
Abstract
Test article (TA)-induced seizures represent a major safety concern in drug development. Seizures (altered brain wave [electrophysiological] patterns) present clinically as abnormal consciousness with or without tonic/clonic convulsions (where “tonic” = stiffening and “clonic” = involuntary rhythmical movements). Neuropathological findings following seizures may be detected using many methods. Neuro-imaging may show a structural abnormality underlying seizures, such as focal cortical dysplasia or hippocampal sclerosis in patients with chronic epilepsy. Neural cell type-specific biomarkers in blood or cerebrospinal fluid may highlight neuronal damage and/or glial reactions but are not specific indicators of seizures while serum electrolyte and glucose imbalances may induce seizures. Gross observations and brain weights generally are unaffected by TAs with seizurogenic potential, but microscopic evaluation may reveal seizure-related neuron death in some brain regions (especially neocortex, hippocampus, and/or cerebellum). Current globally accepted best practices for neural sampling in nonclinical general toxicity studies provide a suitable screen for brain regions that are known sites of electrical disruption and/or display seizure-induced neural damage. Conventional nonclinical studies can afford an indication that a TA has a potential seizure liability (via in-life signs and/or microscopic evidence of neuron necrosis), but confirmation requires measuring brain electrical (electroencephalographic) activity in a nonclinical study.
Seizures may occur in humans and animals as spontaneous events or following exposure to various exogenous products (chiefly chemicals, including small molecule drugs). In the United States, the prevalence of seizures (active epilepsy) in 2015 was 1.2% of the general population, of whom approximately 90% were adults. 12 Most cases of epilepsy in humans result from genetic mutations in critical neuronal proteins involved in maintaining cell health or facilitating signaling pathways needed for intercellular communication. Sporadic convulsions or seizures in various species including humans may reflect anatomic disruption of the parenchyma (e.g., chemically induced degeneration/necrosis, space-occupying lesions,42,51 or physical trauma20,50) but more often result from hyperthermia (e.g., fever or heat stroke 20 ); hypoxia20,50; metabolic imbalances (e.g., altered electrolyte [hypocalcemia, hypomagnesemia, hyponatremia]18,25,27 or glucose [hyperglycemia, hypoglycemia]6,25 levels); or arise from unknown causes. Nonclinical models of epilepsy are typically caused by chemicals (e.g., kainic acid, N-methyl-D-aspartate [NMDA], pentylenetetrazole [PTZ]41,50); electroshock20,24; genetic mutations (e.g., various genetically engineered or inbred mouse24,52 and rat24,40 strains); sensory barrage (intense acoustic or visual stimuli)20,50; and spontaneous events of unspecified provenance (e.g., epilepsy in colony-bred Beagle dogs22,26).
Nonclinical assessment of test article (TA)-induced seizure liability is a complex enterprise. Conventional methods for nonclinical safety screening include in-life endpoints (commonly limited to once- or twice-daily clinical observations and intermittent application of a functional observational battery [FOB], in some cases supplemented by a formal neurological examination); pharmacokinetic (PK) and/or biodistribution (BD) data, as appropriate for the TA class; and pathology evaluation (typically clinical pathology analysis of hematological and serum chemistry analytes, sometimes expanded by inclusion of coagulation and/or urinalysis parameters, as well as documentation of macroscopic and microscopic findings as well as selected organ weights). Taken together, these methods can identify and characterize many functional and structural abnormalities indicative of TA-related neural tissue damage. However, the integrated data set for conventional nonclinical toxicity studies at best implies a potential TA-related seizure liability without providing definitive evidence proving that brain electrical function has been disturbed. Ancillary tests like electroencephalography (EEG) are necessary to detect a disturbed brain wave pattern (i.e., a seizure signature).
The current paper adapts the content from a ½-day continuing education course entitled “Identifying and Understanding Seizure Liability in Pharmaceutical Development” to focus on the utility of neuropathology evaluation in the nonclinical investigation of TA-related seizure liability during biomedical product development. The content represents a truncated version of the information communicated on two occasions, first as a face-to-face session at the annual meeting of the American College of Toxicology (ACT) in November 2022 and again as a virtual course held under the auspices of the Society of Toxicologic Pathology (STP) in April 2024. The objective of this article is to explain what the neuropathology evaluation can, and cannot, contribute to nonclinical assessments of seizure liability. The concepts discussed herein will position possible pathology endpoints in the appropriate context for a meaningful and rational interpretation of the integrated nonclinical data set.
