may occur after multiple doses, and in such cases the pattern (after all doses, after several consecutive doses but only for initial treatments, after several consecutive doses but only for later treatments, or randomly distributed over the entire course of treatment) should be described to better characterize the hazard. Clinical evidence of seizures that arises after multiple doses may indicate a toxic accumulation of the parent TA or a neuroactive metabolite, which in turn may limit how frequently the drug can be administered to patients. The incidence (number of affected animals) is typically provided to understand how pervasive the effect is in the treatment group; in doing so, however, a distinction should be made between one animal that has three convulsions versus three animals that each experience one convulsion. The severity of TA-induced clinical signs typically is assigned over a range extending from detectable to an observer but with no apparent effect on the animal to full incapacitation or even death of the animal. As examples, severity of clinical signs can be captured as detailed descriptive narratives of each event, such as “self-limiting convulsion lasting less than 10 seconds with full recovery within 10 minutes,” “full body convulsion lasting 60 seconds with full recovery after administration of an anti-convulsant medication,” or “full body convulsion lasting for ≥2 minutes with incomplete recovery after administering anti-convulsant medication leading to a decision to euthanize for humane reasons.” Severity of any given sign and the entire cluster of related signs is interpreted in the context of event incidence, frequency, and duration as well as the TA dose and any information on PK levels. Importantly, death may follow a single massive seizure or repeated seizures of variable severity; may be related to severe and/or prolonged changes in breathing (i.e., apnea) and/or cardiac regulation (e.g., cardiac arrest or arrhythmia); or for unknown reasons. Apnea and cardiac dysfunction may decrease blood oxygenation levels, which in turn results in tissue hypoxia, neuronal distress (and often cell death), and ultimately the induction of seizures. Importantly, balance is necessary in regarding the relevance of in-life observations as clinical signs indicative of a possible seizure liability are often ambiguous, and have been demonstrated to be inconsistently associated with abnormal EEG signatures.
35

Pharmacokinetic Analysis in Assessing Seizure Liability
For small molecule TA, PK data on the parent and known neuroactive metabolite(s) may bolster the evidence indicating that clinical signs indicative of a seizure liability are in fact TA-related. Therefore, the nature, onset, offset, frequency, and persistence of clinical effects should be correlated to the PK profile of the parent TA and its active metabolite(s) to understand which molecule(s), if any, might be responsible for the seizurogenic signature. In our experience, clinical signs indicative of a seizure liability are correlated more often with the time of maximum plasma concentration (Cmax) and terminal half-life for the neuroactive molecule. 3
If available, knowledge regarding the distribution of the TA and neuroactive metabolite(s) in brain tissue is useful in characterizing seizure risk. However, absorption, distribution, metabolism, and excretion (ADME) data for nonclinical species are generally not expected in the IND-enabling package to support a Phase 1 clinical trial. Hypothetically, systemic exposure (i.e., blood levels) may be low or variable at the time seizures are observed. In such cases, TA-related relationships between tissue levels and in-life neurological effects may be inferred by indirectly establishing plausibility based on the nature of the TA (e.g., lipophilic, neutral charge), known class effects and MoAs, generation of a dose-response curve, and reproducing any clinical effects within or across species.
The establishment of the dose-response relationship is a critical component in scientifically-based justifications for drug-related effects, including seizures. In general, an increase in the TA dose is expected to result in a higher incidence and/or severity of clinical signs like convulsions and myoclonus while also yielding shorter onset times.36–38 The shape of the dose-response curve (e.g., shallow or steep, early or late onset) is important for fully characterizing a seizure liability. For example, a steep dose-effect curve may occur when the next incrementally higher dose that follows an ineffective dose produces a marked effect. For this reason, the toxicologist may be more conservative when managing effects related to steep dose-response curves or fast onsets. Regardless of the magnitude of a drug-related effect, its relevance should not be dismissed if only observed in a single animal. This point is important given that occurrence of the hazard itself suggests that an adverse effect may occur, unless proven otherwise.
Pathology Evaluation in Assessing Seizure Liability
Routine pathology endpoints are available in data sets for nonclinical pharmacology and toxicology studies but are relatively insensitive in identifying seizure liabilities. 33 Typical data include hematology (complete blood count and leukocyte differential count); serum chemistry panel (analytes evaluating function of many organs, but usually not brain); gross observations of organ appearance at necropsy; weights for the whole body and usually selected organs (including brain); and microscopic evaluation of many organs (generally including multiple regions of the brain). This section outlines the utility of these principal pathology endpoints in investigating nonclinical seizure liabilities.
For clinical pathology, the most useful routine endpoints are analytes in the serum chemistry panel. In particular, measurements of electrolytes (calcium and sodium) and glucose can exclude several metabolic causes of seizures (hyperglycemia, hypocalcemia, hypoglycemia, hyponatremia) that may or may not be related to TA exposure. Such indirect effects may pose a serious risk to patients, but they can be monitored by relatively non-invasive means and managed by improved nutrition and/or medication. Hypoxia (low blood oxygen levels) is also associated with seizures, but analysis of arterial blood gas levels is rarely performed in nonclinical pharmacology and toxicology studies. In recent years, interest has been burgeoning in the translation value of biomarker measurements in fluid specimens (blood and sometimes CSF) from animals. To date, biomarkers are typically analyzed during discovery studies, but where warranted such endpoints may be undertaken in development studies as well. Biomarkers of epileptogenesis (i.e., the process by which neural circuits are altered toward a state of increased seizure susceptibility) identified in animal models of epilepsy include high-mobility group box 1 protein (HMGB1) 39 and various miR.40,41 In terms of detecting TA-related neural injury, NfL, NSE, and Tau (for neuronal effects) as well as GFAP (for secondary glial reactions) assessed in plasma (non-invasive) or CSF (invasive) have shown some success in demonstrating CNS effects in rats.42,43 Their relevance in characterizing seizure liability will depend on correlative data demonstrating electrical disturbances in the brain (EEG) and one or more foci of damaged brain tissue (detected by microscopic [histopathological] evaluation).
For standard nonclinical studies, anatomic pathology endpoints to examine brain structure include brain weights and gross and microscopic evaluations. Of these, microscopic analysis is the only technique that may demonstrate tissue changes indicative of seizures; the classical macroscopic changes that occur in various brain regions of humans with chronic epilepsy are generally not evident in test animals. For conventional nonclinical studies, histopathological examination is performed often for pharmacology studies and almost always for toxicology studies. The microscopic evaluation will attempt to address three major questions: (1) can morphological evidence be identified that helps define the origin and/or etiology of the seizures, (2) is morphological evidence present indicating that seizures have happened previously, and (3) did the seizures contribute to or cause death. The scope of the microscopic examination may need to be adjusted depending on answers obtained in addressing these three questions. In general, conventional anatomic pathology methods used in nonclinical toxicology testing are less informative compared to standard safety pharmacology techniques (EEG) since alterations to brain function are only inferred, not directly measured from the pattern of microscopic changes. Nonetheless, the microscopic examination in nonclinical studies should be constructed so that any morphological changes related to seizure activity might be detected.
