
Abstract
Excipients are essential components within drug products that contribute significantly to their overall quality, effectiveness, and safety. There is a lack of global, harmonized guidance relating to the non-clinical testing of novel excipients which is perceived to create uncertainty and strategic risk, potentially hindering innovation and disincentivizing their use. To test these perceptions, the IQ Novel Excipient Working Group surveyed member companies regarding their main concerns and prior experience regarding the non-clinical evaluation of excipients. Of the 19 respondents, 13 provided, collectively, 33 non-clinical program examples supporting the development of novel excipients. Programs were distributed across a range of therapeutic areas and included a variety of drug modalities and administration routes. Package designs were variable, but where possible, employed the use of existing data, supplemented with new toxicology studies as appropriate. Of the programs which had submitted data to regional health authorities, only three received feedback requesting additional studies or that demonstrated differences in regional opinion. In addition, companies provided recommendations on how the current (or new) guidance related to non-clinical excipient evaluation (and other areas, such as Chemistry, Manufacturing, and Controls and databases) may be improved.
Keywords
Introduction
The U.S. Food and Drug Administration (FDA) define an excipient as any ingredient intentionally added to a drug product (including a biological drug product) that is not intended to exert therapeutic effects at the intended dosage, although it may improve product delivery. 1 The FDA further defines a “new” excipient as one not fully qualified by existing safety data with respect to its proposed context of use (e.g., proposed exposure level/duration or administration route). 1 International Council for Harmonization (ICH) and European Medicines Agency (EMEA) guidance define a “novel” excipient as one being used for the first time in a drug product or by a new route of administration.2,3
Excipients may represent the major constituent of any drug product (80–90%) 4 and contribute to the overall drug product performance. 5 Commonly, excipients function to enhance an active pharmaceutical ingredient (API) solubility and bioavailability and control (e.g., sustain) its rate of release. Other roles may relate to drug product identification, palatability, stability, and manufacturing. Given the evolving landscape in drug development (e.g., introduction of new modalities and advanced drug delivery technologies), the need for new excipients is expected to increase with the global pharmaceutical excipient market projected to exceed $8 billion by 2023. 6
The contribution to the overall safety, effectiveness, and quality of a drug product must be recognized. Existing regulatory guidance (FDA, EMA, ICH, etc.) is useful though somewhat dated; FDA guidance for the non-clinical safety evaluation of excipients was issued in 2005. 1 There is no internationally harmonized guidance regarding the appropriate level of non-clinical testing to support qualification of novel excipients. Coupled with the relatively limited industry experience relating to the development of novel excipients, this is perceived to create uncertainty and strategic risk which may hinder innovation and disincentivize companies regarding their use/development. Consequently, patients may be denied access to new and valuable medicines and/or those which could significantly improve the patient experience for vulnerable groups such as geriatrics and pediatrics. 7 This may also result in the conduct of non-clinical studies with limited or no value.
To test these perceptions and capture pharmaceutical industry experience, the IQ Consortium Novel Excipient Working Group (NEWG) conducted a pharmaceutical company survey to identify the main concerns or limitations regarding the non-clinical evaluation of novel excipients. Objectives included: • Understand companies’ perspectives and drivers regarding the development of novel excipients, including perceived barriers. • Understand how the current regulatory landscape influences the design/conduct of non-clinical studies/packages for novel excipients. • Understand variations in regulatory non-clinical feedback and expectations across different global health authorities. • Learn how companies successfully qualified novel excipients. • Review opinions on current guidance and value of a future, harmonized global guidance.
8
Materials and Methods
The survey (Appendix A) was reviewed by the IQ Consortium NEWG and Drug Product and DruSafe leadership groups and ran between 11th November 2021 and 23rd February 2022; follow up responses were received by 19th August 2022. The survey was distributed to all IQ consortium member companies.
Definitions were provided for the key terms (API, excipient, novel, and new excipient) (Appendix A). The term “novel excipient” was used in the survey to describe an excipient used for the first time in any drug product or in a context different to that already approved (e.g., new administration route and longer duration).
Respondents were asked if they had developed or were currently developing any drug product containing novel excipients for which a non-clinical package was conducted. A non-clinical package was defined as one comprising new non-clinical studies and/or a review/assessment of “existing” data which may be used in-lieu of new studies (e.g., literature, approved use in another context, and data for related molecules). Companies that did not include novel excipients were asked for further details including a rational for this position. Companies that included novel excipients were asked to provide up to 5 non-clinical program examples (between 2010 and 2021) including information such as the type of API for which the excipient(s) was included, administration route, use duration, and therapeutic area and if the excipient(s) was entirely novel or not. For each example, respondents summarized the non-clinical studies conducted (type, species etc.), the key objective of the package, the regions for which the drug product was intended and the health authorities which had reviewed data (e.g., via a New Drug Application (NDA), Biologics License Application (BLA), and Investigational New Drug (IND) application or briefing documents), and if health authority feedback was consistent across regions.