Approaches to the Nonclinical Assessment of Seizure Liability
In the nonclinical setting, seizure liability is detected most frequently following exposure to small molecule TAs (i.e., pharmaceuticals [“drugs”]) rather than biomedical products of other classes (e.g., biologics [nucleic acids and proteins], cell and gene therapies [including genomic editing], medical devices, vaccines, etc.). Nonetheless, the principles appropriate for assessing seizure liabilities of small molecules are also suitable for a similar purpose when applied to other TA classes.
Prior to first-in-human (FIH) clinical trials, nonclinical data that suggest a potential seizure liability are typically restricted to in-life clinical signs in pivotal nonclinical studies that include standard safety pharmacology and/or short-term (up to 13 weeks) repeat-dose general toxicology endpoints. In general, EEG analysis needed to definitively demonstrate seizurogenic potential is not a standard endpoint in nonclinical studies performed for routine drug development but can be implemented when warranted in common test species. The remainder of this section describes the various options for assessing seizure liability in the nonclinical setting, distinguishing between methods that are commonly used for screening and those that are technically feasible but impractical for various reasons.
In-Life Investigations
In nonclinical studies of a novel TA, a potential seizure liability is usually first identified by cage-side observations that detect one or a pattern of clinical signs known to presage or suggest seizures. In recording such events, precise terminology is required to ensure that the potential implications may be interpreted and translated accurately.14,36,39,43,53 Seizures proper are complex electrophysiological phenomena characterized by sudden bursts of aberrant, usually excessive electrical activity arising in the brain. Clinical presentations for seizures range from a standalone shift in consciousness (e.g., the “blank stare” that is the hallmark feature of an absence seizure) to unconsciousness accompanied by involuntary muscle movements (i.e., the classic convulsions of tonic-clonic seizures [where “tonic” means stiffening and “clonic” means rhythmical jerking or twitching of torso and/or limb muscles]) or even death from status epilepticus (i.e., a sustained seizure lasting 5 minutes or longer, or a prolonged series of seizures without complete recovery between seizures). In-life signs that can be mis-interpreted as seizures include severe ataxia, which manifests as uncontrolled, uncoordinated, and often hypermetric (exaggerated) whole body movements; conversion disorder, which is a psychiatric condition characterized by sensory or motor dysfunction; dystonia, which presents as abnormal muscle tone leading to muscle spasms and postural abnormalities; narcolepsy, which appears as a pronounced tendency to sleep; and tremors, which are evident as shaking of one or more body parts (usually a limb). An essential point to remember is that seizures commonly manifest as convulsions, but not all convulsions are seizures. Therefore, when reviewing clinical data, it may be important to request definitions of clinical terms used for a study as well as a detailed narrative of clinical events for which no universal definitions are available.
Clinical presentations of seizures proper differ based on their pathogenesis. Focal seizures (alternatively “partial seizures”) arise by electrical disturbances that begin in a defined brain locus, although they may spread over time to involve the whole brain (a process termed “secondary generalization”). Generalized seizures affect the whole brain simultaneously. In nonclinical studies, neuroactive TAs and/or their neuroactive metabolites tend to induce generalized seizures or in some cases partial seizures that rapidly generalize. Drug effects restricted to partial seizures are rare in nonclinical studies. The nature of any epileptiform activity in the brain can be characterized by EEG monitoring.
