
Abstract
A BioSafe-sponsored survey investigated how vaccine companies (n = 12) perceive the value of the repeat-dose toxicity studies for safety assessment of vaccine candidates. As all major vaccine developers were part of the survey, it was considered representative for the industry practices up to 2022. Vaccine developers indicated that they see scientific value in performing repeat-dose toxicity studies with vaccines, especially when novel components (e.g., adjuvant) or technology is being used. However, a few (3/12) also indicated that repeat-dose toxicity studies could be replaced by a pharmacology study with additional toxicity parameters. For the majority of companies (9/12), findings from the repeat-dose toxicity studies never prevented or postponed a first-in-human (FIH) trial. In the remaining 3 companies, a total of 4 occurrences of postponement or prevention of clinical development occurred and in only 2 of these cases was the finding considered related to the vaccine. A platform approach has been successfully implemented for influenza vaccines already in 2016, and an outline of the regulatory requirements for a platform approach has been recently documented in the latest infectious disease mRNA-LNP vaccine guideline, as well as in the guidance on the development and licensure of COVID-19 vaccines presented by the FDA. Vaccine developers are seeking to extend this platform approach to the development of new vaccines, building on established technologies and using well-defined manufacturing processes. This approach could support reduction of animal use (a principle of 3Rs) while still providing reassurance of the nonclinical safety of these products.
Introduction
It is only ∼30 years ago that the nonclinical safety evaluation of infectious disease vaccines was implemented following the publication of the first guidance on nonclinical development of vaccines by the European Medicines Agency (EMA): Note for Guidance on Preclinical Pharmacological and Toxicological Testing of Vaccines. 1 This Guidance focused on the need to have animal studies conducted before the first-in-human (FIH) clinical investigation. Later, the World Health Organization (WHO) took the initiative to write global guidelines on the nonclinical evaluation of vaccines, 2 on nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines, 3 and most recently on the quality, safety, and efficacy of RNA-based vaccines. 4 The toxicology evaluation of a new vaccine candidate is based on a GLP-compliant repeat-dose toxicity study usually conducted in a single relevant animal species with the intended final clinical formulation. The repeat-dose toxicity study is designed to mimic the clinical regimen (or a more accelerated regimen, dosing every 2–3 weeks) and route of administration, allowing the evaluation of the local reactogenicity (e.g., injection site inflammation) and the systemic toxicity (i.e., toxicity that is occurring at sites distant from the site of initial administration, including potential toxicity related to the development of an adaptive immune response and possible intrinsic toxicity to the non-antigenic components of the vaccine). To better understand how vaccine developers currently approach nonclinical safety evaluation of their vaccine candidates prior to FIH, a survey was performed in 2022 through BioSafe, focusing on the design and information retrieved from these repeat-dose toxicity studies, notably their translation to safety in clinic, to get a snapshot in time of the current practices.
Since vaccines are diverse biological products including, but not limited to, inactivated or live attenuated organisms (bacteria or viruses), purified pathogen subunit, nucleic acid-based, etc., it could be expected that the nonclinical testing programs are adapted based on vaccine-specific features and clinical indication. However, the components (antigens, adjuvants, or excipients) of most candidate vaccines are frequently manufactured with well-defined and characterized processes. This has led to production of multiple vaccines that utilize similar types of constituents or technologies (such as polysaccharide conjugates, recombinant proteins, viral vector vaccines, and mRNA or DNA plasmid vaccines), with only the target antigen (which should not have pharmacological activity itself) being changed, suggesting that translation of the nonclinical safety information from one product to another may be relevant. This so called “platform approach” of vaccine development was also a subject of the survey, deciphering the strategies followed by the sponsors to set the criteria that may lead to established vaccine platforms.
Methods
A questionnaire related to current practices in vaccine nonclinical safety assessment and supported by the BioSafe Vaccines Specialty Biologics Expert Working Group was conducted from May to July 2022. The survey was sent to 36 companies (members of Biotechnology Innovation Organization [BIO]), and the responses from individual companies were kept confidential and anonymized by the BioSafe Coordinator before being provided to the survey authors.
The survey included 37 questions with the aim to receive feedback on the vaccines in development, on the occurrence of nonclinical findings that potentially impacted vaccine development, and more specifically on the companies’ perspectives on the scientific value of the repeat-dose toxicity studies.
In addition, the companies were asked for their experience with a platform approach to vaccine development and the criteria used to set this platform. The survey questions are listed in the Supplemental Materials.
Of the 37 questions, this paper focuses on and summarizes the outcome of 23 questions (bolded in question list), which are considered to be the key to consideration of the impact of the toxicity studies on proceeding to FIH, namely, the information retrieved from the repeat-dose toxicity studies. Briefly, these questions included basic company demographic information, types of vaccines developed, if a repeat-dose toxicity finding impacted FIH initiation, under what circumstances would a study be repeated, use of a platform approach, and how much data is needed to establish a platform. The remaining 14 questions were related to the development of adjuvants or the conduct of other studies (developmental and reproductive toxicity (DART) or biodistribution studies) and were therefore excluded from this paper.
Results
General Information
Total of Complete/Incomplete Survey Responses From Companies According to Their Location.

aIn one case, two representatives from the same company responded to the survey. The responses were consolidated though individual location-based data has been retained and is broken out here for accuracy/transparency.