Multiple factors may influence the design of a neuropathology evaluation for investigating seizures in the nonclinical setting. Although neurological signs may be used in any species to help localize the potential nidus at which seizures arise in the brain, anatomic sampling of brain for nonclinical studies is generally determined by industrial best practices and/or institutional standard operating procedures. Current globally recognized “best practice” recommendations for brain sampling during nonclinical studies, 44 which calls for 7 coronal sections in rodents and non-rodents, provide a suitable minimal screen for common sites at which structural damage in the parenchyma may develop as an inciting cause or secondary consequence of any seizure activity. Principal structures to examine in studies with test article-related seizures include the neocortex (especially the frontal, parietal, and temporal cortices); limbic system (particularly the entorhinal cortex, hippocampus, and amygdala); basal ganglia (caudate and putamen); thalamus (dorsal nuclei); cerebellum; and brainstem. 26 Further brain sampling and processing (i.e., an expanded neurohistopathology evaluation45,46) may be desirable to ensure that these regions have been adequately evaluated. 47 In such cases, papers describing options for more extensive brain sampling have been published for many nonclinical species including rats, 48 rabbits, 49 dogs,50,51 minipigs, 49 and nonhuman primates (NHPs).47,52
In nonclinical toxicology studies, microscopic findings in brain related to TA exposure frequently represent consequences rather than the cause of seizures. Acute changes in seizing animals typically include hemorrhage in the cerebellum with or without concurrent unilateral or bilateral (asymmetric or symmetric), multifocal to diffuse neuronal degeneration and/or necrosis distributed variously in the brain, but usually recognized in the neocortex (middle layers), hippocampus (especially the CA1 and CA3 fields), and cerebellar folia (Purkinje cells). 53 With some time, gliosis microgliosis with variable astrocytosis and perivascular mononuclear cell infiltration may develop in affected regions. Animals enrolled in nonclinical studies generally do not survive long enough to develop the macroscopic findings noted in human patients with chronic epilepsy.
Study Design Parameters for Definitive Nonclinical Assessment of Seizure Liability
Given the numerous factors that influence seizure liability, characterization and management of drug-related seizures may be complicated. Toxicologists address the complexities of assessing seizure liability by designing nonclinical studies to mimic, as closely as possible, the conditions that are expected to be encountered by patients in the clinical setting. This approach helps to ensure that the findings observed in the treated animals can be translated effectively to help predict and manage potential risk in humans. This section considers major factors that may impact the study design and provides practical guidance on various nonclinical study design options that will produce a definitive assessment of seizure liability.
Study Design Parameters that Influence Data Accuracy and Quality
Susceptibility to seizure induction during nonclinical studies may vary depending on many design features. The most substantial factors are the model (test system), dosing schedule, and environmental conditions (Figure 4).
Model
Selection of a relevant test system (animal species) is a key aspect of hazard identification and characterization. The choice ensures that clinically relevant data are generated, which are essential for risk assessment. The model is typically selected based on multiple criteria including expression of the molecular target for the TA in the desired target organ(s), an ADME profile comparable to that in humans, and demonstration that the target organs of toxicity in animals are equivalent from an anatomical and physiological perspective to the same organs in humans. In general, ICH M3(R2) recommends using two species for general toxicology studies with at least one being a non-rodent species. 54 Moreover, for small molecule TA at least one test species should be pharmacologically relevant as defined by such attributes as target expression, binding, and pharmacology and equivalent PK (including metabolite) profiles. In general, there is sufficient concern for off-target effects of small molecules that only one pharmacologically relevant species is sufficient for safety testing, and when feasible NHPs are discouraged as the non-rodent species for general toxicology testing of small molecule TAs (including assessments of seizure liability) due to their limited availability.55,56 Notably, ICH S6(R1) for safety evaluation of biotechnology-derived pharmaceuticals states that non-relevant species can provide misleading data regarding biologic products designed to engage human (primate)-origin molecular targets with high specificity and are thus not recommended. 57 For this reason, NHPs are often the only relevant nonclinical species for examining the safety of biologic-derived TAs.
In general, nonclinical studies for small molecule TA are performed with healthy, young adult, wild-type animals. The genetic homogeneity of such models may be essentially total (e.g., inbred mouse and rat strains); partial (e.g., outbred mouse and rat stocks, Beagle dog strains, minipig strains, and rabbit strains); or unpredictable (e.g., cynomolgus macaques from diverse geographic origins). Human populations typically possess some degree of genetic heterogeneity, which dictates that nonclinical safety studies in rodents is typically performed using an outbred stock to embrace the potential influence of “hybrid vigor” on the response to a TA. In some instances, TA may have an intended pharmacologic effect that manifests as a profound imbalance in either electrolyte (e.g., calcium, chloride, sodium) or glucose homeostasis, which can induce seizures. However, this lowered seizure threshold in animals with normal metabolic checks and balances diverges from the physiological state expected of patients with a disease in which the pharmacologic effect of the TA is to restore an abnormally altered analyte to normal levels. For example, study designs to support nonclinical safety testing might include administration of supplemental glucose to wild-type animals to counter the extreme TA-related declines in serum glucose levels or using a diabetic animal model so that the TA effect would bring elevated glucose levels back to the normal range.
Published data demonstrate the impact of animal age on seizure susceptibility to various drugs. Age may influence the induction of drug-related seizures based on many factors, which may not be fully profiled or understood. For example, drug-induced seizures may develop in immature animals based on the lack of an intact blood–brain barrier to exclude entry of a toxic molecule (parent TA or metabolite) or the presence of incompletely differentiated liver that is incapable of metabolizing toxic molecules into inactive metabolites. Seizures kindled in rats by administration of pentylenetetrazole (PTZ, a GABA receptor antagonist) decrease with the age of exposure. Interestingly, the same study also demonstrates that older rats appear to be more sensitive to drug-induced anatomic lesions and kindling-related impairment of cognitive functions compared to younger rats. 58 Data from numerous studies have demonstrated that seizures arise at lower TA doses in young animals compared to older animals.59–61 Taken together, these data illustrate the importance of considering age when interpreting the implications of seizures and related events observed during nonclinical safety testing.
In nonclinical safety testing of small molecule TAs, human metabolites should be produced in one or both test species because metabolites may carry a seizure liability. That said, in some instances metabolites in animals may induce seizures that have no relevance for human risk assessment if they occur uniquely in the affected test species but are not generated in other animal species and humans. An example of this phenomenon is nabilone (Cesamet™, a medication devised to combat chemotherapy-induced nausea), in which convulsions seen in dogs were not deemed to have clinical relevance given the substantial differences in metabolism between dogs and other species. 62 The contributions of individual nabilone metabolites to the seizure liability in dogs were known and, based on the low production of the seizurogenic metabolite in humans, the seizure liability in humans was judged to be negligible.