All companies were asked if the current non-clinical guidance for the assessment of novel excipients could be improved (and if so, how) and whether a harmonized, global guidance would be useful (and if so, why).
No data were collected against specific company names and all responses were confidential and anonymized by an independent third-party secretariat (Faegre Drinker Biddle & Reath LLP) prior to being provided to the authors. The survey did not include any questions specific to any excipient (e.g., name and type).
Follow Up Questions and Interpretation of Responses
During data review, some weaknesses in the survey were identified, some of which required follow up questions. For example: • A free text field to add more context to the non-clinical packages (design/drivers) would have been useful. • The term “existing data” was not defined; the authors considered (but did not define) source of existing data as the FDA inactive ingredient database (IID) and Generally Regarded as Safe (GRAS) database and Drug Master Files (DMFs) and United States Pharmacopeia (USP). In retrospect, existing data may also comprise of in-house data or data in the literature. • An option to provide more detail regarding the status of the program would have been useful, for example, a “complete” program may represent a one that was finalized (e.g., achieved approval) or stopped for other reasons.
Following initial data review, the secretariat sought clarification for some responses (e.g., responses omitted or contradicted other answers); not all queries were answered/clarified. In some cases, based on the follow up responses and/or answers to other questions, the authors interpreted responses to some unanswered questions which are summarized in Appendix B; three case studies were excluded from the analysis as it was not possible to fully interpret the responses.
Results
Demographics
Nineteen (50%) of 38 member companies responded to the survey. Of these, 13 (68%) confirmed that they had developed or were developing products containing a novel excipient(s) for which a non-clinical package was conducted. Six companies (32%) responded “no” to this question. The 13 companies which included novel excipients provided up to five case studies, such that a total of 33 program examples were received.