The 12 companies that provided evaluable responses were further categorized according to the number of employees that directly work on vaccine development, with 7/12 companies having between 100 and 1000 employees and 5/12 companies having >1000 employees. Although BioSafe did not reveal the names of the vaccine companies that provided the evaluable responses, they nevertheless confirmed that all major vaccine developers, based on those companies with the highest number of approved or authorized vaccines in the United States or Europe (centralized process) as of 2022, were part of this group. It can thus be assumed that the conclusions drawn from this survey are representative of current industry practices.
Intended Clinical Use of Vaccines in Development
The majority of companies (10/12) are working on prophylactic vaccines, and it was noted that over half of them (7/12) are also working on therapeutic vaccines. The most common vaccines under development are subunit/conjugate vaccines and mRNA-based vaccines (both 9/12). Approximately half of respondents are working on inactivated or attenuated (7/12) or DNA-based (6/12) vaccines (viral vectors or plasmids), while very few (2/12) are developing other types of vaccines (not disclosed).
Potential Impact of Nonclinical Toxicity Findings on the Start of First-in-Human (FIH) Clinical Investigation
One of the major questions in this survey concerned prevention of or delays in the start of an FIH clinical trial that vaccine developers faced due to findings from a repeat-dose toxicity study (Figure 1). A total of 9/12 companies answered that this has never occurred, 2 companies indicated that this happened once, and 1 company reported that this had occurred twice. When asking if the toxicity finding was related to the vaccine, it appeared that 2 cases (subunit/conjugate and adjuvanted vaccine) ultimately were not considered related to the vaccine, while the remaining 2 cases were related to the vaccine with 1 case, an mRNA vaccine, due to local reactogenicity, and the other, an inactivated or attenuated virus vaccine (not specified which type it was), due to systemic toxicity (Figure 1). Prevention or postponement of FIH studies and potential relationship to nonclinical toxicity findings. The majority of survey respondents (9/12) indicated that results from a repeat-dose toxicity study had never postponed or prevented an FIH trial. In the minority of respondent companies (3/12) where this did occur, there were only 4 total cases (1 company provided 2 cases and 2 companies provided 1 case each). Of the 4 total cases, only 2 cases were determined to be related to the vaccine.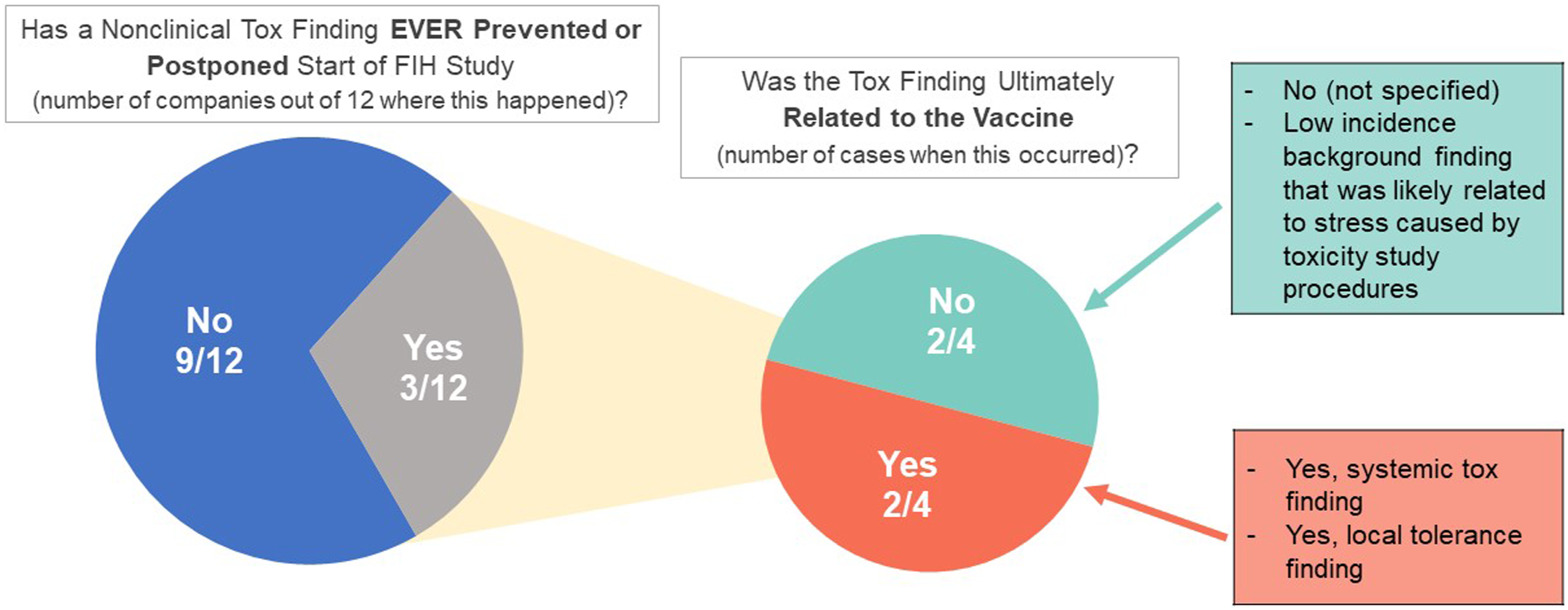
Perspective on Scientific Value of the Repeat-Dose Toxicity Studies
Historically, most toxicity studies with intramuscular vaccines produced very similar nonclinical safety profiles (inflammation at the injection site, increased cellularity in lymphoid organs, and related clinical pathology changes) indicating an expected pharmacological response at the injection site and immunostimulatory effects.5-9 With this in mind, we asked companies for their perspective on the scientific value of the repeat-dose toxicity studies.