Dosing Schedule
The dosing schedule (or dosing regimen) for a TA may impact seizure induction. A dosing schedule includes not only the dose(s) but also the pharmacological impact of the peak dose (maximum concentration [Cmax]) and/or total dose (area under the curve [AUC]), route of administration (RoA) and treatment duration, frequency (dosing interval), and rate.63–67 Induction of seizures may be dictated by the RoA, particularly if exposure levels of a drug (and its metabolites) vary greatly in their bioavailability. For example, intravenous (IV) dosing typically results in a higher Cmax in blood compared to oral (PO) administration of the same drug and dose due to differing ADME kinetics. If seizures for a specific drug are driven by the peak level (Cmax), then adjusting the dosing frequency so that the total dose is shifted from once-daily (SID) to twice-daily (BID) administration can reduce the Cmax and lower the risk of seizures without changing the overall effective dose. Similarly, if IV dosing is necessary to attain pharmacological efficacy, employing slow infusions rather than bolus dosing may reduce Cmax sufficiently to lower the seizure risk. Seizures may occur after drug withdrawal, such as with alcohol and benzodiazepine dependence. When withdrawal-induced seizures are expected, adequate recovery periods are recommended in general toxicology studies to capture delayed effects from drug withdrawal or to adjust the dosing schedule to avoid a drug-dependence paradigm. Overall, toxicologists must carefully consider the dosing schedule to be used for pivotal toxicology studies to ensure that the resulting nonclinical data will have relevance suitable for assessing potential risk to patients who will be exposed in a clinical trial.
The dosing schedule employed for drugs may vary depending on the indication to be treated, which may further complicate risk management. For example, chronic indications may require multiple and/or frequent treatments over time, which may result in the development of kindling (predisposition to seizures following repeated prior neural stimulation) or tolerance for drugs of various classes. Kindled seizures have been reported following administration of sub-convulsive doses of drugs like PTZ. 68 Drug tolerance has been seen in animals after daily treatment with drugs like caffeine, delta-opioid agonists, and PTZ.69–75 Given the potential impact on seizure induction, the dosing schedule employed in nonclinical studies should mimic insofar as possible the schedule that will be utilized in the clinical trial it is intended to support. This design strategy will ensure that any toxicities observed in animals will have the greatest relevance for assessing potential risk to human patients.
Environmental Conditions
Various attributes of an individual’s habitat may impact the tendency of a drug to produce seizures. Examples include many stressors such as extreme temperature changes (e.g., excessive heat), flashing (“strobe”) lights, perceived threats (e.g., any nearby predators), and various tactile stimuli (handling or restraint).68,76–84 For example, published data have shown in rodents that increased body temperature reduces the time to onset and enhances the severity of drug-related convulsions, associated hippocampal damage, and mortality while conditions that decrease body temperature may be neuroprotectant.77–79 The strength of such stressors may depend on their duration and frequency. This information may be used in two ways to optimize the design of nonclinical studies to assess seizure liability of novel TAs. First, environmental conditions may be adjusted to stay within a desired dynamic range, thus reducing stressful stimuli and their effects in modulating the seizure threshold; this step lowers the no observed effect level (NOEL) as defined by seizures. Second, deliberate imposition of one or more environmental stressors may be used to challenge a TA (or TA class) early during drug development to identify any seizure liability. From a 3Rs (“reduce, refine, replace”) perspective, reducing environmental stressors to optimize toxicology study conditions is a suitable approach to minimizing the likelihood that a pivotal nonclinical study will need to be repeated. However, the decision to adjust environmental conditions in any fashion to minimize any potential seizure liability should be considered on a case-by-case basis as environmental changes might affect data translatability for assessing human risk.
Leveraging Nonclinical Data to Design Effective Nonclinical EEG Studies
The standard nonclinical safety strategy outlined in ICH M3(R2) may appropriately identify a potential seizure liability but is typically not designed to provide a complete characterization of seizure risk. The reason for this discrepancy is that seizures may be evident as abnormal in-life neurological signs and/or neuronal necrosis in tissue sections, which are incorporated as routine endpoints for general toxicology studies, while brain electrical disturbances can be definitively detected only by EEG analysis, which is not a standard component of general toxicology studies. In nonclinical safety testing, EEG studies are performed only when there are data demonstrating that drug-related seizures are plausible and further characterization of these effects are needed to help ensure patient safety. Previously collected nonclinical data (in vitro and in vivo) may offer evidence of a potential seizure liability, and such data should be utilized to inform the design of nonclinical EEG studies. In particular, existing data are instrumental in selecting the test species (including group sizes), dosing schedule, investigative methods to be employed, and the timing of various analytical endpoints. In our experience, EEG studies are helpful in characterizing seizure risk when (1) the safety margins based on clinical signs observed during general toxicology or other nonclinical studies are not adequate; (2) the drug exposure levels at which animals exhibited prodromal signs in the absence of convulsions need to be evaluated in patients; and (3) the patient population may have an increased sensitivity to the drug-induced convulsions due to a potential interaction with another drug expected to be administered concurrently. The quality and clinical relevance of data collected from nonclinical EEG studies depends on how the information from various sources are leveraged to design these nonclinical studies. This section describes various factors that are considered in designing nonclinical EEG studies (Figure 7).
In general, EEG studies for a nonclinical drug development program are carried out using a sensitive animal species. The species is typically selected because it was deemed to be the most sensitive test system based on the exhibition of in-life clinical signs indicative of seizures at an exposure level lower than that measured in other species treated under the same (or comparable) dosing schedule. In the absence of exposure data, the species that exhibits convulsions at the lowest dose level (based on body surface area) compared to the other species tested may also be deemed the most sensitive. The most sensitive species is the most relevant species for assessing potential risk to patients unless proved otherwise with data demonstrating that seizures in this sensitive species arise through a non-relevant species-specific mechanism. Ultimately, selection of the species for the EEG studies may be based on data employing various species tested under different dosing schedules and experimental conditions.
Ideally, the dosing schedule (e.g., dose levels, duration and frequency of treatment, RoA) employed in nonclinical EEG studies should be informed by data from studies that mimicked, as closely as possible, the proposed clinical dosing schedule. Duration of the nonclinical EEG study and the choice of day(s) for the most detailed in-life clinical observations and/or EEG analysis should be informed by prior data delineating the onset and offset of the TA-induced convulsions. In some cases, conducting EEG studies with treatment durations that exceed the one employed in earlier studies that demonstrated TA-induced convulsions in the same species may be helpful in understanding the seizure liability if the pattern of seizure-related events changes during treatment (e.g., by drug-related kindling via repeated administration of sub-convulsive TA doses or development of TA tolerance to the induction of convulsions). The dosing schedule must be carefully selected to optimize the likelihood of detecting any brain electrical changes indicative of seizures, which will ensure that the EEG data are clinically relevant.
Endpoints employed in a nonclinical EEG study should be selected based on the expected toxicology profile established using previously acquired data for the parent TA, any neuroactive metabolites, and molecules that are chemically related and/or interact with the same molecular targets. Relevant data may include records of clinical signs, clinical pathology and anatomic pathology findings, and PK measurements (Figure 6). For example, prodromal or premonitory clinical signs consistent with seizures serve to define the potential onset and offset of abnormal EEG patterns, which helps inform the length of an EEG study. The occurrence of clinical signs in the absence of an abnormal EEG pattern suggests that the signs do not provide evidence of an adverse seizure liability. Common clinical pathology endpoints (e.g., imbalances in electrolytes or glucose) explore some MoAs by which seizures may be induced by TAs. Anatomic pathology data indicating degeneration or necrosis of sensitive neuronal populations may help confirm the presence of seizures; when collected for different time points, such findings may help determine if the brain effects precede or follow the induction of seizures. Various PK assessments inform the seizurogenic risk in relation to TA exposure levels when clinical signs (either prodromal or convulsions) first appear, at the time of peak exposure (i.e., Tmax), and to the estimated time of virtually complete elimination (i.e., 5 to 7 half-lives). Some seizurogenic TAs elicit effects at exposures that are less than 2-fold different between the tested nonclinical species; for other TAs, the seizurogenic doses may differ by 20-fold between the most sensitive test species and the other. Together, these PK data provide insight regarding when seizures may develop and how long they may persist following exposure to a parent TA (and its neuroactive metabolites). Other nonclinical data that may prove useful in designing a nonclinical EEG study are results from safety pharmacology studies to examine cardiovascular and respiratory endpoints as well as studies in which seizures may have been induced via a state of cardiac arrest or hypoxia. The nonclinical data available to devise an EEG study may include all these data types or may be limited to a subset of such information. For this reason, the optimal design for a nonclinical EEG study will involve integration of all available factors.
