
Abstract
Safety assessment of biological drugs has its challenges due to the multiple new different modalities, for example, antibody-drug conjugates, bispecifics, nanobodies, fusion proteins and advanced therapy medicinal products (ATMPs), their different pharmacokinetic and pharmacodynamic properties, and their ability to trigger immunogenicity and toxicity. In the public and in the pharmaceutical industry, there is a strong and general desire to reduce the number of animals used in research and development of drugs and in particular reducing the use of nonhuman primates. Important discussions and activities are ongoing investigating the smarter designs of early research and dose range finding studies, reuse of animals, and replacing animal experiments with in vitro studies. Other important challenges include absence of a relevant species and design of studies and developing genetically modified animals for special investigative toxicology studies. Then, the learnings and challenges from the development of the first ATMPs are available providing valuable insights in the development path for these new potentially transformative treatments. Finally, development of strategies for assessment of immunogenicity and prediction of translation of immunogenicity and associated findings to the clinic. On this, the eighth meeting for the European BioSafe members, these challenges served as the basis for the presentations and discussions during the meeting. This article serves as the workshop report reviewing the presentations and discussions at the meeting.
Introduction
BioSafe is the nonclinical safety committee of the Biotechnology Innovation Organization, whose mission is to identify and respond to key scientific and regulatory issues and challenges related to the nonclinical safety evaluation of biopharmaceuticals. In addition to the annual general membership meeting in the United States, an annual BioSafe meeting is held in Europe. 1 -5 The eighth Annual BioSafe European General Membership meeting was hosted by Novartis on October 30 to 31, 2018, in Basel, Switzerland. The attendees were from the biopharmaceutical industry, small biotech companies, and contract research organizations (CROs) from Europe and United States, representing various disciplines including pharmacology, toxicology and pathology, pharmacokinetics (PK), and bioanalytics. They shared experiences and insights into nonclinical safety assessment of biologics including monoclonal antibodies (mAbs), recombinant proteins, and gene and cell therapies.
The meeting covered the following sessions: Session 1. PK/TK and Toxicology; Use of Flexible Study Designs. Session 2. CMC Development Challenges and Strategies. Session 3. Nonclinical Investigative Studies. Session 4. Advanced Therapy Medicinal Products. Session 5. Challenges in the Toxicity Testing of Highly Potent Biologics Without a Relevant Animal Species. Session 6. Assessment and Management of Immunogenicity—Toxicological and Translational Concepts and Strategies.
Furthermore, the following 3 topics were discussed in breakout sessions
Breakout sessions: In Vivo Characterization of Antibodies/Biologics. Tox End Points in Pharmacology Studies. Recent Health Authority Interactions and Questions.
In each session, presentations were followed by podium discussions. As successfully started during the fifth meeting in 2015, 3 so-called “hot topics” were again selected by the planning committee and discussed during the meeting in breakout sessions in smaller groups, with the feedback being presented to all attendees afterward by the breakout session leads.
Session 1: PK/TK and Toxicology; Use of Flexible Study Designs
Session Chairs:
This session covered how flexibility in study designs of PK and toxicology studies to, for example, reduce animal usage can be achieved in different ways. The session summarized different approaches and provided concrete examples. Topics included PKs including bioanalytics, specifically discussing flexible study designs depending on the format of the biological (eg, comparison of mAbs vs bispecifics vs antibody-drug conjugates [ADCs]), stage of development (early vs late nonclinical development), indication, and so on; United Kingdom National Centre for the Replacement, Refinement and Reduction of animals in Research (NC3Rs)/Association of the British Pharmaceutical Industry (ABPI) 2 species project; and reuse of animals for PK and toxicology studies.
A survey was devised by an international working group (consisting of representatives from pharmaceutical and biotechnology companies, CROs, consultants, academia, and regulatory bodies) to collect data on the incidence of 1 or 2 species use across current portfolios (for different molecule types) and the reduction from 2 to 1 species within the package. This was completed by 18 different organizations (from May to October 2017). Biotherapeutic molecules following International Council for Harmonisation (ICH) S6 guidelines formed 45% of the total database of 172 molecules, including 46 mAbs, 15 recombinant proteins, 10 synthetic peptides, and 6 ADCs. Molecules were being developed for a wide range of therapy areas, including oncology (24), immunomodulatory (anti-inflammatory; 15) and endocrinology (13). These had progressed to different stages of development, covering pre-first in human (FIH) studies (9), FIH package (48), or post-FIH longer-term studies (20).
Toxicology studies were performed in a single species for the majority of mAbs (30 using NHP, 1 using a transgenic mouse model, and another using rat) but only 3 recombinant proteins and 1 ADC (all using NHP only). When 2 species were used (14 mAbs, 12 recombinant proteins, all 10 synthetic peptides, and 5 ADCs), the predominant species were rat and NHP. Five of these molecules (2 ADCs and 3 mAbs) reduced to a single species (the NHP) for the FIH package while 2 further mAbs reduced to a single rodent species for post-FIH chronic dosing studies. Nine other molecules following ICHS6 guidelines retained both species for post-FIH chronic dosing studies (4 mAbs, 2 recombinant proteins, and 3 synthetic peptides).