Of the 12 companies that responded, 7 companies answered that they see added value in these repeat-dose toxicity studies and 5 companies added conditions to their scientific value. The group of companies that agreed to the scientific value indicated that these repeat-dose toxicity studies are needed to give confidence in the nonclinical safety of the product prior to the FIH study, since they provide useful information on parameters that need to be monitored during clinical trials. In addition, repeat-dose toxicity studies could inform on dose response to an adjuvant (if included), local and systemic findings, and prediction of immune stimulation and can help redesign studies for the next vaccine candidates.
Companies that added conditions indicated that these repeat-dose toxicity studies have less scientific value when testing traditional, intramuscular vaccines or vaccines using established adjuvants. However, scientific value is recognized when novel adjuvants/delivery devices are evaluated. In addition, repeat-dose toxicity studies can be beneficial to de-risk any potential toxicities of vaccine platform components such as novel lipids.
Use of Pharmacology Studies for Safety Assessment
Companies were asked if they had used pharmacology studies only to support safety assessment of vaccines instead of toxicity studies (Figure 2). A total of 7/12 companies indicated that they used pharmacology studies to support safety evaluation and 3/12 companies indicated that they used pharmacology studies in addition to but not as a replacement of toxicity studies. The remaining 2/12 companies did not use pharmacology studies to support or replace toxicity studies. The survey only provided a write-in option when “No” was chosen. One company provided feedback; however, the answer could not be clearly interpreted. The use of pharmacology studies to support safety assessment of vaccines instead of toxicity studies. Most companies (7/12 companies) indicated that they used pharmacology studies to support safety evaluation, and 3/12 companies indicated that they used pharmacology studies in addition to but not as a replacement of toxicity studies. The last 2/12 companies did not use pharmacology studies to support or replace toxicity studies.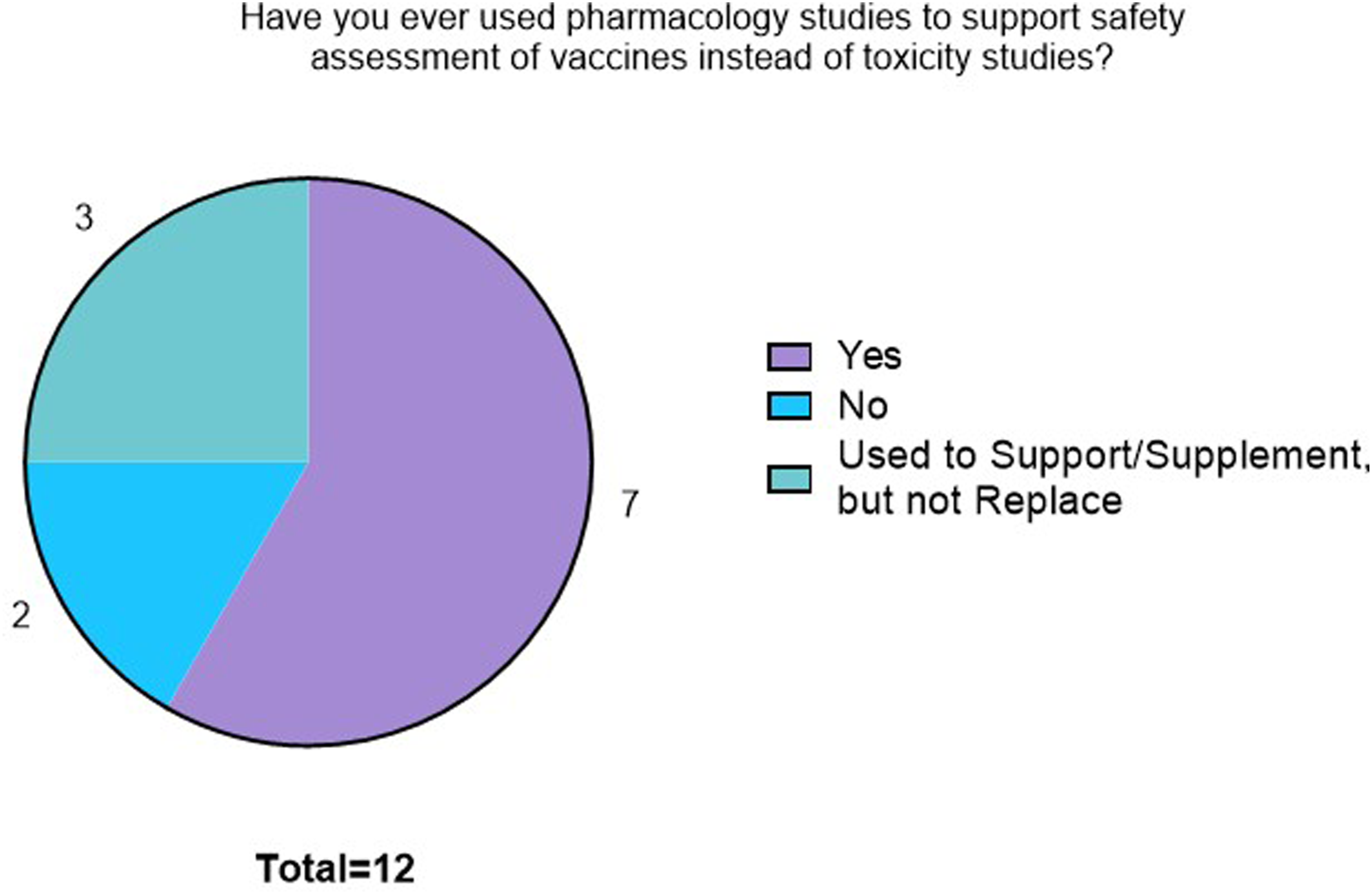