General Design Features of Nonclinical EEG Studies
The success rate for translation of nonclinical data to predict possible clinical outcomes is variable. For neuroactive drugs targeting the CNS, nonclinical data are reported to be reasonably translatable to clinical results when considering regulatory submission documents that synthesize all nonclinical safety assessments. Drug-induced seizures may result from any of many MoAs, but it is not always possible to confirm the exact MoA involved for a given TA.
Figure 8 presents a summary of neuroactive TAs for which nonclinical EEG studies were conducted at a large CRO over a recent 4-year period. These data highlight the diversity of drug products that may be associated with seizure liabilities. Knowledge of the MoA(s) responsible for a seizure liability can contribute to both risk assessment and the development of mitigation strategies, but characterization of seizure activity kinetics relative to drug exposure is essential to assess the clinical risk. Unpredictable seizurogenic effects (i.e., treatment-related epileptiform EEG morphologies) in test animals that are not correlated with higher drug exposures constitute a considerable safety risk for patients.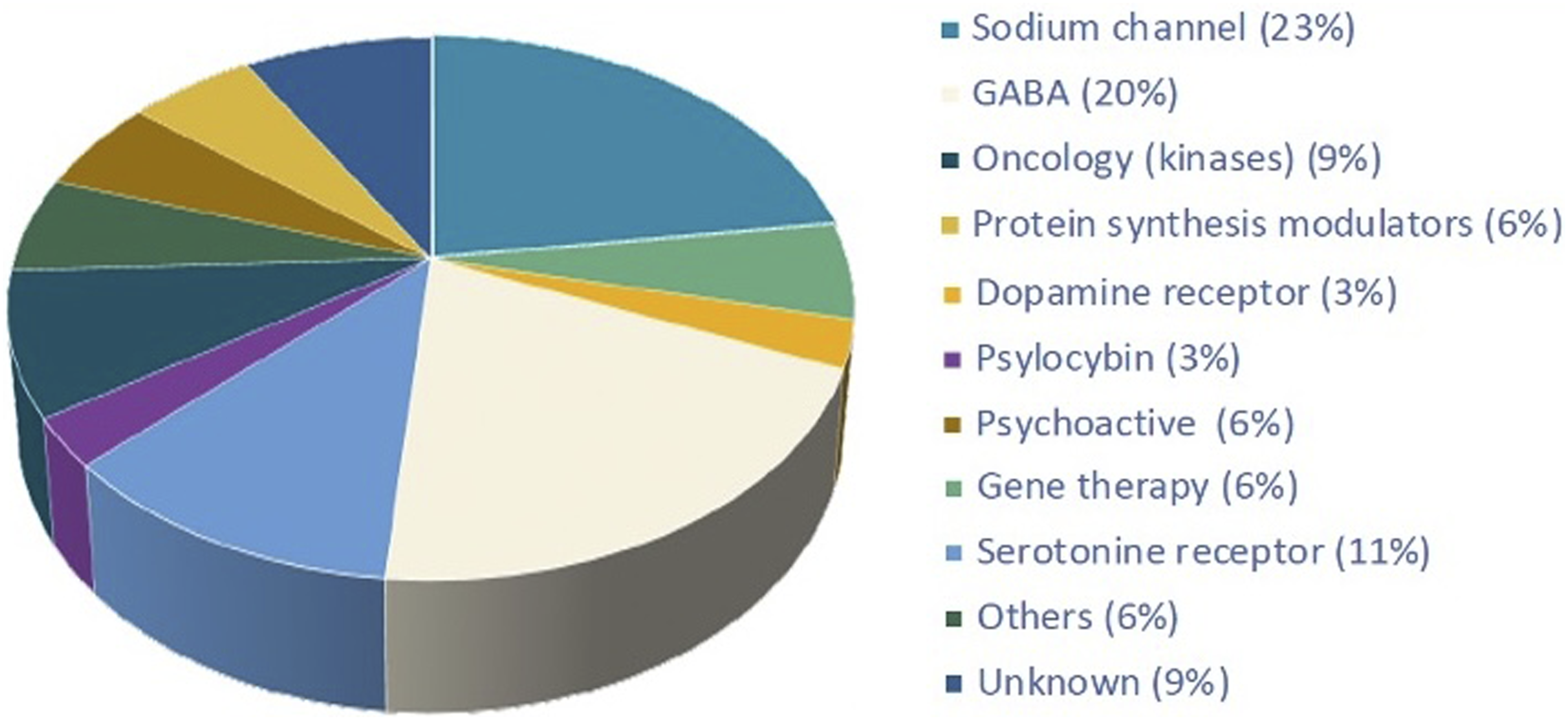
As noted above, the test system of choice is the most sensitive nonclinical species. Beagle dogs are recognized to possess a higher susceptibility to TA-induced seizures compared to other common test species. The magnitude of the vulnerability varies considerably among agents. 85
Nonclinical EEG studies are ideally conducted at an age that is equivalent to that of the relevant clinical population. In humans, the incidence of epilepsy follows a U-shaped distribution with a higher frequency in young (pediatric and juvenile) and very old individuals compared to mature (mid-life) adults.11,86,87 The use of very old animals for EEG studies is rarely considered in toxicology due to their limited availability for commonly used species, the increased variability in brain wave patterns associated with aging, and the scarcity of historical control data. Young animals can be considered for EEG studies, but the size of the telemetry implants used for most EEG studies favors the inclusion of larger animals (e.g., dogs) and limits the use of juvenile rats and NHPs.
Dose level selection for nonclinical EEG studies often follows a paradigm that differs greatly from the approach employed in selecting doses for general toxicology studies. Tachyphylaxis and tolerance (i.e., rapidly and gradually diminishing responses to successive drug doses) are considerations that have been associated with CNS neuroactive drugs for decades, so nonclinical EEG studies need to be designed to avoid under estimating drug toxicology. Adaptive study designs can help reduce animal numbers needed while increasing the EEG no observed adverse effect level (NOAEL). Such study designs will typically include an initial phase in which a range of dose levels (e.g., low, middle, and high) are used to determine the highest exposure at which epileptiform EEG changes are not observed and then a second phase undertaken after a longer TA wash-out (e.g., 1 month or more) or with naïve animals to confirm the EEG-based NOAEL.
Group size in nonclinical EEG studies is influenced by the consistency of an effect across individuals of the test species. In general, nonclinical EEG study designs typically include a minimum of 8 animals per group because susceptibility to drug-induced seizures is notoriously variable among individuals. Some researchers have emphasized the acquisition of longer pre-study baseline EEG recordings to identify individuals with higher variability in brain electrical activity. However, the presence of spontaneous EEG abnormalities is rare in nonclinical species while many animals that exhibit increased seizure susceptibility (especially Beagle dogs) have normal pre-study EEG morphology.