Seizures often progress through a definable series of phases that depend on the seizure type and patient’s demographics.15,35,39,53 In humans, focal seizures often begin with an aura, which is a sensory symptom or mental perception that develops with onset of the altered brain wave pattern. Auras cannot be detected in nonclinical studies because animals cannot verbalize their perceptions. Seizures caused by toxic exposures/metabolic syndromes are typically heralded by a prodromal phase (in which irritability or lethargy are evident) before signs of a full-blown seizure manifest during an ictal phase (marked by altered consciousness and convulsions), and a postictal phase (featuring confusion, fatigue, and occasionally transient paralysis of body parts innervated by affected brain regions). The three phases of seizures caused by toxic exposures/metabolic syndromes may be discerned during nonclinical studies if cage-side observations are undertaken by experienced personnel. Subtle prodromal signs seen in animals may include both neurological effects (e.g., ataxia, head shaking, and/or tremor) and nonspecific signals such as excessive facial movements (e.g., chewing, licking, and yawning); partially closed eyelids; and in some cases vomiting. In nonclinical studies, the significance of prodromal signs is often understood only in retrospect when their occurrence in relation to convulsions can be confirmed. In GLP-compliant toxicity studies, dose-related convulsions in healthy or diseased (even seizure-prone) animals are likely to be perceived by regulatory reviewers as evidence of a seizure liability of concern for human risk assessment unless additional de-risking studies are performed. Until proven otherwise, for pro-convulsant TAs, more benign prodromal signs observed at lower TA doses (with or without convulsions at that dose) are also likely to be perceived by regulatory reviewers as indicating a potential seizure liability, and thus as adverse events. The reason for this regulatory perspective is that the duration and characteristics of epileptiform activity and/or seizures associated with a specific TA often follow a pattern, and this TA-related signature is typically preserved across multiple test species.
Convulsions and tremors in the absence of abnormal brain electrical activity are not seizures. Thus, definitive discrimination of convulsions versus seizures is not possible in routine nonclinical studies since brain electrical activity is not assessed using methods incorporated in conventional study designs. Historical control data for multiple nonclinical species suggests that TA-related convulsions are associated with EEG alterations to a variable extent (ranging from 45% to 100%). The proportion of myoclonic movements associated with EEG alterations is substantially lower than convulsions, and this proportion further decreases when myoclonic movements are observed at sleep onset (i.e. when there is a higher frequency of physiologic myoclonus). When a TA or a dose level induces myoclonic movements but no convulsions, EEG monitoring is essential to characterize the neurological signs and the seizurogenic risk. Tremors alone can be associated with epileptiform morphologies, and when a TA is associated with frank seizures at higher doses, the risk of abnormal EEG morphology during tremors rises at the time of peak drug concentraion (Tmax). For the few instances where the clinical observation data remain inconclusive, additional nonclinical studies may be conducted to clarify the seizure liabilities. Such studies typically combine two elements: an EEG analysis to directly measure brain electrical activity, and 24-hour video-monitoring to clarify patterns of prodromal and ictal clinical signs (in terms of duration, frequency, incidence, and severity) in relation to any altered brain wave pattern. Video-monitoring over time is critical because clinical signs are often intermittent and thus may not be seen at the once- or twice-daily observation periods used for conventional nonclinical studies. Moreover, video-monitoring is essential for EEG morphology interpretation. Further details regarding key design features of nonclinical EEG studies may be gleaned elsewhere.3,4,7 In the absence of adequate safety margins in nonclinical studies in which convulsions or prodromal signs occur at the dose levels tested, EEG studies are needed to confirm the nature of the neurological effects, the presence of seizure activity, safe drug doses, and systemic and tissue exposure levels in the tested species.
Neuropathology Evaluations
Routine pathology endpoints assessed during conventional nonclinical studies are variably sensitive for identifying seizure liabilities but usually are insufficient to definitively confirm that clinical evidence of seizurogenic potential represents a true seizure liability. Types of pathology data collected for standard nonclinical studies include hematology (complete blood count and leukocyte differential count); serum chemistry panel (to examine function of many organs, but usually not brain); gross observations of organ appearance at necropsy; weights for the whole body and selected organs (generally including brain); and microscopic examination of a variable menu of organs (typically including multiple brain domains). The standard nonclinical pathology data that are most informative when assessing a potential TA-related seizure liability are detailed lists of microscopic findings, ideally documented for particularly sensitive brain regions and not just the organ as a whole.
In the clinic, in-life neuroanatomical evidence of brain damage associated with seizures may be acquired using computed tomography (CT) and magnetic resonance imaging (MRI). An MRI study is the gold standard for assessing regional morphology in brain domains with a recognized vulnerability in seizure disorders while CT scans are utilized in some cases to detect skull trauma and acute hemorrhage in the brain meninges or parenchyma that may be responsible for certain seizure states. Macroscopic MRI findings observed in patients may show structural anomalies underlying seizures, such as focal cortical dysplasia, while patients with chronic epilepsy may exhibit cerebellar atrophy (i.e., shrunken cerebellar folia), currently attributed to chronic anti-convulsant exposure, and/or hippocampal sclerosis (i.e., reduced regional size and/or visible condensation [scarring], Figure 1).1,2,23,48 These lesions reflect extensive local neuronal loss with subsequent astrocytic and microglial reactions. Comparable neuro-imaging methods (e.g., micro-CT, micro-MRI [magnetic resonance microscopy] 16 ) are feasible in animals but are almost never employed in nonclinical studies as a routine screening tool due to their high cost (in terms of labor, money, and time).