Companies who Do Not Employ Novel Excipients
Six companies did not include novel excipients. Reasons included that a novel excipient was not required (n = 3), did not improve the drug product characteristics (n = 1), or could not be manufactured in compliance with Good Manufacturing Practice (GMP) (n = 1). Four of these companies avoided the use of novel excipients and cited concerns (barriers) related to higher costs and longer drug development timelines, the risk of regulatory non-acceptance of the excipient and/or drug product, and the large burden to generate non-clinical and clinical safety data. Other concerns related to potential late-stage attrition, while one company stated they were not willing to add additional program risk by inclusion of a novel excipient (even if the program was in a serious life-threatening disease).
Programs Employing Novel Excipients
Therapeutic area, drug modality, route of administration, and duration of exposure
Therapeutic Area
Programs were distributed across ten therapeutic areas (Figure 1), most commonly, “metabolism” (n = 8), “oncology” (n = 5), “anti-infective (n = 5), and “neurology” (n = 4). Distribution of programs by therapeutic area.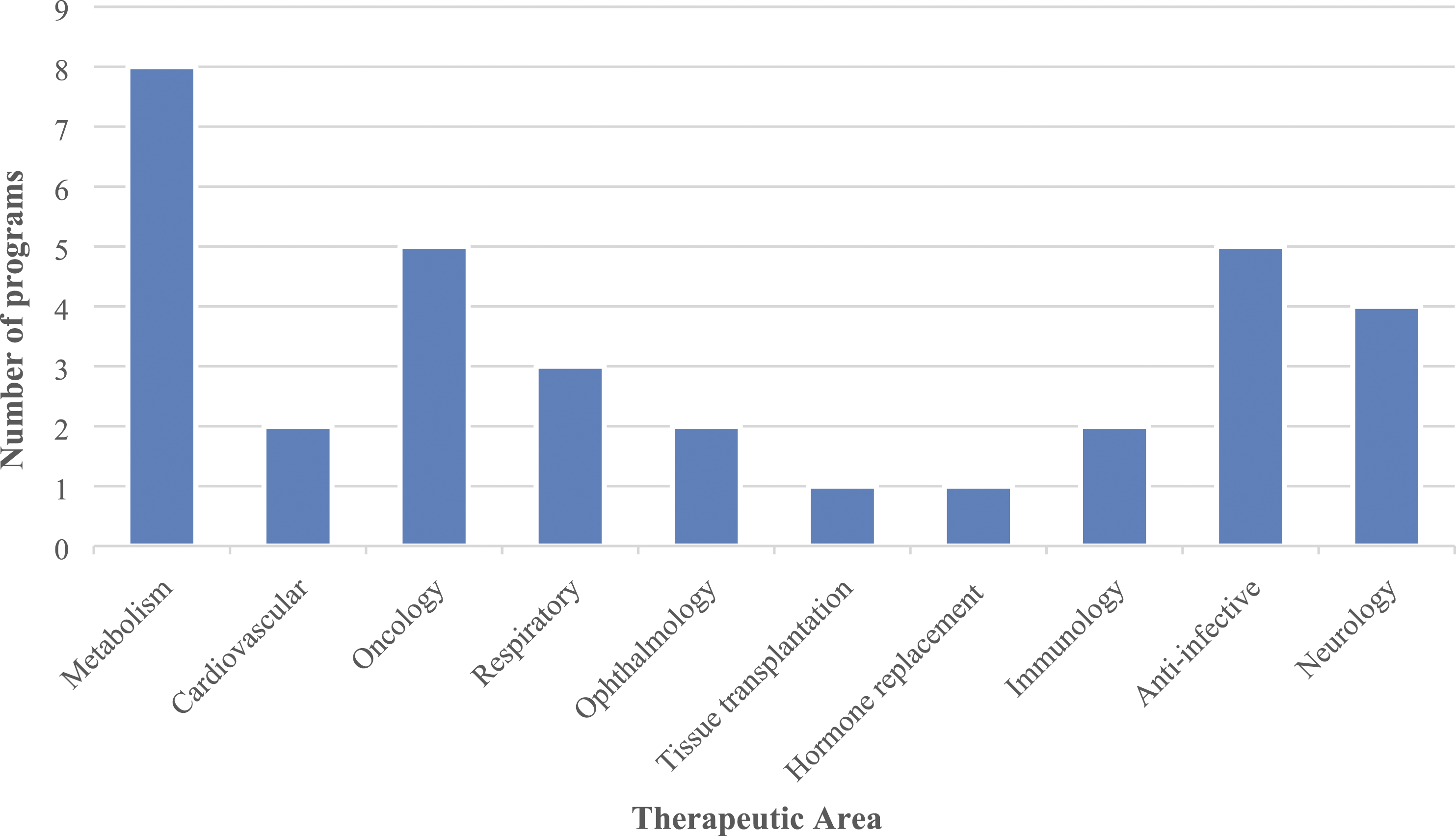
Drug Modality
The most common drug modality for which novel excipients were being developed were small molecules (n = 17) followed by peptides (n = 7) and antibodies (n = 5). One program employed “excipients” to support the ex-vivo storage of human tissue prior to transplantation.
Route of Administration
The most common route of administration was oral (n = 13) followed by subcutaneous (n = 5) and intravenous (n = 4) injection. The majority of peptide programs (71%) and approximately half of the small molecule programs (47%) employed oral delivery. The distribution of administration routes, relative to drug modality is shown in Figure 2. Distribution of routes of administration relative to molecule type.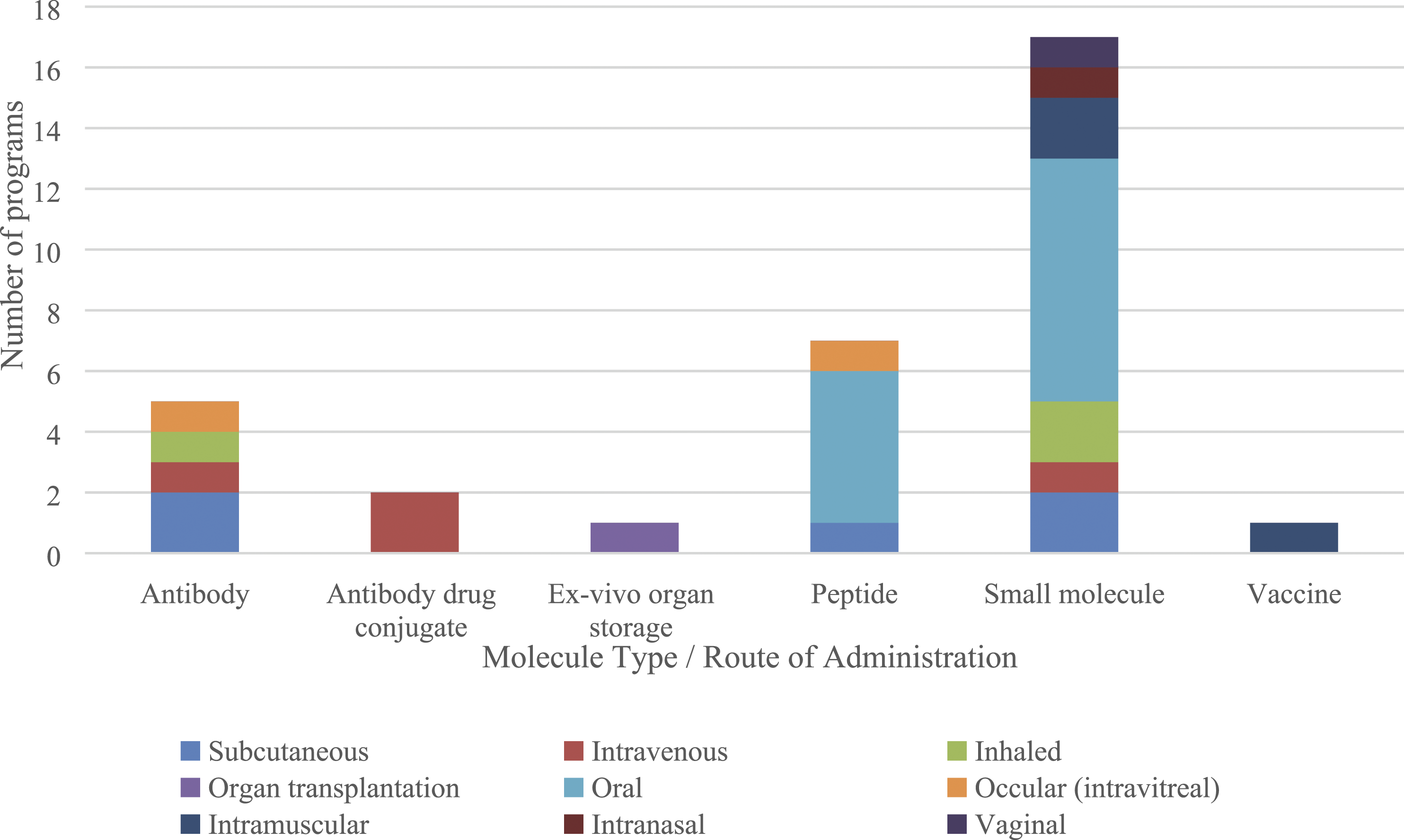
Duration of Exposure
The majority of programs related to long-term administration (n = 27) followed by short (n = 4) or intermediate (n = 2) durations.
Novelty of the Excipients Being Developed