The choice of when and how to collect brain electrophysiological data during a nonclinical EEG study is influenced by several factors. The unpredictable timing of TA-induced seizures is often considered as justification to use continuous EEG recording over a short (i.e., snap-shot) time span from restrained or telemetered animals. Video-EEG (i.e., the simultaneous acquisition of telemetered EEG and visual recording to correlate brain electrical patterns with clinical signs) provides a significant improvement for identifying TA-induced seizures compared to restrained EEG recordings using surface electrodes. Video allows for the identification of premonitory signs that can become important signs to monitor in clinical trials. Video recording also permits consultation with various clinical specialists (including veterinary neurologists) prospectively and retrospectively. Finally, video can facilitate EEG interpretation and in particular discrimination of several more challenging EEG artifacts from genuine TA-related convulsions.
Overall, nonclinical testing for seizure liability is a scientifically driven exercise that takes into account the existing toxicology data and clinical considerations. Consulting with regulators to confirm the design parameters for nonclinical EEG studies is suggested prior to study conduct as it ensures alignment on the scientific approach.
Regulatory Perspectives on Nonclinical Assessment of Seizure Liabilities
Risk/benefit analyses prior to clinical trials and marketing are the province of both project toxicologists in industry as well as regulatory reviewers. These analyses utilize nonclinical data to anticipate potential or likely outcomes in clinical trials. In industry, risk/benefit considerations represent the integrated judgments of nonclinical scientists and clinicians, where toxicologists and allied scientists (e.g., pathologists, pharmacokineticists, pharmacologists) translate the various pharmacological and toxicological data generated during nonclinical studies to humans while clinicians provide a level of risk tolerance for a given patient population or disease indication. The toxicologist will take the lead on risk assessment, providing the “risk” portion of the analysis, but may contribute to the “benefit” analysis during early stages of drug development when there is only nonclinical pharmacology data regarding the potential benefits of the drug. Working together, the clinician and toxicologist assess the relative risk of a drug and the risk tolerance of the proposed patient population. This section provides a regulatory perspective on seizure liability, and is organized to provide an understanding of (1) principal points to consider during a risk/benefit analysis and (2) some strategies used in clinical trials to mitigate seizure liability.
Objectives of Risk Assessment
The toxicological risk assessment for pharmaceuticals is designed to examine the potential for a TA to induce adverse effects on the health of an intended human population. Elements of this process include hazard identification and characterization, exposure assessment, and the final integrated risk assessment.
Classically, risk is defined as the hazard inherent in exposure to a particular product, often judged with respect to the hazard incidence and severity relative to a dose-response continuum. Hazard characterization in nonclinical safety testing combines both hazard identification and dose-response assessments. Hazard identification for seizure liabilities that might be encountered during FIH clinical trials often is limited to a few in vitro assays (e.g., secondary pharmacology screens) and selected in vivo studies (e.g., dose-range finding, safety pharmacology, repeat-dose toxicology). Where in-life clinical signs suggest a genuine possibility that a seizure liability exists, nonclinical EEG studies may be added to provide definitive proof regarding the state of brain electrophysiological function. Characterizing a hazard relative to the dose-response is generally limited to repeat-dose toxicology studies and EEG studies that are designed to identify a NOAEL or NOEL.
Exposure assessment is another element of the risk/benefit analysis. For candidate drugs, an exposure assessment is relatively straightforward compared to chemicals within the environment or at the work site. The reason for this simplicity is that exposure to pharmaceuticals is precisely known for both nonclinical studies and clinical trials.
A rational, integrated risk assessment may be made after collecting the necessary nonclinical and early-phase clinical data. Such assessments are based on the TA’s pharmacological class, the results of the standard battery of nonclinical tests, and the outcome of clinical trials conducted in patients with a particular disease treated under a given dosing schedule.
Nonclinical Risk Assessment: Pre-Trial (“Preflight”) Considerations
Testing a prospective new drug in humans is risky and, although rare, serious injury or death of volunteers and patients participating in early clinical trials are within the realm of possibility. A specific example relevant to assessing seizure liability is the disastrous 2016 Phase 1 clinical trial of the fatty acid amide hydrolase (FAAH) inhibitor BIA 10‐2474, in which one volunteer died and four other volunteers experienced SAEs.88,89 One contributor to this tragedy was that the regulatory toxicology package was conducted as a ‘box-checking’ exercise using multiple species (mouse, rat, dog, and NHP, exceeding the two species stated in the ICH M3(R2) guideline 54 ), but the potential for seizures and other adverse neurological (i.e., functional) effects was missed in the routine (generic) analysis.
In our experience, however, the risk assessment is better understood by reference to aviation analogies, where the nonclinical package represents the “preflight check” while the compiled IND submission and subsequent clinical trials are the “in-flight” monitoring. The various ICH and regional guidances for nonclinical studies (e.g., ICH M3(R2), 54 ICH S6(R1), 57 ICH S9,90,91 and the FDA start dose guidance 92 ) serve as a preflight check for sponsors and regulators to address concerns generic to identifying potential pharmaceutical risks. The most convincing way to tackle a line item on the preflight checklist is with data from well-designed nonclinical studies with the proposed pharmaceutical. In some cases, a line item on the preflight checklist may not be relevant for a specific TA, so the omission of this data set from the IND-enabling package will require due justification.
The following elements should be considered as part of the nonclinical “preflight checklist” (Figure 9) to be conducted prior to the FIH clinical trial.
Adequate Nonclinical Test Battery
Verify that the complete battery of standard GLP-compliant nonclinical studies has been conducted. If a study was not conducted, supply a suitable rationale in the regulatory submission (e.g., mutagenicity and metabolism are irrelevant for a biologic, safety pharmacology endpoints were incorporated into repeat-dose toxicology studies). In general, specific kinds of nonclinical information should be communicated in appropriate sections of a regulatory submission so that the nonclinical reviewer can locate the information efficiently. The Nonclinical Overview (Module 2.4) for IND applications in the U.S., the Investigator’s Brochure (IB), and the Investigational Medicinal Product Dossier (IMPD) for the Clinical Trial Applications (CTAs) of the European Union (EU) and Medicines and Healthcare products Regulatory Agency (MHRA) of the United Kingdom are appropriate places to describe the nonclinical testing strategy with justifications for not conducting certain studies provided in the generic product development recommendations in ICH M3(R2).
Nonclinical Data Interpretation
Consider if any safety signals are evident in the nonclinical data that may suggest a potential seizure liability. In the standard test battery, nonclinical signals for seizure liability are typically limited to in-life evidence of brain dysfunction (e.g., premonitory signs, convulsions, and tremors), which may arise as a direct or indirect test article effect. Definitive proof of seizurogenic potential may require demonstration of TA-related alterations in brain function during a nonclinical EEG study if there are insufficient safety margins to the clinical signs in question.
Confidence in Nonclinical Data
Data quantity and quality provide a level of confidence that any seizure liability has been identified and characterized. Ideally, nonclinical tests should be conducted in compliance with GLP. If a safety signal is observed in the standard battery of studies, consider whether additional studies might be needed to better characterize the implications of that signal. For example, if the MoA is known, detection of off-target pharmacological activity and/or in-life premonitory signs consistent with seizures during safety pharmacology or repeat-dose toxicology studies may trigger further investigation to enhance characterization of the TA safety profile.