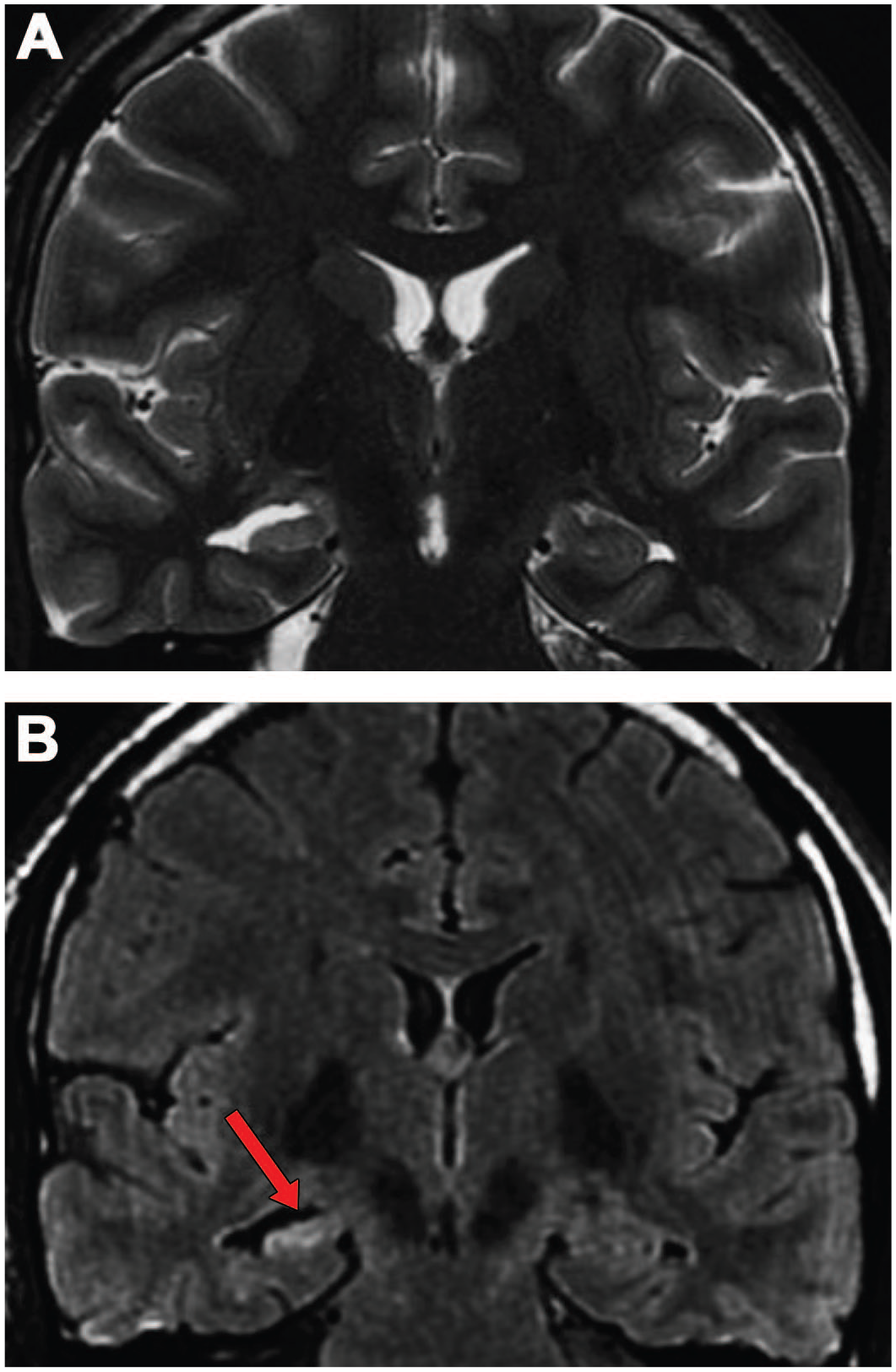
Magnetic resonance imaging (MRI) study demonstrating unilateral hippocampal sclerosis in an adult male with chronic mesial temporal lobe epilepsy. Coronal T2-weighted (Panel A) and fluid attenuated inversion recovery (FLAIR; Panel B) images taken perpendicular to the long axis of the hippocampus. The right hippocampal head (arrow) is diminished in size (“atrophy”) and exhibits a hyper-intense signal (“sclerosis”). Images reproduced from Abud et al 1 under a Creative Commons CC BY 4.0 DEED International license.