Across the programs, nine included entirely novel excipients while 24 included excipients which were novel with regards to their proposed use (mostly used in other drug products by a different administration route).
Objectives of the Non-Clinical Packages
The main objectives of the non-clinical packages were to provide data for an excipient(s): • For which there were no prior data or use precedence (n = 9). • For use in a new context (n = 24) such as a different administration route (n = 9), higher dose (n = 6), longer treatment duration (n = 1), different patient population (n = 1), different region (n = 1), or one or more of the prior reasons (n = 5). For one program, a rationale could not be determined (see Section 2.1).
Non-Clinical Packages Conducted
Fourteen programs were ongoing while 19 were completed; the post-survey status of “ongoing” programs is not known. Approximately half (n = 17) of the programs used existing data in combination with the conduct of new non-clinical studies. Others were evenly divided between those that either conducted new studies alone or relied on existing/available data. Two programs which used existing data alone (both to qualify excipients at higher doses) included use of a “read-across” approach whereby safety data from other molecules were used to support risk assessment; both received favorable feedback from the FDA. No other programs employed read-across (no further questions regarding read-across were asked).
Of the nine programs evaluating entirely novel excipients, 4 comprised of new non-clinical studies alone while five comprised of new studies with existing data. Of the 24 programs for which the excipients were novel in terms of their proposed use, these were qualified using existing data (n = 7), existing data with new non-clinical studies (n = 12) or new non-clinical studies only (n = 5). Summaries for some of the programs are described below.
Excipient Programs for Antibodies (n = 5) and Antibody Drug Conjugates (ADC) (n = 2)
All programs intended to qualify excipients for long-term administration. One antibody and one ADC program employed existing data alone to support use at a higher dose or for a longer duration. Remaining programs were designed to qualify excipients at higher doses or for new administration routes (inhaled or ocular) using new toxicology studies, with or without existing data.