Notably, confidence in the nonclinical data should extend to trusting properly collected negative data. For example, the absence of in-life premonitory signs or convulsions in repeat-dose toxicology studies may occur in combination with evidence suggesting a seizure liability, such as knowledge of the drug class and molecular structure, secondary binding screens, or concerns from observations acquired during other in vivo studies. In such cases, the degree of confidence in the negative data is influenced by various design factors. Was the absence of clinical signs made using cage-side spot checks or by continuous 24 h video-monitoring? Were clinical observations made by technical staff with experience in identifying subtle premonitory signs or merely overt convulsions? Does the timing of convulsions in nonclinical species support any dosing in the clinic, or does a weeks-long delayed onset of seizures in a repeat-dose study permit short-term human exposure? Clearly, the greatest confirmation for negative nonclinical data is a formal EEG analysis to confirm the absence of brain wave alterations and identification of a true seizure NOEL.
Causality of Nonclinical Findings
The Bradford-Hill criteria for demonstrating causality using epidemiological data employ multiple criteria to assess whether exposure to a given TA (e.g., environmental or occupational chemical, drug) is responsible for a particular response (pharmacologic or toxic) in humans. 93 The usual criteria include the response strength, consistency, specificity, temporality, biological gradient (i.e., dose-response), experimental design, analogy (class effect), biological plausibility, and coherence. These same criteria provide a conceptual framework against which nonclinical data may be judged with respect to whether a particular effect in animals is related to TA exposure (as either a direct or indirect consequence). Confirmation of a likely direct TA-mediated neuroactive effect include clear differences in the response between control and TA-treated groups; consistent responses among animals of a given species (for one or both sexes) in a single study or across several studies, or for several test species; a clear dose-response relationship; proven entry of the TA into the CNS; and a known seizurogenic MoA for the TA or similar agents in the class. Creating a matrix that collates data for all the data types noted above should help provide a clear positive or negative biological plausibility for the TA with respect to its seizurogenic potential. An indirect TA-related effect may be indicated if the nonclinical data suggest that seizures are engendered by physiological alterations (e.g., abnormal serum electrolyte or glucose levels, hyperthermia) or stress. Once the spectrum of TA-related effects has been identified, the nonclinical data often will indicate potential means for monitoring the advent and resolution of seizures in the clinic (often without needing to implement universal EEG monitoring in clinical trial participants).
Translatability of Nonclinical Findings
Typically, GLP-compliant repeat-dose toxicology studies are conducted in young healthy animals. If the drug is designed to correct a metabolic imbalance in a disease state (e.g., hypocalcemia, hyperglycemia [diabetes mellitus]), use of the drug in healthy animals may cause seizures that cannot be translated to a patient population with the metabolic imbalance. In these cases, consideration should be given to supplementing test animals with the metabolic substrate (or using a nonclinical model of the disease indication) to mitigate pharmacology-related seizures, and caution should also be used in the clinic when enrolling healthy volunteers.
Safety Margins Based on Nonclinical Findings
The integrated data from the battery of nonclinical studies should define a threshold value (NOEL or NOAEL) by which a safety margin with respect to any seizure liability may be set. While transient prodromal signs (licking, salivation, etc.) and convulsions may not cause any obvious harm to the animal (i.e., are not adverse), their presence may still be associated with abnormal electrical signaling in the brain—which cannot be known definitely unless EEG data are available. Accordingly, a NOAEL should not be automatically interpreted as indicating the absence of convulsions unless the presence of “nonadverse” prodromal signs has also been ruled out. This discrepancy means that regulatory reviewers often will reset the NOAEL to the dose without any prodromal clinical signs, often leading to a low or no safety margin relative to a pharmacologically active dose. To raise the NOAEL, regulatory reviewers may request a nonclinical EEG study to provide definitive confirmation of a seizure NOEL; alternatively, a toxicology study with 24 h video-monitoring may be used to confirm a seizure NOAEL (based on the lack of clinical signs indicating an enhanced incidence or severity of seizures). For drugs, regulatory reviewers generally prefer that a 10-fold safety margin exists between the nonclinical seizure NOEL and the high dose to be used in the clinic. In some cases, reviewers may increase this margin based on concerns with the nonclinical data (e.g., data gaps, inherent uncertainty, variable exposures across nonclinical studies) or with the patient population (e.g., individuals with lowered seizure threshold, difficulty in distinguishing any drug effect from the disease background, scarcity of clinical monitoring techniques). Certain cases may warrant lowering the safety margin to the high clinical dose to below 10-fold the NOAEL or NOEL (e.g., experience with the class of molecules). Given this perspective, preemptive inclusion of a nonclinical EEG study to de-risk a potential seizure liability will often represent a prudent demonstration of due diligence in assembling a nonclinical safety profile for neuroactive TAs.
Nonclinical Risk Assessment: Risk/Benefit Analysis (“In-flight”) Considerations
In a risk/benefit analysis, toxicologists use the nonclinical weight of evidence with input from clinical colleagues to assess several key parameters including the risk tolerance for the clinical population, desirable safety margins, and risk management and mitigation strategies. Risk/benefit assessments for a given TA and clinical trial are undertaken using comparatively standard packages of nonclinical data but a case-by-case analytical approach. In general, consultation with the reviewing division of the relevant health authority is recommended to ensure that the designs of pivotal study designs will successfully support a regulatory submission.
Risk Analysis
When writing the IND, risks for the proposed drug candidate should be well characterized (Figure 9). For a FIH clinical trial, the risk assessment is defined by nonclinical data for the drug candidate as well as information on any related molecules in the class. Primary elements in the risk assessment are to understand how the nonclinical seizure NOEL informs if the seizure risk in nonclinical species might be relevant to patients, and if so how the risk might be monitored and managed in the clinic. Information on the MoA for the TA and drug class may also provide an indication of relative concerns for the new drug candidate; an increased concern is typically faced by new molecular entities (NMEs), but clinical experience with the drug class may minimize concerns for new TAs in the same class. Moreover, the MoA may inform clinical management practices (e.g., which drugs might be used to prevent or stop a seizure) to minimize any potential seizure liability during the clinical trial. All these considerations may aid regulatory reviewers in adjusting the safety margin up or down.
Relative risk (i.e., the risk posed by a proposed new drug compared to the risks of approved drugs in the same class or to other drugs [of any class] to treat the same disease indication) is another consideration. While a proposed new drug may have a seizure liability, this risk may be an unavoidable, and therefore acceptable class effect in approved drugs having a comparable chemical structure and MoA. In some cases, relative risk of a proposed drug may need to be demonstrated with a comparator study with approved reference agents of the same class.
Risk is also dictated by the species exposed to the TA. When available later in development, accumulating human data will supersede nonclinical data. In terms of nonclinical sensitivity, it is generally accepted that Beagle dogs exhibit a background incidence of seizures exceeding those of other nonclinical species, and Beagle dogs are also more vulnerable to drug-induced seizures for small molecule TAs of certain classes (e.g., the GABA receptor antagonist PTZ and the serotonergic antidepressant citalopram).3,85,94–96 That said, this characteristic of Beagle dogs is not broadly applied to new TA classes to justify lower safety margin.