With respect to clinical pathology endpoints, various parameters may provide information regarding the presence and sometimes the cause of TA-related seizures. Useful routine serum chemistry measurements include electrolytes (calcium and sodium) and glucose since fluctuations in these analytes can induce seizures (hyperglycemia, hypocalcemia, hypoglycemia, hyponatremia).6,18,25,27 Careful attention must be given to understand the basis of these shifts, which may or may not be related to TA exposure. Hypoxia (low blood oxygen levels) is also associated with seizures, but measurement of arterial blood gases is rarely undertaken for routine nonclinical studies. Biomarkers of neural injury may be measured in both nonclinical studies and the clinical setting in fluids (blood and/or cerebrospinal fluid [CSF]), which can be collected once or intermittently over time. In general, for nonclinical programs these biomarkers are evaluated more often in drug discovery studies, but where warranted such endpoints may be assessed during safety studies as well. In animals, neural biomarkers of current interest include proteins38,49 released by primary injury to neurons (e.g., neurofilament light chain [NfL], neuron-specific enolase [NSE], tau) or secondary glial responses to parenchymal injury in the brain (e.g., glial fibrillary acidic protein [GFAP]) as well as certain microRNAs.11,47 Importantly, these comparatively noninvasive biomarkers are not specific for seizures,19,21,32,34,37 so their relevance in detecting a seizure liability will depend on corroborative data showing that altered analyte levels or activities are associated with electrical disturbances in the brain (by EEG) and/or visible brain damage (by microscopic evaluation).
Standard anatomic pathology endpoints for conventional nonclinical studies include brain weights, gross observations, and microscopic evaluation. Of these, microscopic analysis is the main technique for detecting tissue changes indicative of seizures. Key considerations assessed during routine microscopic evaluations include whether a pattern of morphological findings can be identified that helps define the origin and/or etiology of the seizures and whether the seizures were the sole or a contributing cause for any seizure-related deaths. In many cases, TA-related microscopic findings in the brains of test animals are an indication that a seizure has occurred rather than evidence identifying the cause of the seizures. Microscopic findings associated with toxicant-induced seizures may include neuronal degeneration/necrosis in various brain regions — most often localized to the neocortex (middle layers), hippocampus (particularly the CA1 and CA3 neurons; Figure 2), and cerebellar folia (Purkinje cells)—as well as acute hemorrhage; these changes range from multifocal to regionally extensive and may be unilateral or bilateral (asymmetric or symmetric). 44 Following exposure to brain-penetrating small molecules, classical features of neuronal death in the central nervous system (i.e., shrunken “red dead” cells featuring hypereosinophilic cytoplasm, condensed [pyknotic] or fragmenting [karyorrhectic] nuclei, and clear pericellular halos) typically develop within 1 day of cell demise, with numbers peaking at 2-4 days 46 before being removed by 7-10 days after the insult. 45 In the authors’ experience, this same pattern holds for seizure-related neuronal death, whether or not incited by a toxic small molecule TA. The timing of neuronal death may vary from this pattern for toxic agents of other classes (e.g., biologics, gene therapy agents) or for neurons in other locations. 5 Gliosis and perivascular mononuclear cell infiltration may develop in affected regions if the animals survive for a sufficient time to permit such processes. For glial reactions in the brain, microgliosis tends to develop earlier (by 7 days post-insult) and more consistently compared with astrocytosis (about 7-14 days post-insult). 8 Subtle lesions may be highlighted with specialized neurohistological procedures to detect cell death (e.g., Fluoro Jade in paraffin sections [Figure 2] or amino cupric silver in frozen sections) or cell type-specific immunohistochemical markers for reactive glia (e.g., GFAP for astrocytes or ionized calcium-binding adaptor molecule 1 [IBA1] for microglia). Animals in routine nonclinical studies are typically culled for humane reasons if acute severe seizures are observed and generally are not treated with the TA long enough to develop any macroscopic findings if repeated sublethal seizures occurred but went undetected during the study. An important consideration to remember when interpreting microscopic data in assessing potential seizure liability is that the neuronal and glial patterns described above for seizures are not pathognomonic for seizures (toxicant-related or otherwise) as other entities (e.g., hypoxia, metabolic disturbances) may cause neuronal necrosis in the absence of electrophysiological anomalies. This lack of specificity underscores the importance of electrophysiological testing when assessing seizure liability in nonclinical studies.