As a follow-on question, the companies were asked whether a pharmacology study incorporating toxicology parameters could replace a repeat-dose toxicity study with the vaccine (Figure 3). Eight companies supported this proposal, whereas four others did not. Both answers had write-in options and because five companies added conditions to their “yes” answer, BioSafe decided to add an additional answer option called “depends.” Company position whether a pharmacology study with additional toxicology parameters could replace a repeat-dose toxicity study with the vaccine. Four of 12 respondents did not agree with this position, 3/12 companies responded positively without conditions, and the remaining 5/12 respondents thought this could be possible depending on certain criteria.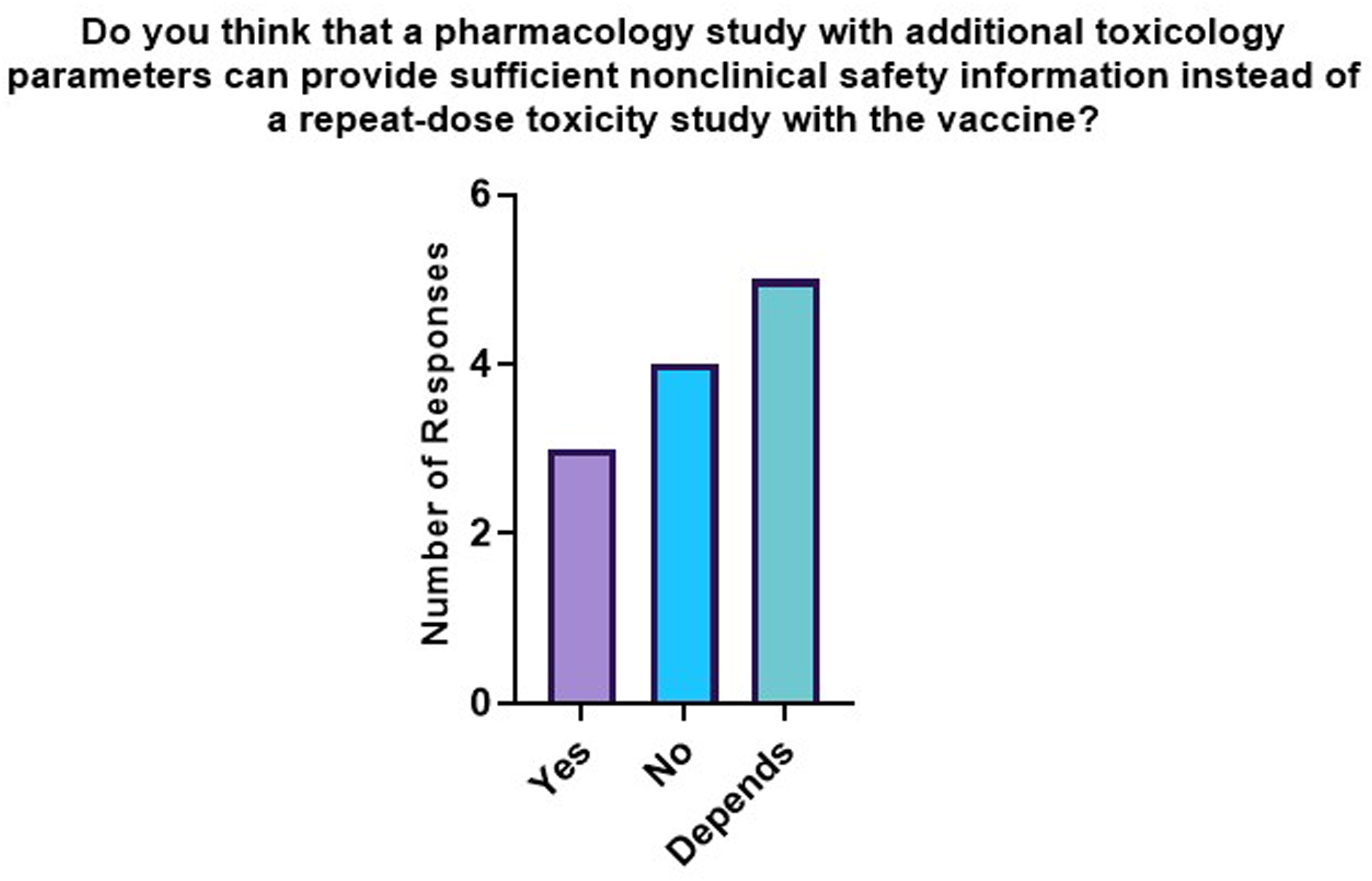
Feedback received from companies that responded “No” pointed to the lack of terminal procedures (histopathology), the lack of historical control data in pharmacology animal models, and the fact that pharmacology studies are usually not GLP compliant, which could lead to regulatory questions on data integrity. Companies that voted “yes” indicated that the addition of toxicology parameters to a pharmacology study could generate the same nonclinical safety information. Companies that responded “depends” indicated that this could work when a pharmacology study is complementary to repeat-dose toxicity studies already conducted in support of a platform approach. One company noted that this approach could be successful, depending on the animal species and design of the pharmacology study because such a study is performed with a pharmacology “mind-set” potentially compromising the quality of the toxicology assessment.