Benefit Analysis
The benefit component of a risk/benefit analysis considers the existing therapeutic landscape as well as the nonclinical pharmacology studies (Figure 9). If there is an unmet medical need for the drug, patients who stand to benefit from taking the trial drug (e.g., terminal refractory cancer patients) may possess a higher risk tolerance; however, this elevated tolerance for risk may not translate to clinical studies with healthy volunteers. Nonclinical pharmacology studies play a much larger role in assessing possible benefits to pediatric patient populations as it is unethical to study ineffective doses in children who cannot provide informed consent on their own behalf.
Another consideration in assessing potential benefits is whether the target patient population exhibits a background incidence of seizures. A principal challenge for clinical trials of such conditions is to distinguish drug-related seizures from other types of seizures (disease-related or spontaneous). If nonclinical data suggest a seizure liability, a logical justification may be needed to permit testing of the new drug in the clinic.
Risk Management
The objective of risk management is to develop an actionable plan to avoid or minimize risk during clinical trials. Several risk management strategies may be deployed during clinical trials for a drug with a potential or known seizure liability (Figure 10). Examples include maintaining an adequate safety margin to the nonclinical seizure NOEL at all clinical doses, modifying the criteria for patient inclusion/exclusion in the clinical trial, altering the dosing schedule, in-clinic monitoring, and seizure mitigation using various medications.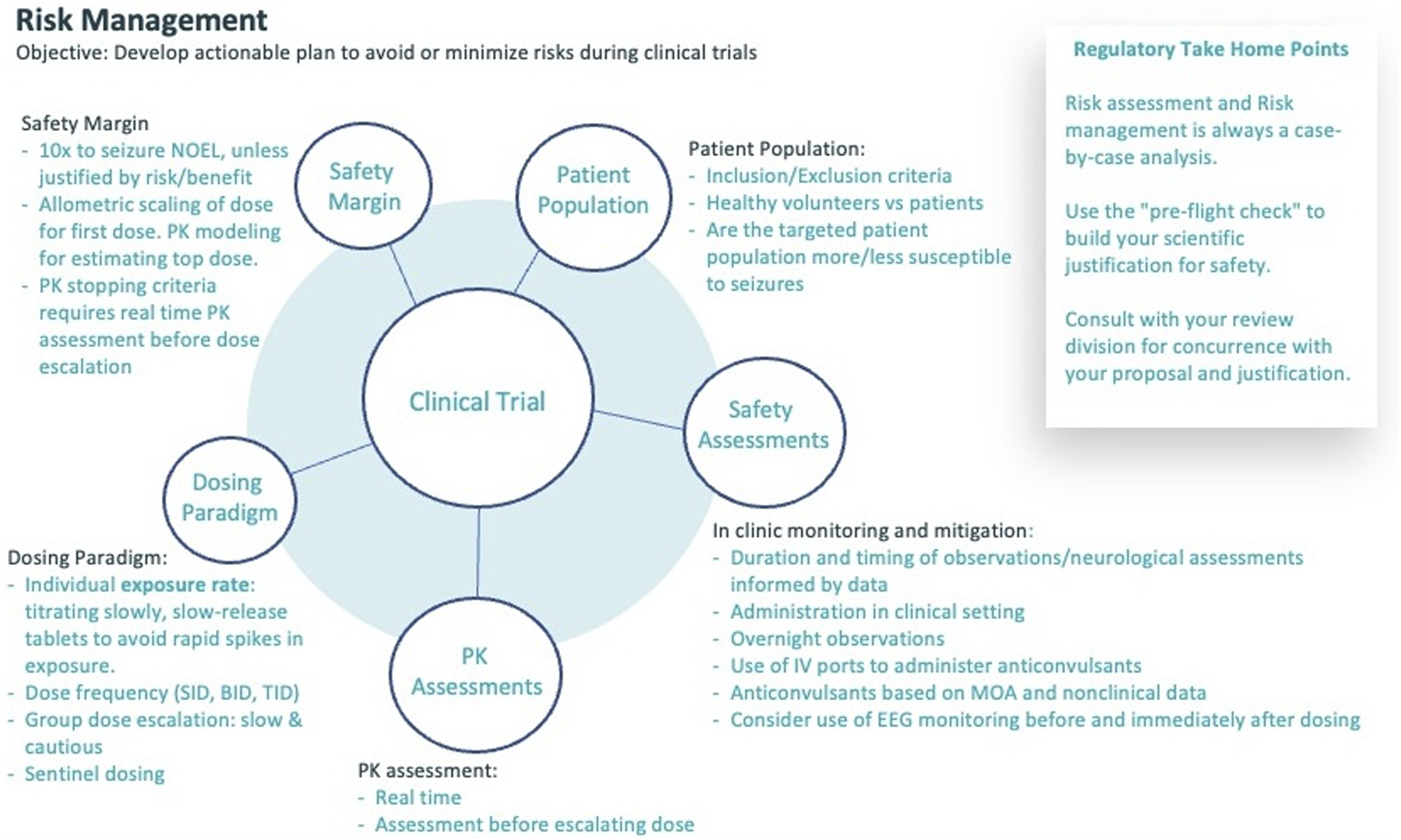
Maintaining an adequate safety margin to the nonclinical seizure NOEL at all doses to be tested in the clinical trial can be accomplished by allometric scaling of the dose and/or use of exposure caps. If exposure caps are in place, PK stopping criteria and real-time PK assessments should be included in the clinical trial design.
Modifying patient inclusion/exclusion criteria is a common approach to risk management in planning clinical trials. Exclusion criteria may reject patients based on a history (personal and/or familial) of prior seizures or head trauma, or prior or current intake of specific medications that may lower the seizure threshold. If a clinical trial intentionally includes patients with a known predisposition to seizures (e.g., people with alcohol use disorders or epilepsy), then additional caution should be applied in the trial design such as increasing the safety margin, altering the dosing schedule, and/or expanding clinical monitoring.
Dosing schedules can be designed to mitigate seizure risk. Common approaches include slowing the pace of dose escalation, adjusting the frequency of dosing, and sentinel dosing. Slower dose escalation is incorporated when the dose-response curve is steep. The frequency of dosing may be increased to two or three times a day, thereby splitting the full-day dose into smaller aliquots to reduce the Cmax and Tmax drug concentrations (which are often related to onset of seizures).97–99 Sentinel dosing is the practice of treating, collecting, and interpreting the safety data for a single subject in a cohort before administering the drug to other patients in the cohort.
In-clinic monitoring is a common and powerful means for managing risk during clinical trials. Useful practices include clinical observations (cage-side checks, functional observational batteries [FOB], and where warranted formal neurological examinations); use of intravenous ports to facilitate PK sampling and rapid administration of anti-seizure medications; and sometimes intermittent EEG monitoring. When designing the FIH clinical trial protocol, the duration and timing of in-life clinical observations and other neurological assessments is based on the available nonclinical data. While the seizure liability is most often encountered at Tmax exposures, 99 the nonclinical data may demonstrate that the liability also occurs after multiple doses or at blood levels of drug that do not reflect the Tmax. These latter situations suggest that seizures result from more gradual accumulation of the drug and/or neuroactive metabolites in the brain.