A key decision for progression of post-FIH chronic dosing studies in 1 or 2 species revolves around the “similarity” of toxicities between the species. A review of the target organ toxicities identified in the FIH package of studies (2-13 weeks duration) was performed to determine how often toxicities were similar or different between the 2 species. For the 40 biotherapeutic molecules that used 2 species for FIH studies (13 mAbs, 11 recombinant proteins, 12 synthetic peptides, and 4 ADCs), toxicities were similar in both species for 85%, 36%, 42%, and 25% per molecule type, respectively. For the 11 molecules that progressed from FIH to post-FIH studies, 6 had similar toxicities at FIH (4 mAbs, 1 recombinant protein, and 1 synthetic peptide), but only 2 mAbs reduced to 1 species. Although this data set was small when individual molecule types were scrutinized, it did suggest that there may be opportunities for more molecules currently following ICHS6 guidelines to reduce to 1 species for post-FIH chronic dosing studies in the future.
Then,
Deficiency in IL-2 signaling and in regulatory T cell (Treg) biology is strongly linked to the pathogenesis of some autoimmune diseases. The studied compound has an half-life extension and improved Treg selectivity in single-agent biologic. Full characterization of PK in early preclinical monkey studies can face difficulties due to the long half-life of mAbs, sparse sampling times, ADA, and low limit of quantification. Including an Espresso design element (within-individual uptitration) can get earlier readouts, more accurate determination of target-mediated drug disposition (TMDD), and possibly assess hysteresis.
The espresso PK study design starts at low dose and fast within-subject uptitration during a 2-week time frame to minimize potential effects from ADA formation. Starting dose at 10% of maximum PD and final dose reaching full receptor occupancy (RO) and 100% response:
1. Low dose has to be low enough to avoid full response (10% applied).
2. Highest dose is determined by reaching 100% response (eg, full RO through continuous response monitoring).
3. As safe as conventional study design: Same maximum concentration, same area under the curve.
The main modeling outcome was as follows: Starting dose was established using minimum anticipated biological effect level (MABEL) approach (targeting minimal [10% smallest measureable] Treg increase), Efficacious dose in man (PAD) was predicted for a targeted Treg increase, and Dose escalation will be guided by emerging PKPD data in trial.
Finally,
Session 2: CMC Development—Challenges and Strategies
Session Chairs:
The increasing complexity of new biopharmaceuticals and the accelerated regulatory filing time lines request that we rethink our routine drug development and delivery. This session focused on the increasing demand for collaboration and interaction of chemistry manufacturing and control (CMC) and nonclinical safety to address various safety aspects in drug product development. The session included case studies discussing a broad variety of challenges (eg, particle formation, aggregation, pain on injection, immunogenicity, use of new excipients, etc), and how those have been successfully managed.
Local pain after subcutaneous (SC) injection is a common side effect of antibody treatment, and development of formulations with reduced nociceptive effects is a significant benefit for patients. Therefore, we evaluated the Paw flinch and visceral pain rodent models, for their ability to predict pain upon injection. Paw flinch and visceral pain rodent models were tested using 2 formulations known to produce a difference in pain perception in patients, measured by the Visual Analog Scale (VAS) pain score in the clinical setting. Results from both models did not correlate with results in humans, so neither model was considered to be suitable for differentiating the pain of injected formulations in humans. A promising new approach utilizing spinal reflexes after pain stimuli was tested. 8 This assay, using the electromyogram (EMG), recorded after intraplantar injection in anaesthetized rats which allows for quantification, and thus a potential differentiation, of SC injection pain. A proof-of-concept study with the EMG model was conducted to compare the same 2 formulations with known VAS pain scores, and the method was able to differentiate between pain related to the 2 formulations in agreement with findings in clinical trials.
Then,
A strategy based on the biological role and endogenous concentrations of L-methionine, its status as established excipient for various routes of administration, 9 and toxicity studies in rabbits and monkeys was applied. The strategy was supported by health authorities and use of formulation in phase III studies was approved. L-Methionine is a widely used stabilizer in therapeutic protein formulations 10,11 and was added to a late-stage clinical formulation in order to enable a robust commercial formulation with a long shelf life.
The systemic safety of L-methionine was justified by its role in the metabolism as an essential amino acid and that it is an already established excipient for various routes of administration of approved drug products. 12 In contrast, L-methionine is so far not included in any approved commercial product or within any Roche development program. Furthermore, L-methionine was not qualified as part of the regulatory nonclinical development program of the respective molecule.
Methionine plays an important role in the synthesis of proteins and essential biomolecules. Its systemic safety has been well established in animals, including absence of ocular toxicity following application of high methionine concentration diets. 13 In humans, hypermethioninemia (a condition defined as elevated plasma methionine) is not associated with obvious ocular effects. 14,15
Methionine is naturally present in the eye; ocular tissues and ocular fluids physiologically contain methionine. 16 -18 Ocular methionine may serve as antioxidant through reversible protein oxidation, 19 therefore playing an important role in protective mechanisms maintaining transparency and refractive properties of ocular tissues.
In support of its safety for intravitreal use, Roche has conducted toxicity studies up to 10 weeks in rabbits and cynomolgus monkeys in which L-methionine at different concentrations was a component of the vehicle and/or the test article formulation. In these studies, no ocular effects attributable to methionine were reported at the various methionine concentrations tested (5-25 mM dosed in 50 µL/eye, corresponding to a 0.25-1.25 µmoL dose). Taken into account the differences in vitreous volume of about 1.5 and 2 mL in rabbit and monkey, respectively, 20 ocular methionine concentrations in nonclinical studies are considered to have exceeded those estimated in humans following a single intravitreal application of 50 µL of drug product containing 7 mM methionine.