Vaccine Formulation Changes That Require Performing an Additional Repeat-Dose Toxicity Study
The companies were asked about the nonclinical testing strategy when the vaccine formulation is changed after the pivotal repeat-dose toxicity study has been completed (Figure 4). Addition of a preservative or an established buffer or excipient would generally not require an additional repeat-dose toxicity study, as 10/12 and 11/12 companies voted for this option, respectively. However, the addition of a novel adjuvant to the formulation would generally (11/12) require a dedicated repeat-dose toxicity study. When asking whether the addition of a novel chemical entity adjuvant (synthesized adjuvant) would necessitate a repeat-dose toxicity study, 9/12 companies indicated the need for such a study in rodents. An additional repeat-dose toxicity study in non-rodents was considered necessary by 5/12 companies. A similar result was obtained when a novel natural adjuvant is added in the formulation as 8/12 voted for performing a repeat-dose toxicity study in rodents, whereas only 4/12 companies saw the need for an additional study in non-rodents as well. Company position on performing an additional GLP repeat-dose toxicity study when changing the vaccine formulation. Respondents strongly agreed with conduct of an additional GLP repeat-dose toxicity study when a new adjuvant (11/12), a different adjuvant (8/12), or a novel buffer or other excipient (8/12) was included in the vaccine formulation. Respondents also strongly agreed that a new study was not needed when an established buffer or excipient (11/12) or a preservative was added to the vaccine formulation (10/12). However, respondents were evenly divided (6/12—yes and 6/12—no) on if an additional study would be required with a change to the formulation that altered the immune response to the antigen(s) in the vaccine.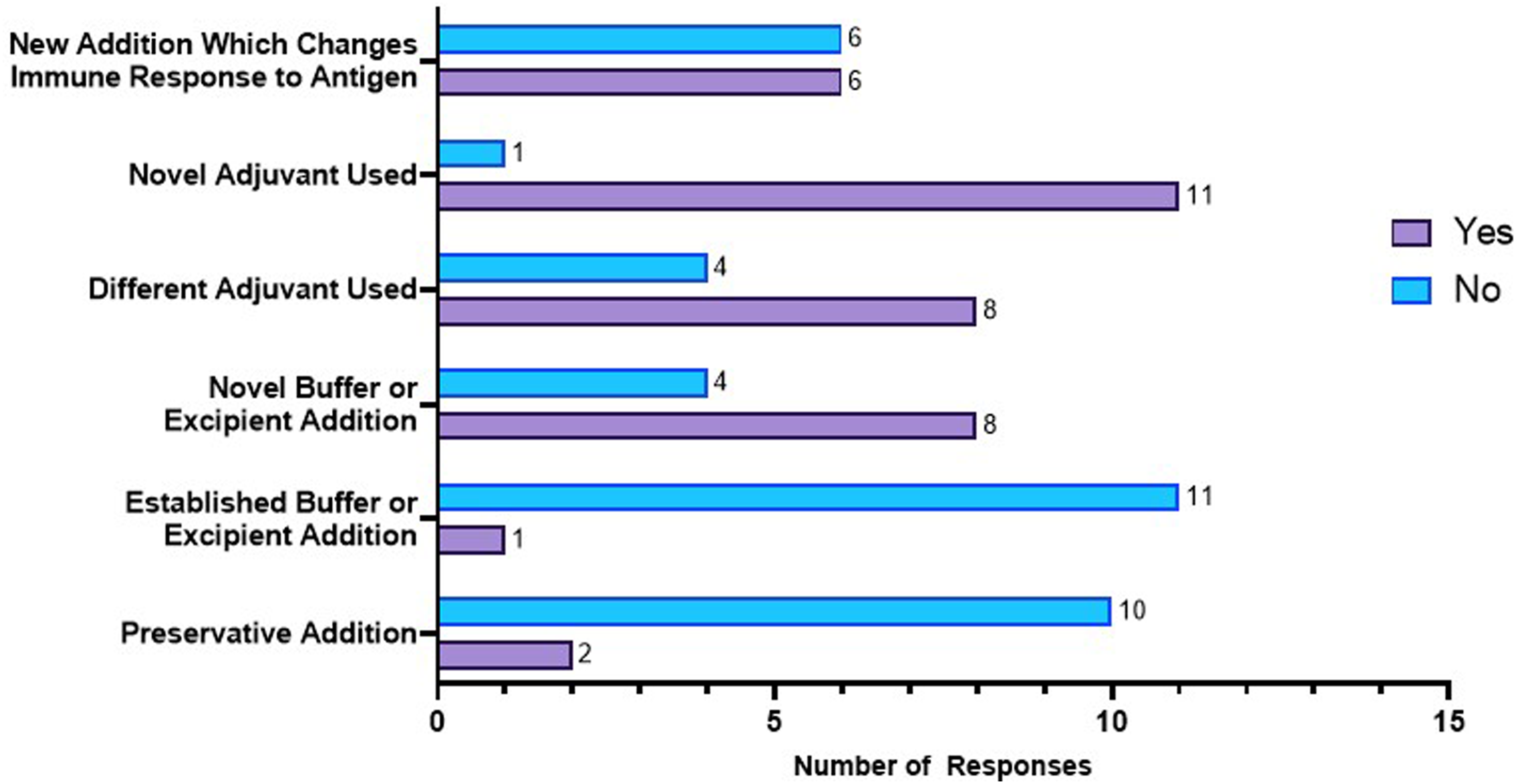
There was also less agreement on performing a dedicated repeat-dose toxicity study when adding a novel buffer or a different but established adjuvant to the formulation as 8/12 companies took the position to perform a new study. When asking if an additional repeat-dose toxicity study needs to be conducted when adding any component (novel or well-established) that would change the immune response, a mixed response was received as half of the companies responded either “yes” or “no.”