One antibody and one ADC program intended to qualify novel excipients using existing data with limited additional studies. For the antibody, these included pharmacology (rat) and repeat-dose toxicology (rat and dog) while for the ADC, repeat-dose toxicology, toxicokinetics (TK), and Absorption, Distribution, Metabolism, and Excretion (ADME) studies in rats were conducted alongside in vitro (metabolism and drug product toxicology) and in-silico (genetic toxicology) studies.
The other three programs conducted safety pharmacology and repeat-dose toxicology studies in one or two species (one program employed transgenic mice).
None of these programs included carcinogenicity studies.
Excipient Programs for Peptides (n = 7)
All programs intended to qualify excipients for long-term administration by either oral (n = 5), subcutaneous, or ocular (intravitreal) routes.
Oral: Two programs for novel excipients comprised non-clinical packages including safety pharmacology, genetic, repeat-dose and reproductive toxicology, TK, metabolism, and carcinogenicity studies (one or two species). Two programs sought to qualify excipients at higher doses than already approved; one program was comparable to those described above (excluding carcinogenicity studies) while the other program included additional metabolism studies (to evaluate the effect of a new enteric coating and to explore the etiology of an observed target organ toxicology). The final program sought to qualify an excipient with prior use in drugs (not as an excipient) and food using a literature-based approach (agreed with regional health authorities).
Subcutaneous: Sought to qualify a new route of administration via use of existing data and conduct of additional local tolerability studies (two species).
Ocular: Sought to qualify an excipient by a new route (intravitreal) through conduct of mini-pig, repeat-dose toxicology (with TK) studies with drug product.
Excipients Programs for Small Molecules (n = 17)
Generally, programs sought to qualify novel excipients or established excipients in a new context in drug products intended for long-term administration across a range of therapeutic areas (oncology, respiratory, metabolism, anti-infective, cardiovascular, neurology, and hormone replacement) and routes of administration (Figure 2). Programs generally reflected full toxicology packages using two species (e.g., broadly in-line with ICH M3(R2) 9 ), comprising of genetic toxicology, safety pharmacology, single and repeat-dose studies, reproductive toxicology, and occasionally, carcinogenicity studies. Some programs employed the use of existing data.
There were some examples of smaller (e.g., bridging) toxicology packages, related to the qualification of established excipients in a new context (e.g., new route) which generally included the use of existing data and a small number of additional studies, often in one species; one program used existing data alone to qualify an excipient at a higher dose than already approved.
Some programs qualified excipients intended for short (n = 2) or intermediate (n = 2) use in another context over those already used (e.g., new route and higher dose). Three of these programs employed existing data alone while for one program (intended for short-term use); existing data was supported by new genetic toxicology, safety pharmacology, repeat-dose toxicology, reproductive toxicology, TK, and metabolism studies, in one or two species.
Other Programs (n = 2)
One program qualified a novel excipient for use in a vaccine and used existing data, in-silico genetic toxicology, repeat-dose toxicology, reproductive toxicology, and metabolism studies in rats.
The constituent(s) of a tissue preservation fluid used for ex-vivo tissue storage prior to transplantation was considered to be an excipient by one company; this material was approved in another drug product and the program employed existing data and a single dose toxicology study in rats.
New Approach Methods
The use of NAMs in the safety assessment of excipients is described by the IPEC federation. 5 NAMs are comprised of non-animal methods such as in silico (e.g., quantitative structure analysis relationships or physiologically based models) or in vitro (e.g., organoid cultures) systems.
Of the 33 programs, only 2 employed the use of NAMs. In both programs, the excipients were novel and intended for small molecule (oral) or vaccine (intramuscular) drug products. NAM data for both programs was used alongside a series of in vivo studies and disclosed in the appropriate regulatory documentation. The survey did not ask further questions related to NAMs and so it is not possible to comment further on the importance of these data or the regulatory position in terms of its review (both programs reported favorable health authority feedback across a range of territories).
Regulatory
Companies were asked to summarize the regions to which they intended to market the drug product containing a novel excipient. On review, this question was considered not to offer any value to the overall survey and the results are not discussed.
Across the 33 programs, 12 had data submitted data to the US and/or European health authorities. A further 15 programs had submitted to at least one further other region. Data for six programs had not been submitted to any authority.
Only three programs received feedback which disagreed with the sponsors’ original proposal (e.g., requested additional studies) or which varied across regions. These examples related to excipients in small molecule drug products intended for long-term administration in either oncology (n = 1) or respiratory (n = 2) indications.