Human ocular methionine concentration data have been summarized recently. 18 Mean vitreous concentrations in human control eyes of 22.3 to 24.7 µM 21,22 up to 84 µM 23 have been reported. Based on these concentrations, an intravitreal application of 50 µL of drug product (containing 7 mM methionine) at the planned maximum once monthly dosing regimen is expected to result in a slight local increase in vitreous methionine concentrations of approximately 85 to 90 µM only considering an average human vitreous volume of 4 mL. 20 This corresponds to an increase of ∼2- to 4-fold compared to reported mean vitreous concentrations in human control eyes mentioned above.
Considering its established systemic safety profile and natural presence in the eye together with the small amount of added methionine not substantially increasing ocular background levels, there are no safety concerns foreseen with the planned concentrations of methionine to be administered intravitreally once monthly as part of the drug product.
Health authorities supported the safety of L-methionine as a novel pharmaceutical excipient for intravitreal use for long-term treatment based on the scientific rationale and the nonclinical data provided. Therefore, no dedicated additional nonclinical studies were required to qualify the safety of L-methionine, which is currently being evaluated in clinical phase III studies.
Finally, the formulation change could be implemented, and the phase 3 clinical studies could be started without running a long-term term toxicology study in rodent and mammalian nonrodent species.
Next,
UCB has developed an in vitro cellular assay for assessing the potential for triggering immunogenicity of drug candidates based on the analysis of potential T lymphocytes proliferation induced by therapeutic antibodies. In the assay, the drug is directly incubated with cells isolated from peripheral blood mononuclear cell. The investigation is focusing on the impact of CMC and formulation-related conditions on the immunogenicity potential through the evaluation of the most common stress factors encountered during drug manufacturing (temperature, agitation, and freeze/thaw) as well as the impact of different formulation candidates.
A case study demonstrated the link between the presence of protein aggregates (generated through various stresses) and a high immunogenicity potential, represented by the proliferation of T-cells in the in vitro model.
Finally,
Breakout Sessions (“Hot Topics”)
In Vivo Characterization of Antibodies/Biologics (Guenter Blaich, AbbVie )
The group discussed the fate of a biotherapeutic upon and after injection and whether a characterization is of that is needed and/or adds value. Metabolites from a biologic can be produced artificially but so far none of the participating companies were looking into metabolism and tissue distribution. There was consensus that there are no good assays to predict immunogenicity in humans, but some companies are using certain models for ranking of drug candidates. Regarding SC bioavailability in humans, studies in cynomolgus monkey and minipig were not considered predictive regarding SC bioavailability in humans. However, a few companies were using in vitro skin assays for candidate selection. Regarding excipients and their metabolic stability, the group had agreement that this is a topic which currently gains more and more interest from regulatory agencies (ie, Tween 20 and 80 stability). Finally, the question around pain on injection after SC injection was discussed (see also Session 2, Talk Preethne Boeser); there was agreement that based on literature pH, injection volume/speed and buffer components are playing a major role.
Toxicology End Points in Pharmacology Studies (Andrea Kiessling, UCB )
The value and feasibility of implementing toxicological end points in early pharmacological studies were discussed.
As standard, healthy animals are used in the definitive toxicity studies. However, healthy animals most often lack important aspects of the relevant disease indication and thereby do not reflect all pharmacology-driven important safety risks.
The discussion forum agreed that in such cases, it is of high importance to address these risks in close collaboration between the project pharmacologist and the toxicologist. Agreed actions in such cases may include in vitro studies that closely mimic the conditions in the patient, review of literature data on competitor drugs, and human and animal data informing about the intended target and associated pathways or mechanistic in vivo studies in pharmacologically relevant animal species using the drug or a relevant surrogate.
It was felt that for the latter, a tight connection of the pharmacologist and the toxicologist is required in the design of such studies to ensure that the relevant end points are included based on deep understanding of the pharmacology and animal model. This will allow for proper conclusions and decisions from the investigations. Caveats can result from potential differences in sensitivity for the toxicology effects between the animal model and the patient, as well as for animal models where the disease develops often in an accelerated manner not reflecting the chronic development of the disease in humans and associated safety risks. In some cases, the model itself may be associated with safety consequences itself that confound a specific toxicological effect of the drug. Lack of historical controls for some models may also make interpretation of these more difficult. Nevertheless, carefully designed studies in relevant pharmacology models can help to derisk a safety concern in early clinical development and help to develop an appropriate risk-mitigation/management plan and monitoring in clinical studies.
Recent Health Authority Interactions and Questions (Adam Hey, Novartis )
Hot topics from interactions with regulatory agencies were discussed. The first topic was Juvenile toxicity and an increasing demand for up to 1 year juvenile toxicity studies with mAbs. This despite a full toxicology program without any issues and toxicities and including chronic toxicity and enhanced pre- and postnatal development (EPPND) studies. Experiences showed the European Medicines Agency (EMA) being more flexible with a more holistic and scientific approach in case of clean safety studies whereas US Food and Drug Administration (FDA) being insistent in requesting a specific juvenile study. In that context, a case of a request for a juvenile study in cynomolgus monkeys where they were not a relevant species was reported.
Another topic was requirement from several health authorities for condom use in males participating in FIH trials with mAbs. The requirement was repeated and confirmed despite submission of detailed calculations on the absolute minimal to no potential transfer and large margins to proven no-observed-adverse-effect levels in animals.