Platform Approach for Vaccine Development
In the last set of questions, we asked the companies how they perceive the platform approach for nonclinical safety vaccine development. All companies agreed to our formulated definition of a vaccine platform: “When using a platform for vaccine development, companies can develop multiple vaccines in a short period of time by using the same mechanism, cell line and/or delivery method (e.g., vector, lipid nanoparticle etc.). In the example of an adenoviral vector, a gene of interest can be inserted and tested in a nonclinical toxicity study. For subsequent other vaccines, the gene of interest will be replaced by another gene.” In addition, most companies (9/12) indicated that they had used a platform approach for vaccine development, primarily for mRNA-LNP vaccines.
The survey next investigated how many repeat-dose toxicity studies are needed to establish a vaccine platform. The majority (7/12) of respondents indicated that three or less studies would be sufficient to establish a platform approach. A few companies indicated that between 3 and 5 repeat-dose toxicity studies with different vaccine candidates would be required, while none indicated that more than 5 studies would be needed.
When asked if clinical data are needed to validate the platform approach or if nonclinical data are sufficient, a variety of responses was received; however, most companies (8/12) indicated that clinical data are always or sometimes required (Figure 5). Companies that voted for “always needed” (5/12) indicated that it is important to show that nonclinical findings translate to the clinic before considering a platform as being established. Company position for the need of clinical data to validate a platform approach. When queried about the need for clinical data to validate a platform approach, responses were mixed; however, most companies (8/12) indicated that clinical data are always or sometimes required.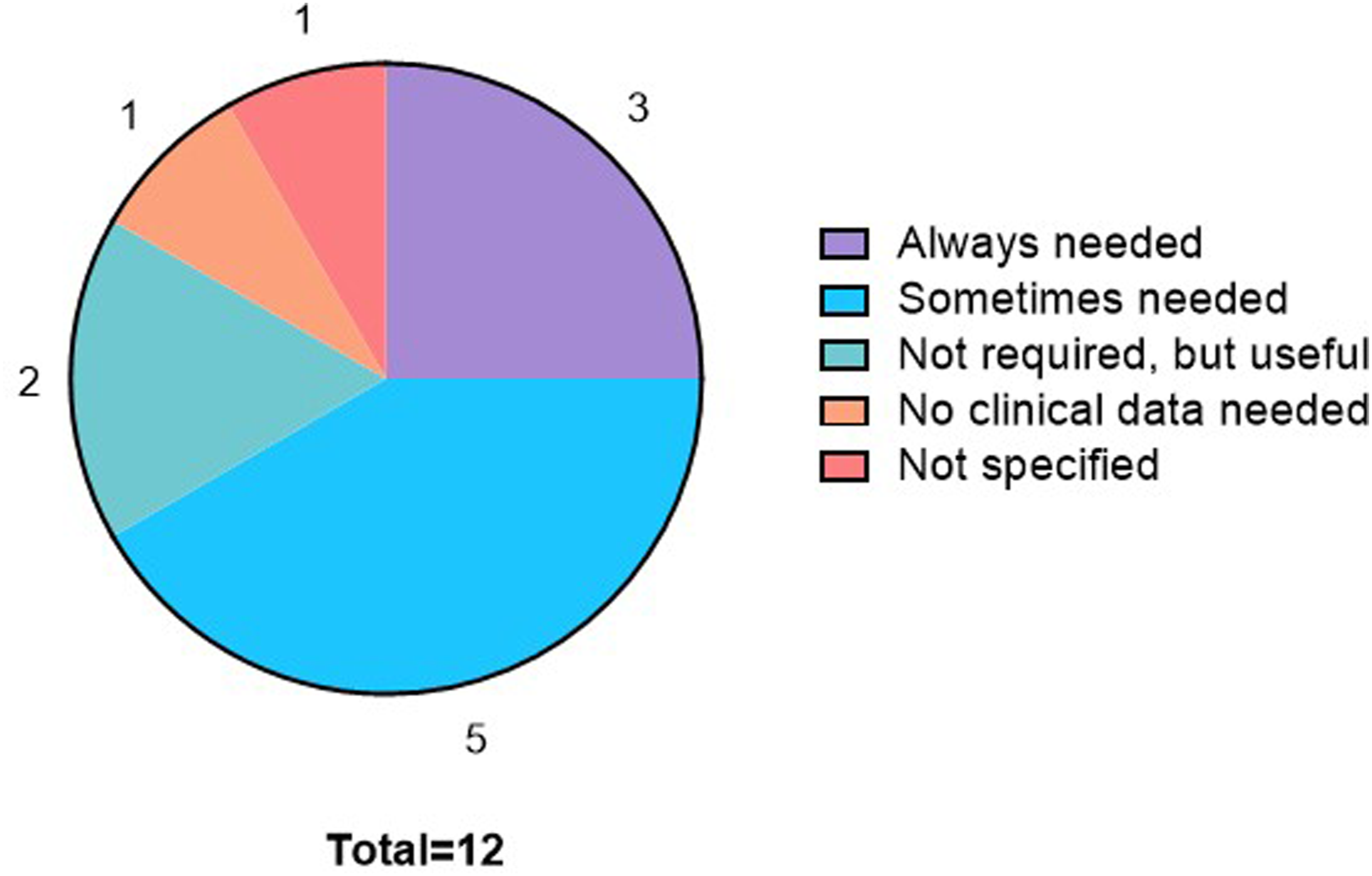
Companies also indicated that if they felt a repeat-dose toxicity study is redundant, they would ask health authorities for concordance with this approach, provided the health authority consultation does not impact project timelines.
Discussion
A survey related to current practices in nonclinical safety evaluation of vaccines was sent out to 36 companies globally, all members of BIO. Twelve companies provided a complete set of evaluable answers. This is a reasonable number of responses given that only a subset of BIO companies are involved in vaccine development and only those with a vested interest in vaccine development were likely to respond. The survey is considered representative of the industry practices up to 2022, since the major vaccine developers, based on the number of approved or authorized vaccines in the United States or Europe (centralized process) as of 2022, were part of the responding companies, mainly based in the Western World.