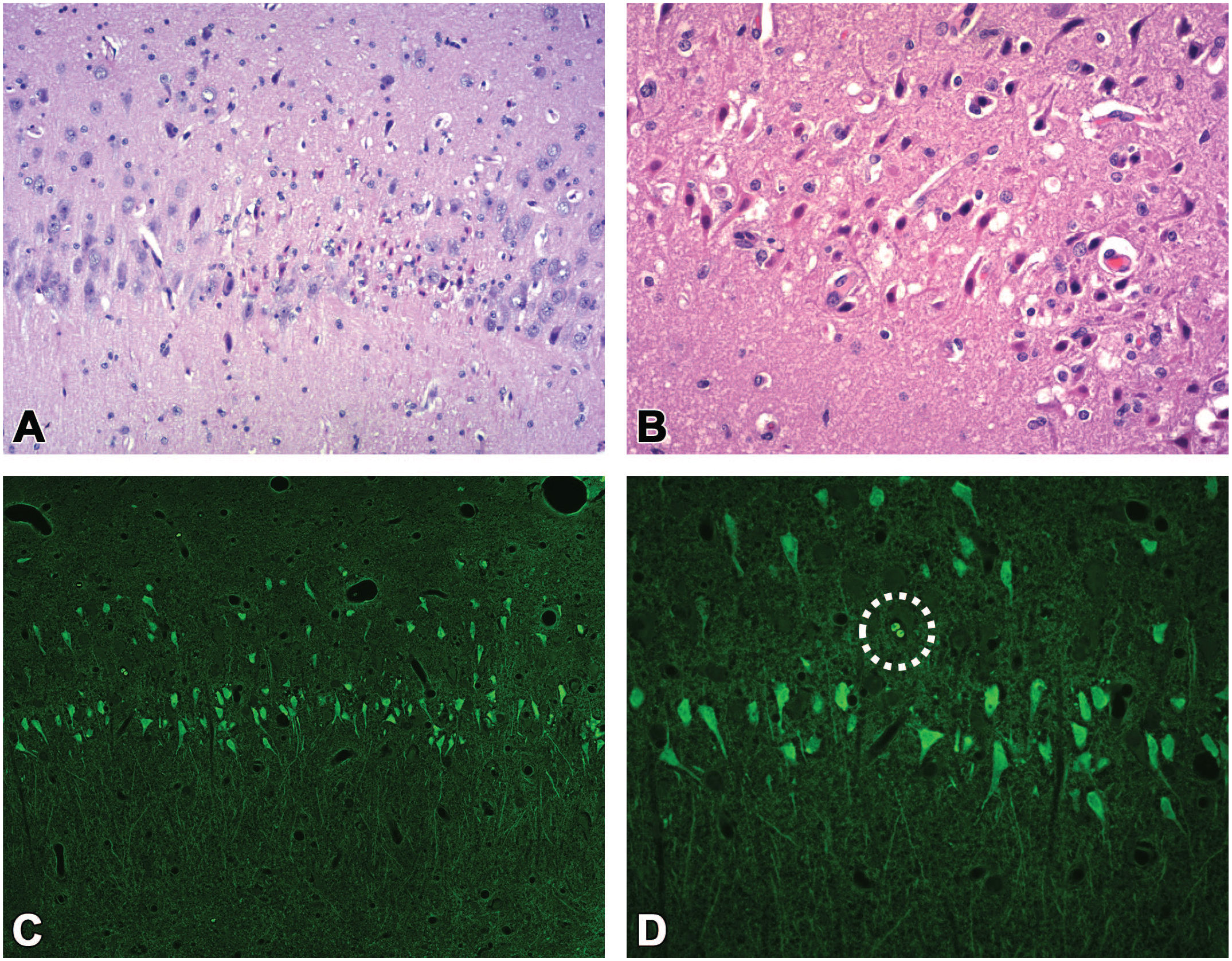
Acute neuronal degeneration and necrosis in the CA1 field of a kainic acid-treated, adult Sprague-Dawley rat indicating peri-mortem tonic-clonic seizures. In routine hematoxylin and eosin (H&E)-stained sections (Panels A and C), affected neurons possess condensed, hyperbasophilic nuclei and hypereosinophilic cytoplasm (i.e., classic “red, dead” morphology) while the nearby parenchyma is pocked by many small, clear, colorless vacuoles. In site-matched sections (Panels B and D) stained with Fluoro Jade B (a cell death marker), affected pyramidal neurons and their proximal processes fluoresce bright green against a dark green parenchyma. The dotted white circle in Panel D denotes a capillary containing two erythrocytes, which exhibit bright green autofluorescence if viewed using a fluorescein isothiocyanate (FITC) filter.