Finally, examples of mAb programs where rodent carcinogenicity studies were conducted were reported but also that these due to questionable species relevance were not impacting labeling of the tested compounds.
Session 3: Nonclinical Investigative Studies
Session Chairs:
In this session, 2 cases of nonclinical toxicological issue resolution were presented covering the investigative activities involved as well as the translational aspects for patients. Furthermore, a new public–private consortium was introduced that promotes translational safety assessment approaches for immunomodulatory therapeutics with a special focus on human relevance.
First,
Clinical trials with nonsilenced anti-CD40L (anti-CD154) mAbs in patients with systemic lupus erythematosus and idiopathic thrombocytopenic purpura were halted after unexpected fatal thromboembolic events (TE). Previous results have shown that soluble CD40L/anti-CD40L mAb immune complexes (ICs) can activate and induce proaggregatory effects on platelets in vitro via FcgRIIa. Pulmonary thrombi consisting of platelet aggregates and fibrin and thrombocytopenia were found in humanized FcgRIIa transgenic mice after injection of preformed IC consisting of mouse soluble CD40L and antimouse CD40L mAb. To date, no such studies have been performed with IC of mouse sCD40 and anti-mouse CD40 mAbs.
Humanized FcgR transgenic mice were injected with single mouse proteins (soluble CD40L, soluble CD40), antibodies (isotype controls, antimouse CD40L mAb, antimouse CD40 mAb), or IC of soluble protein with the respective antibody.
In addition, in vitro platelet aggregation was performed using blood from FcgR transgenic mice and human healthy donors. Whole blood samples from FcgR transgenic mice were treated with the same proteins and antibodies as in the in vivo study. For human whole blood, isotype controls, soluble CD40L, anti-CD40L mAb including IC in different ratios, soluble CD40, and aCD40 mAbs (eg, CFZ533) including IC in different ratios were used.
In the humanized FcgR transgenic mice, evidence of thromboembolism (thrombi formation in the lung shown by histopathology) and thrombocytopenia with the soluble CD40L/anti-CD40L mAb IC was observed, but not with the soluble proteins or antibodies alone, nor with the soluble CD40/anti-CD40 mAb IC.
The in vitro platelet aggregation assays performed using blood from FcgR transgenic mice and human healthy donors with recombinant proteins, mAbs, and IC from respective species confirmed these findings.
These data provide in vivo and in vitro evidence that all anti-CD40 mAbs, either alone or IC with soluble CD40 protein, do not induce thromboembolism, indicating that the thromboembolism associated with anti-CD40L mAbs is target related but not costimulation pathway specific. Combined with clinical evidence, these data further support the notion of minimal risk for patients developing life-threatening TEs following administration of a pathway blocking anti-CD40 mAb.
Then,
The expected impact of this initiative is to ultimately help deliver safer medicines to patients via: provision of new tools and models to enable a better understanding of the inherent safety risks of immunomodulatory therapeutics; improvement of drug development processes and regulatory assessments for immunomodulatory therapeutics; better definition of limitations in the translatability of nonclinical test systems to patients and will also contribute to the principles of the 3Rs.
Finally,
Session 4: Advanced Therapy Medicinal Products
Session Chairs:
The field of Advanced Therapy Medicinal Product (ATMP) is quickly progressing and this session covered a number of key nonclinical development aspects. The session began with a review of current hot topics in this field and then focused on the potential safety mitigation strategies that could be employed to reduce the potential of on-target/off-target normal cell recognition, examined the potential of chimeric antigen receptor T cells (CAR-T) maternal microchimerism (MM) in newborns, and the final presentation shared the findings of the assessment of potential genomic integration sites (IS) of Tisagenlecleucel-T (CTL019).
First,
The Committee of Advanced Therapies (CAT) at EMA hosted a special session “EMA/CAT regulatory aspects of advanced therapy medicinal products (ATMP).” Martina Schuessler-Lenz (CAT chair) and Ilona Reichl (CAT) introduced into the session with presentations by EMA/CAT experts on regulatory aspects of CAR-T cells (Marcos Timon), AAV vectors (Hans Ovelgönne), and genome editing (Matthias Renner). During the subsequent panel discussion, more valuable information was provided: Simplified environmental risk assessment procedure is available for ex vivo gene-modified medicinal products. Patric Celis (CAT Secretariat) encouraged sponsors to go for mutual acceptable CTAs: FDA and EMA have regular interactions and are willing to support. Fast access to market can be substantially supported by early discussion with Health Technology Assessment bodies. Draft guideline for ex vivo gene-modified cells is open for comments (prolonged comment window July 31, 2019, due to EMA move to Amsterdam).
Then,
Next,
Then,
The final presentation by
Cells were transduced ex vivo with a replication-deficient human lentiviral vector harboring an EF1α promoter and a transgene encoding a CAR with a CD8α leader sequence, a murine anti-CD19scFv a CD8 hinge and transmembrane region, and 4-1BB/CD137 and CD3Ζ/TCRΖ signaling domains.
Adverse events were previously reported in human gene therapy in which γretroviral vectors integrated near the 5′ ends of cancer-associated genes, the injected cells proliferated, and accumulated genetic lesions, which eventually evolved to frank leukemia. Clonal expansions without clinical consequences have been observed for lentiviral vectors in HMGA2; a recently reported integration event in TET2 might have assisted CAR-T19 therapy for CLL.