For the oncology drug product, the program intended to qualify an excipient mixture, the components of which were proposed for use at higher doses and via a new route of administration (intravenous) than used in other drug products. The excipient mixture comprised of components A (counter-ion in an already approved product) and B (polymeric mixture). The drug product was formulated as a nanoparticle. To support phase 1 and 2 studies, health authorities in Europe and US agreed with the limited non-clinical package (repeat-dose toxicology studies in two species only). However, the level of testing requested by these authorities to support phase 3 was different. Requests for component A include particle distribution studies in two species (Europe), systemic exposure relative to the approved drug product (Europe), and genetic toxicology assessments (US) while for component B, genetic toxicology and photo-toxicology assessments were requested (Europe).
Two inhaled programs (one completed and one ongoing) had significant non-clinical packages. For the completed package, the European and US authorities provided different opinions to the sponsors proposal to conduct a 3-month product toxicology study in dog and to not conduct carcinogenicity assessments (based on the nature of the excipient). While the FDA accepted the absence of any carcinogenicity studies, the European agency requested a carcinogenicity study in one species. The European agency also requested a longer drug product toxicology study in dogs (six months), while in addition to the 3-month product toxicology study in dogs, the FDA requested an additional, equivalent study in rats. The company highlighted that the health authority discussions focused on the drug product as a whole (with more than one API); it was therefore not possible to determine to what extent the requests related to the excipient alone and/or the combination of active ingredients. In a subsequent interaction, the Japanese authority considered the “excipient” package as deficient and requested further chronic toxicology (two species), carcinogenicity assessments (two species), and reproductive toxicology assessments (only a limited reproductive package had been proposed).
For the ongoing package, while the proposed non-clinical package was accepted by the US and Japanese health authorities, additional excipient metabolism, in vitro secondary pharmacology, and safety pharmacology data were requested by the European agency.
Comments on guidance for the non-clinical assessment of excipients
When asked if the currently available guidance regarding the non-clinical assessment of novel excipients could be improved, 17 companies responded “Yes” while only two stated “No.” Similarly, when asked if a harmonized guidance for the non-clinical development of excipients would be of value, 18 companies responded “Yes” with only one responding “No.” Additional comments from respondents are summarized below:
Several respondents felt a harmonized guidance (such as ICH) was important to align and streamline requirements across health authorities, noting that non-US authorities tend to follow ICH M3(R2) 9 rather than the FDA 2005 guidance. 1 The age of the FDA guidance (2005) was also recognized, noting that it may not sufficiently address the more modern use of excipients, for example in some advanced drug delivery systems. Some respondents felt that the current guidance was subject to divergent interpretation and recognized that some regional authorities may require more data, potentially driving companies towards a conservative approach and resulting in the conduct of possibly, unnecessary studies.
Recognizing the high cost and time burden associated with excipient development, it was suggested that any updated or future harmonized guidance should include details regarding which studies are required relative to specific phases of development (e.g., a staged approach), potentially expediting delivery of formulations to the clinic. Further, any guidance should provide clarity on the types of data considered acceptable to qualify excipients (e.g., data from literature, read-across, and NAMs). One company required more clarity on the non-clinical studies required to qualify an excipient in an oncology product, relative to those required for the API. Another suggestion was to increase clarity on the requirements/acceptance of data generated with the drug product vs excipient alone (including the conduct of specific bridging studies).
Further comments related to how existing data for an excipient could be more widely accessed and leveraged to qualify its use in a new context (or how data which may already have been reviewed by a health authority may become more accessible in general). One respondent highlighted the benefit of creating an independent regulatory pathway for qualifying excipients (akin to the FDA’s Novel Excipients Review pilot program).
In addition, any new/updated guidance would benefit from including details on toxicology study requirements (what and when), design and safety margin calculation and expectations.
Excipient development: other areas in which difficulties are experienced
Twelve companies responded to the question “in addition to your experience with non-clinical data packages for excipients, are there any other areas in relation to novel excipients for which you experienced difficulties and/or may require further focus/guidance?” Of these, one responded “No” while 11 identified one or more areas of difficulty/focus (summarized below).
Given there is no global, harmonized definition of a “novel excipient,” sponsors can find it difficult to identify if an excipient is novel (or not) in a particular territory; it was noted that an appropriate system to define excipient use by quantity and route of administration existed in Japan.