In conventional nonclinical studies, anatomic sampling of brain for microscopic evaluation of brain is generally determined by rote application of recognized industrial best practices and/or time-tried institutional standard operating procedures. Internationally accepted recommendations for sampling during nonclinical general toxicity studies, 10 which calls for 7 brain blocks in both rodents (full coronal sections) and nonrodents (coronal hemi-sections), provide a suitable baseline screen for major brain regions in which seizure-related lesions may develop. Structures to assess in nonclinical studies when evaluating a potential seizure liability include the neocortex (especially the frontal, parietal, and temporal cortices); limbic system (particularly the entorhinal cortex, hippocampus, and amygdala); basal ganglia (caudate and putamen); thalamus (dorsal nuclei); cerebellum; and brainstem (Figure 3). 48 Further brain sampling and/or processing (i.e., expanded neurohistopathology evaluation9,13) may be desirable to ensure that brain regions associated with specific in-life clinical signs not localized to this generic list of sensitive sites will actually be sampled. Well-annotated neuroanatomic atlases have been released to show trimming schemes for the baseline screen (7 levels) and expanded surveys (9 to 16 levels depending on the species) for several nonclinical species including rats, 33 rabbits, 30 dogs,17,28 minipigs, 31 and nonhuman primates (NHPs 29 ).
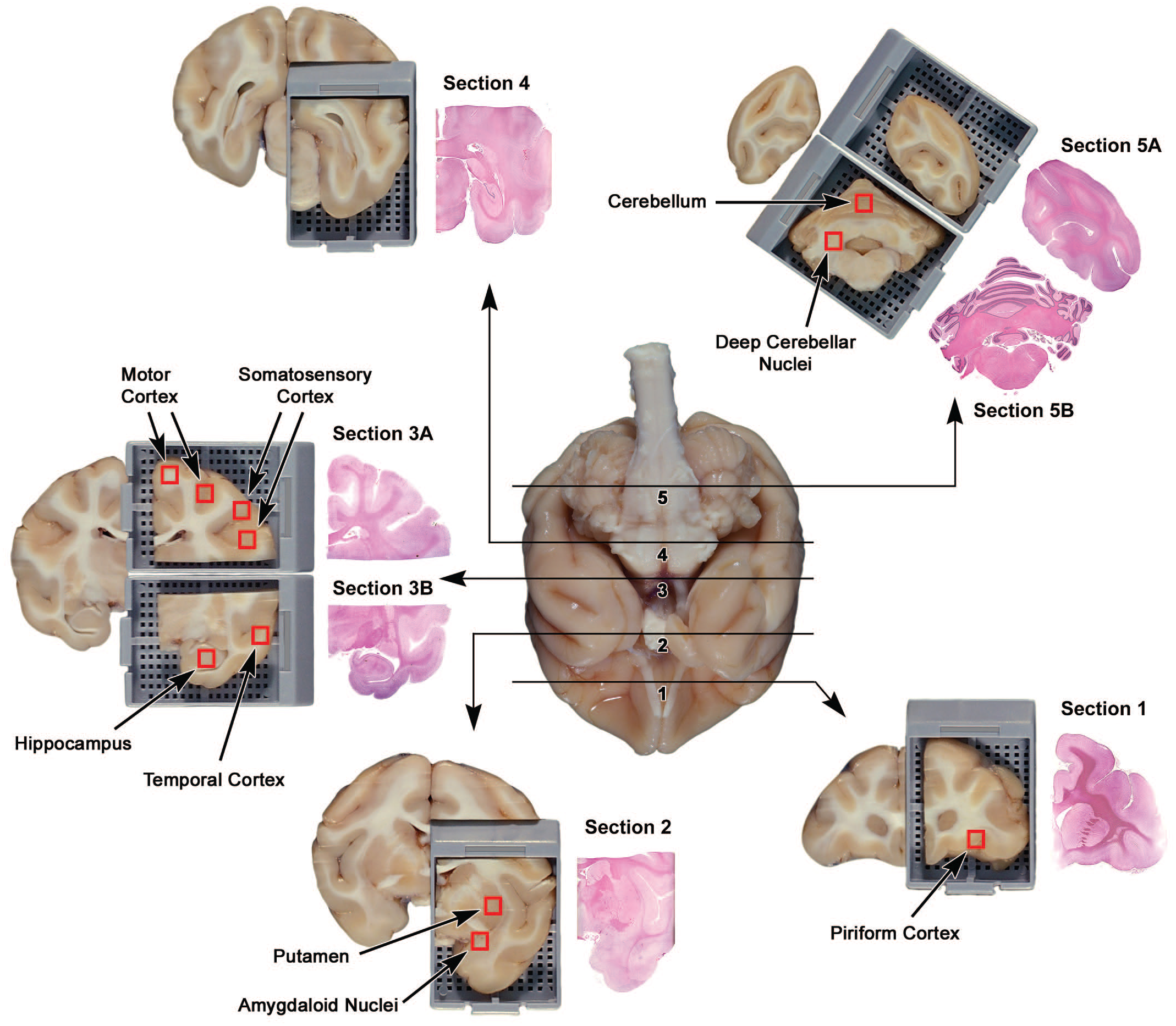
Brain trimming scheme for a cynomolgus macaque showing that the widely recognized 7-level sampling scheme for nonclinical general toxicity studies will effectively incorporate numerous brain regions (annotated red boxes) that have been linked to spontaneous and test article-related seizures in animals and humans. Image reproduced with expanded annotation from Bolon et al, by permission of Sage Publications. 10
Conclusions