Genomic IS from 12 treated cancer patients and 2 healthy controls were determined (qualitative polymerase chain reaction [PCR]: 0.04-0.71) and transgene expression (flow cytometry: 3.7%-42.3%). Integration sites were characterized by 2 protocols:
Shearing extension primer tag selection ligation-mediated PCR followed by deep sequencing (MiSeq; Illumina) and analyzed using adapted Gene-IS pipeline at Genewerk GmbH (Germany).
Nonrestrictive linear-amplification mediated PCR followed by deep sequencing (MiSeq; Illumina) and analyzed with INSPIIRED pipeline at Bushman Lab (UPenn)
Total and unique IS numbers were concordant across protocols and in line with the transduction percentage. The IS distributions were as expected for lentiviral integration in human T-cells, showing integration in gene rich regions, within transcription units, and near epigenetic marks associated with active transcription. All samples showed polyclonal scores, either by traditional metrics (richness, evenness, Shannon, Simpson or Gini indexes, and S. chao population size) or the recently developed ones (UC50 at UPenn and pmdIndex at Genewerk).
Specific assessment of IS targeting LMO2, CCND2, MECOM, IKZF1, and HMGA2 was performed. The CCND2, MECOM, and IKZF1 gene loci harbored less than 5 IS, whose relative abundance was far below 1%. Therefore in conclusion, no hotspots of vector IS were identified and the CTL019 project samples showed conventional lentiviral IS in T-cells, without evidence for preferential integration near genes of concern.
Session 5: Challenges in the Toxicity Testing of Highly Potent Biologics Without a Relevant Animal Species
Session Chairs:
This session covered toxicity testing of highly potent biotherapeutics with a focus on CD3 bispecific T-cell engaging antibodies. The session provided a broad overview on the topic with a summary of a recent FDA/HESI-ITC workshop on safety assessment of CD3 bispecifics as well as a case example for a toxicity testing strategy in absence of a relevant animal species.
A 2-step strategy for assessment of these and off target effects was presented.
First step:
To tackle these challenges, a nonclinical strategy for selectivity testing was developed, combining “state of the art” complementary technologies, to power the probability to identify risks. In an early stage of development, the strategy mostly relies on the elucidation of binding mode and insilico predictions of potential off-target peptides and in vitro (killing assays), using T2 cells and HLA-typed primary cells expressing the off-target.
Second step:
Once the clinical lead candidate is selected, the second part of the strategy relies on an unsupervised approach, comprising 3 pillars used in an iterative way: (1) ligandome assays in which the lead candidate is used to “fishing” the complexes with MHC-class I—epitope in organ lysates, (2) immune-competent in vitro models (2-dimensional/3-dimensional microphysiological systems), and finally (3) an in vivo study in B6 HLA transgenic mice using a hemisurrogate.
This selectivity testing strategy combining a wide range of analytical chemistry methods, cellular and molecular assays using human tissues, and cultured cells together with the use of a hemisurrogate in an animal model offers a potential guidance for TCR-like T-cell bispecifics.
The discussions resulted in the following recommendations: Affinities to tumor target and CD3 play a key role for efficacy and toxicity. A detailed target liability assessment is considered to be important but may not be able to predict all side effects encountered in FIH studies. T cell-dependent cellular cytotoxicity assay formats differ between companies and do not necessarily mirror the in vivo situation such as the effector to T cell ratio, effector cell dynamics, cytokine exposure, PK, and soluble receptor effects. It is sufficient to derive CR data from cytotoxicity assays and no dedicated CR assays are required as the community and the FDA understood that CR is a liability for T cell engagers. New mouse models might provide a case-by-case support for translation into patients. Step dosing can be included in toxicity studies to mitigate CR and allow exploration of higher doses and tumor-target specific toxicity. However, use of pretreatments (ie, dexamethasone or anti-IL-6) to block CR in regulatory toxicity studies is not advised. It was presented that dexamethasone would be the best cytokine blocker, but there is no clear agreement on which cytokine(s) is/are most important. Tissue-based findings can occur with and without expression and care should be taken to optimize terminology when comparing programs for each modality. For some targets (eg, MHC-presented peptides), there is no relevant toxicology species available and the nonclinical package and liability assessment must be based solely on in vitro data. Minimum anticipated biological effect level calculations differ considerably between companies, but receptor saturation is well below a critical threshold and plays no role for MABEL. More emphasis should be put on PK/PD/Tox modeling and there can be quite a difference between FIH doses dependent on the modeling approach taken. The clinical starting dose should not be too low, but safe, and companies and regulators are still wrestling with the fact that MABEL does tend to set a rather low clinical starting dose. The paper of Saber et al
35
[Reference citation 33 has been changed to reference 35 as the author name “Saber” matches with ref 35 in reference list. Please approve.] was presented in detail. Single-patient cohorts can be considered for phase 1 cohorts, and different dose-escalation schedules and durations of the DLT period were discussed. Options for management of CR syndrome in the clinic would be step dosing as well as dexamethasone and anti-IL-6 treatment. As examples for clinical safety and potential neurological adverse effects of T cell engagers, Blincyto and CD19 CAR T cells were presented.
Session 6: Assessment and Management of Immunogenicity—Toxicological and Translational Concepts and Strategies
Session Chairs:
Session covered a wide range of immunogenicity-related aspects including consequences of an ADA response observed in toxicity studies such as immune complex-related safety findings and their relevance to human safety, and how to understand at a preclinical stage what safety consequences may be encountered clinically when therapeutic proteins with an endogenous counterpart are neutralized or inactivated by ADA.