Several comments related to whether excipient databases (e.g., the FDA IID) could be enhanced or created to better visualize/share existing safety data: • Make data for an excipient in the FDA IID (e.g., by the intended route and dose) available and citable in lieu of further toxicology studies (e.g., if the sponsor does not own the prior toxicology data). • Broader access to non-clinical data for excipients would be valuable; perhaps best served through an industry consortia-owned database. • Excipients employed in biologic drug products should be included in the FDA IID.
A further respondent referenced limitations in using the FDA GRAS database and its translation to drug products (database specifically relates to substances intentionally added to food) while another commented on the limited resources regarding excipient use in drug products for pediatrics.
The difficulty (and sometimes, inability) to obtain DMFs from excipient manufacturers was noted recognizing that this may lead to sponsors duplicating studies already completed by others. The absence of a DMF system for sharing confidential excipient data in regions such as Europe and Japan was also recognized.
Several comments related to CMC such as excipient specification, analysis, quality, and supply. • Given that many excipients are heterogeneous mixtures (either by design or due to the nature of their manufacture), it is not always easy to know how to define or quantify impurities and understand if indeed any impurity (which may be an inherent excipient component) itself contributes to the routine functionality of the excipient. Furthermore, quantification of the individual functional components can be limited by the capability of analytical methods in terms of their separation (e.g., chromatographic limitations) or quantification (detector capability). • Aspects relating to complex novel excipients which require chemical synthesis are unclear (e.g., CMC development requirements, regulatory agency expectations for filing content and detail, GMP manufacturing requirements). • Lack of clarity regarding what constitutes a “major” or “minor” change to an excipient (e.g., changes to its synthetic route, analytical methods, specification, and manufacturing site) and what is required to establish comparability. • Concern regarding excipient manufacturers committing to a sustainable supply of high-quality material, particularly for less commonly used materials. • Unclear what non-clinical data required to qualify excipients which may be added to a formulation employed only in a clinical trial (e.g., to ensure sufficient blinding, such as a colorant). • Unclear how/if to bridge data for programs in which the same excipient is being developed for different purposes.
Finally, one company commented on the FDA’s Pilot Program for the Review of Innovation and Modernization of Excipients (PRIME). PRIME is intended to allow excipient manufacturers to obtain FDA review of certain novel excipients prior to their use in drug formulations and was initially available for novel excipients not previously used in FDA-approved drug products and which did not have established use in food. 10 While noting it is an exciting step forward, they considered the initial pilot to be disappointing due to its restricted scope and suggested that a lower hurdle for entry, in terms of the data required and time (e.g., to include materials previously used in food or via new routes of administration), would have been beneficial.
Discussion and Conclusion
The survey sought to gain insights into pharmaceutical industry experience relating to the non-clinical evaluation of novel excipients. Objectives were included to gain a better understanding of perceived barriers as well as non-clinical program drivers and approaches and experiences with regulatory authorities. The survey also asked for company’s thoughts on current guidance.
While 4 companies avoided the use of novel excipients due to concerns regarding development cost, duration, and regulatory risk, the majority of companies employed novel excipients in their drug products. Excipients were employed across a broad range of therapeutic areas with the largest number of programs directed toward oral delivery of small molecules and peptides.
Most programs intended to qualify established excipients for use in a new context (e.g., different administration route). In general, companies sought to use existing data where possible, but commonly supplemented these data with new non-clinical studies. Existing data alone were used in approximately one third of programs, with most other programs also including non-clinical studies even when some existing data were available. The number and type of non-clinical studies varied significantly across the programs, ranging from a few short and small studies (e.g., in vitro and/or limited studies in a single species) to far more robust assessments (e.g., repeat dose in at least 2 species and sometimes including reproductive toxicology and carcinogenicity studies). The rationale for the conduct of additional studies cannot be determined through this survey, but likely reflects the variety of potential scenarios faced by any sponsor (e.g., the amount of existing data owned or accessible to a company and the overall objective(s) of the program). Companies commented that further clarity regarding the use and acceptance of existing data (including literature) would be valuable and that improved guidance and data access may increase industry confidence to use such information and possibly result in fewer animal studies.
For most programs (irrespective of therapeutic area or drug modality), health authority feedback was positive; only three programs reported negative or divergent feedback across different health authorities. One of these cases was however complicated by the fact the company were developing a complex excipient mixture while another was complicated given that the health authority feedback was directed toward the drug product as a whole, making it difficult to ascertain excipient specific feedback. While overall, the reported non-clinical data packages were generally accepted by regulators across different regions, these few examples demonstrate the value of health authority engagement and alignment during the excipient/drug product development program. Overall, the low number of programs which received challenging feedback from health authorities was lower than the expected considering the diversity of the programs and suggests that pharmaceutical companies are generally successful in building appropriate non-clinical packages to qualify excipients based on current guidance. Consequently, it could be argued that the current guidance is “fit for purpose” while recognizing that alignment with regional regulatory authorities will be important.