Many drug candidates and some marketed drugs possess the ability to induce seizures under certain dosing schedules. In general, seizure liability is difficult to investigate in the nonclinical setting since none of the conventional assessment methods detect the electrical disturbances in the brain that represent the indispensable attribute of seizure-inducing agents. Instead, common seizure-related findings that may be detected by routine nonclinical methods include convulsions and prodromal signs such as tremors during life and in some instances acute neuronal death in tissue sections. If these standard endpoints are insufficient to fully characterize a seizure liability, additional nonclinical analyses may be undertaken to confirm a TA’s seizurogenic potential. For an ongoing study, expanded neurohistopathological evaluation on embedded brain specimens and/or archived fluid samples (CSF or serum) may be performed to detect sites of parenchymal damage and cell type-specific markers of neuronal death or glial responses. However, definitive demonstration of a seizure liability will typically necessitate a nonclinical study in which in-life neurological signs and anomalous electrical patterns in the brain are investigated using both EEG analysis and video-monitoring.
Footnotes
Acknowledgements
The authors gratefully thank Ms. Beth Mahler for assistance in optimizing the figures.
Author Contributions
The authors are solely responsible for the contents and crafting of this paper. Katie Sokolowski conceptualized and developed the learning objectives of the continuing education course and developed the regulatory perspective presentation. Judy Liu develped and presented the clinical perspective presentation. Marcus Delatte develped and presented the nonclinical perspective presentation. Simon Authier developed and presented the nonclinical EEG presentation. Owen McMaster presented the regulatory perspective presentation. Brad Bolon conceptulaized and develped the evaluation of seizure liability from a histopathological perspective and drafted the manuscript. All authors assisted in editing the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the views or policies of their employers or global regulatory agencies including the U.S. Food and Drug Administration (FDA).