To enter an FIH trial with any new vaccine candidate, a GLP repeat-dose toxicity in a relevant animal species is one of the prerequisites.2-4 This is a consequence of the recognition of vaccines as human pharmaceutical products in the early 1990s, which led to a transposition of the regulatory guidelines in place for the development of drugs to vaccines, and in particular, the toxicological evaluation of new vaccine candidates in in vivo animal models. Toxicological evaluation of vaccines took into consideration the pharmacological specificities of the vaccines and their clinical use. This resulted in a limited number of in vivo toxicity studies being required for approval, in most cases a repeat-dose toxicity study, and potentially a developmental and reproductive toxicity (DART) study. The objective of the repeat-dose toxicity study conducted with vaccine candidates aims primarily to evaluate their tolerability instead of their intrinsic toxicity.
Answers to the survey clearly demonstrate that most companies (7/12) find scientific value in the repeat-dose toxicity studies, providing confidence in the product and information on parameters to be monitored in the FIH study. However, the impact of the information that can be retrieved from these studies is clearly associated with the novelty of the vaccine formulation, such as the addition of a novel adjuvant, the introduction of a new lipid to a lipid nanoparticle (LNP), or other novel excipient, sometimes leading to the conduct of studies in both rodent and non-rodent species. For vaccines based on established technologies (such as polysaccharide conjugates, recombinant proteins, established adjuvants contained in approved vaccines, or approved egg- or cell-based influenza vaccines), the impact of the repeat-dose toxicity study was considered far less significant. However, when the option of substituting the repeat-dose toxicity study with a pharmacology study with additional safety parameters was presented, only a few companies (3/12) agreed without conditions. The other companies indicated that such an option could be considered a supplement to a repeat-dose study or was inadequate to reach the objectives expected for a repeat-dose toxicity study.
The importance given by the companies to perform a repeat-dose toxicity study, when related and relevant data to address nonclinical safety is available, also seems to be motivated by the risk of delay to programs if the health authorities question the sufficiency of the nonclinical package. This was reflected in the survey responses where companies indicated that even if they felt a repeat-dose toxicity study was redundant, they would only approach health authorities with a proposal to not conduct it if the consultation would not impact project timelines.