Other topics were potential in vivo models to overcome immunogenicity in traditional toxicity studies. Finally, approaches to measure immunogenicity in the context of PK and PD as an integrated assessment and novel proposed ways to determine ADA were discussed.
Case study 1: The weight of evidence was in favour of ADA-mediated ICD and included postdose infusion reactions (facial/inguinal erythema) after multiple repeated doses, glomerulopathy (ultrastructural changes confirmed by transmission electron microscopy) with associated proteinuria, vascular inflammation in multiple tissues, and presence of granular deposits (containing human IgG [hIgG] indicating the presence of drug, monkey IgG [mIgG], monkey IgM [mIgM], and complement C3) by immunohistochemistry (IHC), and detection of ADAs.
Case study 2: A serious, adverse infusion reaction concurrent with ADA in a high dose animal which led to its early termination as well as glomerulopathy with associated clinical pathology changes, vascular inflammation in multiple tissues, and presence of granular deposits (containing drug, mIgG, mIgM and C3) by IHC was consistent with ADA-mediated ICD. Vascular inflammation observed in a coronary artery in 1 male monkey in the low dose group was less obvious in the absence of clinical signs and ADA and insufficient tissue containing the lesion with which to conduct IHC. Since the lesion occurred at a similar location and with similar features compared to the lesion in the coronary vessel of the unscheduled decedent high dose female, it was considered likely to be ADA-related ICD.
Case study 3: In a 26-week study, vascular inflammation was observed in several tissues in multiple animals following treatment with a humanized, wild-type, antichemokine mAb. The nature and distribution of the microscopic inflammatory lesions at known predilection sites for IC deposition, the presence of ADA in some affected monkeys, and the immunohistochemical demonstration of deposited test article, monkey IgG, IgM, and/or C3 at some sites of vascular injury and tissue inflammation are characteristic of IC disease. However, due to the high incidence and dose responsiveness of the inflammatory changes in skin (noninjection site), the large number of monkeys affected, and poor correlation of affected monkeys with detectable circulating ADA and the lack of granular deposits in skin, the mechanism of IC formation is unlikely to be solely attributed to ADA formation. Based on the weight of evidence, it cannot be excluded that other mechanisms besides ADA, for example, pharmacology or chemokine binding to endothelial glycosaminoglycans, may be playing a role in mediating the inflammatory changes.
Case study 4: Arterial inflammation in multiple tissues of 1 low dose female and in the bronchial artery of 1 high dose female was observed in a 1-month study, without evidence of ADA or clinical signs in either monkey. Granular IC deposits containing mIgG, mIgM, and/or C3, but not hIgG, were detected at the vascular lesions in the low dose monkey. However, the severity and distribution of the inflammation were much greater than the numbers of associated granular deposits. In the high dose monkey, the lesion was not present in the IHC sections. Although findings were consistent with IC pathogenesis, the weight of evidence does not support that the changes were associated with ADA. The lesions were morphologically similar to spontaneous arteritis reported in cynomolgus monkeys. 36,37 Cynomolgus monkeys have a high incidence of spontaneous IC formation compared to humans. 38,39 Immune complex disease was not observed in a 6-month study at the same doses.
This resulted in the following recommendations: In the absence of discriminatory biomarkers for ADA-mediated ICD pathogenesis, it is necessary to take an experimental weight-of-evidence approach. Include the conduct of analytical assays to determine ADA and/or circulating immune complexes (CIC); IHC analysis of pathological lesions to detect granular deposits containing human IgG and/or monkey IgG, IgM, and C3; and measuring serum biomarkers such as complement cleavage products, cytokines, and c-reactive protein (CRP). Optimal timing of sampling of these markers is critical for these analyses, for instance, CIC (15 minutes-48 hours postdose), biomarker analysis (cytokines IL6 and MCP1 1-4 hours), complement cleavage products C3a and Bb (15 minutes-4 hours), and CRP (24-48 hours postdose). Including considerations of sampling procedures in the protocol is advised. Immune complex formation continuously occurs in healthy organisms without adverse consequences.
38
However, the normal clearance process is saturable and in an excess of large CIC or impaired clearance mechanisms, the complexes can deposit in vessel walls resulting in local inflammation. Biopharmaceuticals that deviate from endogenous counterparts may be more immunogenic and likely to lead to ICD. It is important to understand the mechanism of vascular inflammation to contextualize the relevance to human safety.
Then,
Such an assessment should include an in silico assessment of literature data of knockout animals (if available) and available human data in diseases that lack partial or complete function of the protein of interest.
In some cases, such data may not be available, for example, due to infertility of constitutional knockout models. The risks for generation of neutralizing antibodies to the endogenous counterpart depend on many factors such as the nature of the target, the drug characteristics (such as the format, the presence of mutations and linkers, impurities or contaminations, or drug aggregates), the route of administration, the therapeutic indications, and concomitant medications, all impacting the risk for immunogenicity to both the drug and its endogenous counterpart. Safety consequences regarding a neutralization of the endogenous counterpart depend on many factors such as the existence of functionally redundant proteins, the human expression pattern of the protein, and the availability of rescue medications, or can be learned from other drugs aiming for therapeutic administration of the same or a homologous protein. If the available in silico data are insufficient, in vitro investigations of potential proinflammatory and immunomodulating effects could be done or even in vivo studies mimicking neutralizing immunogenicity or studies in newly generated knockout models and their consequences might be warranted.