An important consideration in mitigating seizure concerns for humans is that the nonclinical data, MoA, and/or drug class may offer insights regarding the most effective anticonvulsants if a patient experiences a seizure during the trial. In such cases, a clinical trial protocol may include specific guidance for which anticonvulsant and dose(s) to use as a first-line treatment. In general, facilities participating in a clinical trial should be equipped with appropriate anticonvulsants conforming to current clinical practice and as informed by the nonclinical data.
At present, EEG monitoring during clinical trials is controversial, and thus not required by regulatory agencies. The current thinking is that EEG monitoring does not prevent seizures, and that a patient experiencing a seizure can be identified by clinical signs without EEG monitoring. For these reasons, EEG is not considered as a helpful, let alone essential mitigation strategy for use in clinical trials.
Conclusions
Seizures are defined by abnormal electrical activity in the brain. Seizures are evaluated in the clinical setting using a variety of functional and structural endpoints. Clinical trials to develop new drugs (and especially neuroactive agents) include a genetically and physiologically diverse population of patients, some of whom may have heightened seizure sensitivity. For example, epilepsy is common in the human population, may be the disease indication for which the drug is being developed, and may exist as an undiagnosed pre-existing condition in healthy volunteers. Seizures can present clinically as a progression from prodromal (premonitory) to ictal to postictal phases for general seizures. In contrast, partial seizures can be subtle and may not be easily recognized. If seizures occur during a clinical trial, discrimination of drug-related events from undiagnosed epilepsy from sporadic background seizures may be challenging. Medical evaluation is standard for individuals who have experienced a seizure, and the diagnostic work-up and any subsequent treatment course will be conducted by the supervising clinician for the clinical trial in consultation with a neurologist to ensure that patient safety is protected.
The cumulative nonclinical data from standard in-life clinical observations (and where warranted neurological examinations), in vitro and in vivo pharmacokinetic assays, and clinical pathology and anatomic pathology evaluations sculpt a narrative that may identify and partially characterize the seizure liability for a given test article. Nonclinical assessments of seizure liability should evaluate all drug-related findings that occur before, during, and after any prodromal signs or convulsions are observed. This amalgamation involves documenting the duration, frequency, incidence, and severity of seizures for individual animals and for all animals across the study as well as correlating the full range of in-life and post mortem findings with the dosing schedule (and especially the dose-response relationship). Together, these and other nonclinical data collected under various experimental conditions are combined to create a profile for drug-related seizures (Figure 11). The seizure liability narrative ultimately provides a clear and scientifically-based, integrated discussion of all nonclinical data and their implications for predicting potential seizure risks to patients in clinical trials.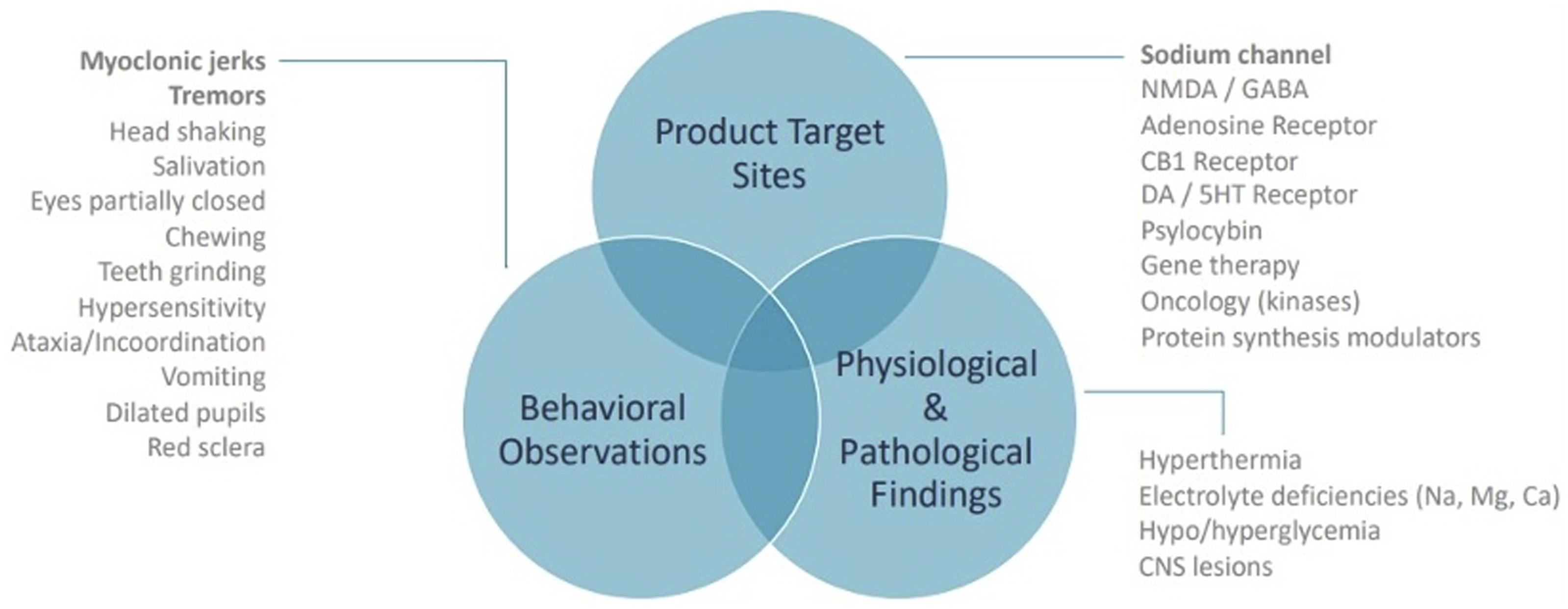
Nonclinical findings that are indicative of seizure liability are generally limited to clinical signs (e.g., convulsions, prodromal signs). Convulsions (irregular body and/or limb jerking) and various prodromal signs (e.g., ataxia, head shaking, and sometimes non-specific effects including altered facial movements and vomiting) are not always seizures (i.e., incited by abnormal brain wave activity), but convulsions and prodromal clinical signs will be considered to represent a seizure liability unless demonstrated otherwise. In the absence of convulsions, clinical signs observed during nonclinical studies can be challenging to interpret as a seizure liability. The difficulties arise because clinical signs can be subtle, the timing of observations may not capture the events, and study sites may describe signs differently. Harmonizing terms used for clinical signs associated with seizure liability is an area open for future standardization efforts. Other nonclinical data including PK and pathology data provide context for characterizing the seizure liability. However, definitive confirmation that a test article is inducing seizures requires functional testing of brain electrical activity, such as EEG analysis. In general, EEG is not included as an endpoint in the standard battery of GLP-compliant safety pharmacology and toxicology studies, so a nonclinical EEG study is usually designed as an additional experiment, and the key design parameters are set on a case-by-case basis using the data from a prior nonclinical study that identified the onset and offset of seizures in the most sensitive nonclinical species. Such EEG studies often include 24 h video-monitoring to document the pattern of in-life clinical signs indicative of a seizure liability. Integration of all these data types is useful for characterizing or de-risking the nonclinical seizure liability.
Supplemental Material
Supplemental Material - Identifying and Understanding Seizure Liability in Drug Development
Supplemental Material by Katie Sokolowski, Laura Erwin, Judy Liu, Simon Authier, Owen McMaster, Brandon Pressly, Brad Bolon and Marcus Delatte in International Journal of Toxicology
Footnotes
Author Contributions
The authors are solely responsible for the contents and crafting of this paper.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.