Only two programs incorporated the use of NAMs while only two programs employed a read-across approach. The programs employing read-across, which sought to qualify excipients at higher doses than those already approved, did not conduct additional non-clinical studies but did reference existing data. Positive health authority feedback was received for both programs (submitted to FDA only) as well as those including NAMs (multiple regions). The survey did not ask further questions related to NAMs or read-across and therefore it is not possible to comment further on these data or their relative importance to the specific programs. While the low uptake of these non-animal methods may, in-part, reflect the age of some of the programs (potentially dating back to 2010), it suggests that there is an opportunity to increase their use. This is supported by the fact that several respondents recommended that any updated/new guidance should address the use of NAMs and read-across approaches (including use of published literature).
Several respondents recognized that a global, harmonized guidance for the non-clinical evaluation of excipients would be of value to drive consistency and provide a common framework to excipient manufactures and users. A series of other comments made reference to the age of the FDA guidance (2005) and that this may not account for significant advances in technology since its issue.
Survey respondents recognized other challenges/areas for improvement in the development of novel excipients such as improving current databases and accessibility and wider regulatory acceptance of DMFs. Several CMC-related comments related to the need to harmonize approaches to excipient specification, challenges in impurity qualification, and how to demonstrate excipient comparability in the event of a change in manufacture processes.
In conclusion, most surveyed companies develop novel excipients across a wide range of therapeutic areas, drug modalities, and routes of administration. The respective non-clinical packages typically comprised of existing data and supplemented, as necessary, with new study data. Almost all respondents indicated that the current guidance regarding the non-clinical development of excipients could be improved and that a global, harmonized guidance would be of value. Contrary to this however, there were only a few examples of challenging or variable health authority feedback and these related to challenging programs. The survey suggests that any now or updated guidance should include the generation and acceptance of data generated using NAMs and read-across as well as the acceptance of existing data (including literature). Other comments recognized that other systems could also be improved, such as improving data accessibility and content/usefulness of excipient databases and CMC aspects such as excipient specification and impurity qualification.
Supplemental Material
Supplemental Material - Pharmaceutical Industry Perspective on the Non-Clinical Evaluation of Novel Excipients: Results From an Industry Survey Conducted by the IQ Novel Excipient Working Group
Supplemental Material for Pharmaceutical Industry Perspective on the Non-Clinical Evaluation of Novel Excipients: Results From an Industry Survey Conducted by Paul S. Giffen, Lorrene A. Buckley, and Jason Pinkstaff in International Journal of Toxicology
Footnotes
Acknowledgments
The survey was developed under the auspices of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ), a not-for-profit organization of pharmaceutical and biotechnology companies with a mission of advancing science and technology to augment the capability of member companies to develop transformational solutions that benefit patients, regulators, and the broader research and development community. The survey was sponsored by the IQ novel excipient working group (NEWG), which is a member of the Drug Product Leadership Group (DPLG). The mission of the respective groups is to improve awareness of the importance of novel excipients in facilitating new therapeutic concepts into products for patients (NEWG) and to influence the strategic direction of drug product development and manufacturing for the benefit of industry and patients (DPLG). Both groups reviewed the survey prior to its distribution. The survey was also reviewed by members of IQ DruSafe Leadership Group, whose mission is to advance non-clinical safety sciences and impact the global regulatory environment. The authors would also like to thank the following individuals who reviewed the survey and/or final manuscript: Vered Dror (Teva), Alan Christensen (Novo Nordisk), Patricia Parris (Pfizer), Maryam Rafie-Kolpin (AstraZeneca), and Philip Sherratt (Bristol Myers Squibb). We thank all companies that responded to the survey. The following companies agreed for their organization names to be disclosed: AbbVie, Amgen, AstraZeneca, Eli Lilly & Co, Genentech, Gilead Sciences, GlaxoSmithKline, Johnson & Johnson, Merck & Co., Novo Nordisk, Pfizer, Seagen, Theravance Biopharma Inc., and UCB.
Author Contributions
Giffen, P. contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Buckley, L. contributed to conception and design, contributed to acquisition and interpretation, and critically revised manuscript; Pinkstaff, J. contributed to conception and design, contributed to acquisition and interpretation, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.