The latter may include monitoring of safety events in ADA-positive antibodies in toxicology studies, dosing of pharmacologically relevant animals with neutralizing antibodies, and conditional knockout models when constitutional knockout models are not vital. Such an assessment should always be embedded in an overall risk–benefit evaluation.
Various transgenic mouse models have been used as potential predictive tools to assess immunogenicity as predictive tool. The major principle is to express a human therapeutic protein in a transgenic mouse model to suppress a xenogeneic immune response against the therapeutic protein. We explored a human IgG transgenic mouse strain expressing a minirepertoire of human IgG1 antibodies as model for long-term toxicity testing of an IgG coupled cytokine. In contrast to wild-type mice, these mice showed no ADAs at low dose and only low titer ADAs at mid dose. Exposure and PD decrease to some extend over time due to ADA (at mid dose) and/or target upregulation/TMDD at both doses. This human IgG to mouse model may be a suitable and by regulators accepted model for some toxicity testing.
Circulating immune complexes and their contribution to ICD in nonclinical toxicity studies are poorly characterized. Immune complexes can be involved in toxicity findings of varying degrees of severity. The formation of ADAs against human or humanized therapeutic mAbs in nonclinical toxicity studies is typically responsible for ICD-related toxicities. 40 -42 Providing a link between ICD and ADA formation provides evidence that the ICD is likely ADA related and not, for example, the result of drug: target complexes or pharmacological mechanisms; subsequently derisking the potential for pharmacology related ICD in clinical studies. Detection of ADA typically involves the use of the drug as both the capture and detection reagent. However, these drug–drug bridging assays are susceptible to interference from several sources, the most common of which are circulating drug and instances where the drug target is multimeric in nature, preventing detection of the circulating ADAs or generating false positive signals when no ADAs are present, respectively.
The assay was based on capture of the human mAb drug and detection with an antimonkey IgG detection reagent that does not crossreact with human IgG and thus is drug tolerant. The alternative approach to traditional drug–drug bridging assays has been developed for the measurement of ADA in nonclinical cynomolgus monkey toxicology studies (note 1), which is drug and multimeric target tolerant and assay was based on published reports using a similar assay format. 43 -45 The method described here is based on capture of the human mAb drug that may or may not be complexed with other proteins in circulation (ie, target, ADA, etc) and detection with an antimonkey IgG detection reagent that does not crossreact with human IgG. This unique assay format allows for the accurate detection and measurement of human mAb drug-specific CICs in serum without the common interference issues associated with traditional ADA bridging assays which can aid in understanding loss of drug exposure and any relationship to observed toxicities. Building on the uniqueness of the assay format, we have developed a novel sample analysis workflow, followed by statistical trend analysis that allows for characterization of the form of ADA present in the sample over the course of a nonclinical toxicology study. To determine the form of the ADA present in test samples, they are analyzed in duplicate before and after the addition of 3 sequential ascending drug spikes. The concentrations of these 3 sequential ascending drug spikes are highly dependent on the concentration and frequency of the preclinical toxicity study doses. Optimizing the 3 sequential ascending drug spikes in relation to the study dose and frequency is important to establish sufficient antihuman IgG antibody capture capacity corresponding to the analytical range of the assay, which, in turn, enables the generation of interpretable trend analysis results. For most studies, the most appropriate drug concentration spikes, to allow for acceptable assay capacity and interpretable responses for trend analysis, are 100, 1,000, and 10,000 µg/mL (concentrations in terms of 100% serum). After analysis, the resulting data are evaluated for trends by examining the fold change between and across unspiked and spiked normalized sample response values. The derivation of the statistical fold change threshold is determined from analysis of naive animal assay variability. Through the use of this alternative assay format, novel sample analysis workflow, and statistical trend analysis from the spiked and unspiked sample results, the form of ADA present in the sample can be understood as either “complexed,” “free,’ or a combination of both complexed and free ADA (“mixed”). When comparing this method to traditional ADA bridging methods and correlating to PK data from animal sample profiles with varied levels of toxicities, this interference-tolerant ADA method demonstrates superior ADA detection over the traditional bridging method format. The ADA form is also identified which can be informative when considering loss in drug exposure concentrations resulting from ADA-mediated drug clearance or incidence and severity of observed preclinical toxicities.
Examples were presented from cynomolgus monkeys dosed intravenously with 30 mg/kg a humanized mAb weekly for 4 weeks. Each monkey demonstrated varying degrees of toxicities, impact on drug exposure, and ADA responses. A comparison of drug concentration and ADA using both the described interference-tolerant ADA method and a traditional bridging ADA method was performed which confirmed the validity of the method, based on both predose and postdose samples from days 1, 8, 15, and 22.
Example 1: Animal “A” exhibited no observed drug concentration changes and minimal toxicity consisting of mild edema after day 15. The form of the ADA measured in the interference-tolerant ADA assay was fully complexed in both the pre- and postdose day 15 and 22 samples, with no ADAs detected using the traditional ADA bridging assay at these same time points likely due to drug interference.
Example 2: Animal “B” exhibited a loss in drug exposure at the day 22 predose sampling time point with no observation of toxicity. The form of ADA measured in this animal was fully complexed in the pre- and postdose day 15 and the postdose day 22 sample with mixed ADA in the day 22 predose sample. Although drug tolerance limitations remained an issue for detection of ADA with the traditional bridging assay and ADAs were not detectable in the day 15 pre- and postdose samples and the day 22 postdose sample, ADA was measurable in the day 22 predose sample resulting from the reduced drug concentration from clearance and/or deposition correlating to the mixed ADA form and diminishing interference in the traditional bridging assay from circulating drug.