In the literature, there are several publications5-10 summarizing the observations collected in repeat-dose toxicity studies with vaccines where no systemic adverse effects have been reported. The observations are mainly limited to inflammatory changes at the injection site as well as increased cellularity in the draining lymph nodes or other lymphoid organs, and some alterations in clinical pathology parameters, consecutive to the expected pharmacological activity of vaccines (parameters linked to the inflammatory reaction/immune response) or to the nature of some of their components (e.g., aluminum salts and emulsion) that drive the changes. This suggests that the vast majority of the vaccines are well tolerated with no safety concerns raised during their nonclinical development, and this was confirmed by the answers collected in the survey. However, the feedback received from a few companies indicates they experienced a delay, if not prevention, on 1 or 2 occasions in the clinical development of a vaccine candidate that was related to a nonclinical safety issue, linked to local reactogenicity with an mRNA vaccine, and systemic toxicity with an inactivated or attenuated viral vaccine. It was however not clarified in these two cases if this resulted in just a postponement of FIH initiation or if this nonclinical safety finding prevented the program from moving into the clinic. In addition, the exact number of FIH studies that were considered by the companies to answer the questionnaire was not asked; therefore, the relative number of FIH studies that were postponed/prevented because of a nonclinical safety finding in the repeat-dose toxicity study could not be calculated. However, one company did provide more context and reported that a finding in one out of at least 50 repeat-dose toxicity studies delayed the FIH initiation, but this finding was ultimately found not to be related to the vaccine; thus reaffirming that overall vaccines were well tolerated non-clinically, especially in the case of this particular company with a large sample size of repeat-dose toxicity studies. Translation of this benign nonclinical safety profile into the clinic is supported by a recent clinical review which confirmed that safety issues are rare with vaccines. 11
With this in mind and in consideration of the 3Rs, we questioned whether a repeat-dose toxicity study is needed for each new vaccine candidate. There are already examples where it is the accepted practice of health authorities to not require new repeat-dose toxicity studies. Despite the frequent reformulation of seasonal influenza vaccines following the bi-annual recommendations by the World Health Organization (WHO) of the viral strains to be included in the vaccines, it is accepted that toxicity evaluation is not required when the same or similar influenza strains and manufacturing processes as the initially approved vaccine are used to develop the next seasonal influenza vaccine. This is a type of platform approach, although this concept and specific terminology was not defined as such at the time of guidance implementation. 12 Of note, the original toxicity studies must be of adequate scientific value and quality, and a justification on the relevance of the extrapolation to the candidate vaccine must be provided. 12 It was also this underlying philosophy that led to the first position of Global Regulatory Workshop of the International Council of Medicines Regulatory Authorities in March 2020 with regard to the nonclinical data to be required for new vaccines against COVID-19, 13 indicating that the extent of preclinical data to support the clinical trials with COVID-19 vaccines “depends on the vaccine construct, the supportive data available for the construct and data from closely related products.” This point was reiterated in the 2022 WHO guidance on “Evaluation of the quality, safety and efficacy of messenger RNA vaccines for the prevention of infectious diseases: regulatory considerations” 4 and 2023 FDA guidance for “Development and Licensure of Vaccines to Prevent COVID-19.” 14
It was clear from the survey that most companies (9/12) are trying to utilize a “platform approach” (as defined in the survey), especially in the case of mRNA-LNP vaccines. However, there are diverse opinions as to how many repeat-dose toxicities need to be conducted, and if or how much clinical data would be required by health authorities to establish a platform. The companies also indicated they are trying to expand the use of this platform approach, relying on representative data, to vaccines based on established technologies using well-defined manufacturing processes. While not covered by this publication, it is also the authors’ opinion that a platform approach would also apply to the DART assessment of vaccines, if needed.
It is the opinion of the authors that an update of the current regulatory guidelines is needed to expand the use of the platform approach. This approach would offer the possibility to bring new vaccine candidates into the clinical development more quickly without compromising vaccine safety assessment while minimizing the use of animals.
Supplemental Material
Supplemental Material - Are Repeat-Dose Toxicity Studies Informative for Safety Assessment of Vaccine Candidates? A Survey of Vaccine Developers
Supplemental Material for Are Repeat-Dose Toxicity Studies Informative for Safety Assessment of Vaccine Candidates? A Survey of Vaccine Developers by Cynthia M. Rohde, Eric Destexhe, Jan Willem van der Laan, Sarah Gould, Rachel Coe, and Bert Haenen in International Journal of Toxicology.
Appendix
Abbreviations
Biotechnology Innovation Organization
Committee for Medicinal Products for Human Use
Committee for Proprietary Medicinal Products
developmental and reproductive toxicity
first in human
good laboratory practice
International Council of Medicines Regulatory Authorities
lipid nanoparticle
Replacement, Reduction, and Refinement
messenger ribonucleic acid
World Health Organization
Footnotes
Acknowledgments
We would like to acknowledge the contributions on this topic of the Biotechnology Innovation Organization (BIO), a US trade association. We thank our colleagues and subject-matter experts for their critical feedback and editorial input.
Author Contributions
Rohde, C.M. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Destexhe, E. contributed to conception, contributed to interpretation, drafted manuscript, and critically revised manuscript; van der Laan, J.W. contributed to interpretation, drafted manuscript, and critically revised manuscript; Gould, S. contributed to conception, contributed to interpretation, drafted manuscript, and critically revised manuscript; Coe, R. contributed to acquisition and analysis; and Haenen, B. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Bert Haenen, Cynthia M Rohde, Eric Destexhe, and Sarah Gould are employees and/or shareholders of Janssen Research & Development, LLC, Pfizer, Inc, GSK group of companies, and Charles River Laboratories, respectively, companies involved in vaccine development. Jan Willem van der Laan and Rachel Coe declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data Availability Statement
Anonymized survey data are available upon request.
Supplemental Material
Supplemental material for this article isavailable online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.