Example 3: Animal “C” exhibited a loss in drug exposure in both the day 15 and 22 pre- and postdose samples as well as demonstrating a severe toxicity reaction after the fourth dose. The form of the ADA measured for this animal was mixed (both free and complexed ADAs were found to be present in the sample) in the day 15 and 22 predose samples and complexed at the day 15 and 22 postdose samples. This trend from mixed to complexed ADA from the predose to postdose sampling time points is indicative of increased clearance and/or deposition of drug-specific ADA complexes and thus believed to be related to the observed severe toxicities. In addition, the interference-tolerant assay was able to detect ADA at all 4 sampling time points whereas the traditional bridging assay was only able to measure ADA in the day 15 and 22 predose time points. The reduction in drug exposure and mixed form of the ADA allowed for ADA detection using the traditional bridging method due to reduced interference from circulating drug.
Example 4: Animal “D” exhibited reduced drug exposure at both the day 15 and 22 predose and postdose sampling time points and severe toxicities which ultimately led to early termination of the animal. The form of the ADA for animal D was similar to the ADA described for animal C (example 3) with the exception of the day 22 predose sampling time point, where the form of the ADA measured was free indicating additional clearance and/or deposition of the drug–ADA complexes where the observed toxicities required euthanasia of the animal. Also, as observed before with animal C (example 3), ADAs were detected in all 4 of the day 15 and 22 pre- and postdose sampling time points with the interference-tolerant ADA assay, whereas the traditional bridging assay was only able to detect and measure ADA at the day 15 and 22 predose sampling time points when the form of the ADA was either mixed or free, and the drug exposure was nonquantifiable.
In conclusion, the interference-tolerant ADA analytical method described represents a novel robust drug tolerant preclinical immunogenicity method with the ability to measure predose sampling time points and early postdose time points when infusion reactions are often observed. This confirms not only the occurrence of ADA in the presence of high drug concentrations but also the form of the ADA detected. Although additional experience with the assay is necessary to better understand the toxicological significance of the form of ADA detected in this assay, the improved ability to detect ADA will aid in interpreting causal relationships with toxicities observed in nonclinical studies, thus reducing unnecessary delays in compound development cycle times. Furthermore, the nature of this novel interference-tolerant approach allows for a common format which can be applied for all human biotherapeutic mAbs using similar reagents for any nonclinical species. Measuring ADA and drug-specific ICs in a different nonclinical species can be easily accomplished by identification of the appropriate antispecies detection reagent that does not demonstrate crossreactivity with human IgG and applying a similar sample workflow as previously described.
In contrast to small molecules, toxicity of biologics is mainly due to exaggerated pharmacology rather than off-target effects. Therefore, the validity of toxicity studies for biologics relies upon the demonstration of active drug exposure throughout the study. However, for many biologics this active exposure in animals is impaired by the formation of ADAs. Immunogenicity can either change the exposure via antibodies enhancing (clearing ADAs) or reducing the clearance (sustaining ADAs). Although clearing ADAs usually go along with reduced PD, sustaining antibodies do not necessarily lead to enhanced PD (as they might be neutralizing to a certain extent). Neutralizing antibodies impair the binding of biologics to their targets, thereby reducing their pharmacological activity. It is crucial for the interpretation of safety data to assess whether and to what extent ADA-bound biologics retain pharmacological activity.
This highlights the importance of using an assay format capable of detecting pharmacologically active drug. The choice of the right PK assay format to assess TK in repeated dose toxicity studies is therefore utmost important. It is strongly recommended to use an assay format being capable of detecting pharmacological active drug. Such an assay usually involves the target to capture the biotherapeutic. Some companies did embark into a cell-based potency assay to show active exposure. However, to achieve a decent sensitivity, this approach can only be used for rather potent drugs and in addition suffers from low throughput and high associated costs. A “total” PK assay as standalone solution is discouraged as it is blind to the effect of neutralizing antibodies but might be used in conjunction with a suitable PD marker or a dedicated neutralizing assay.
According to ICHS6R1, ADA samples should be drawn but only analyzed based on evidence of altered PD activity, unexpected changes in exposure, or evidence of immune-mediated reactions.
Neutralizing antibodies should be assessed, if ADAs are detected in the absence of a PD marker to demonstrate sustained activity in the animals.
According to the presented strategy, neutralizing capacity can also be assessed indirectly using a PK assay capable of detecting pharmacological active drug. Immunogenicity assessment as described in the ICHS6R1 is only meaningful if at least ADA and TK can be assessed in the same animal. This is currently feasible in NHPs and (theoretically) rats but not mice. In the future, microsampling technologies (eg, Gyros) might allow collecting all information within the same rodent animal (preferably from main group). In addition, as ADA assay validation is usually the time-consuming step, the strategy outlined before does only pay off for subchronic or chronic toxicity studies in which ADA sample testing significantly contributes to the workload.
Conclusions
Nonclinical safety assessment of biologics is optimally done using an integrated approach based on the overall toxicology, PK, PD, pathological, and modeling data.
The complexity and the specific challenges of biologics with respect to the multiple different formats, their PD and related pharmacological and toxicological effects, PKs, and potential for eliciting immunogenicity merits discussions of this in a dedicated forum of specialists in biologics safety assessment.
The annual BioSafe meetings in European Union and United States serve that purpose, gathering experts from a range of pharmaceutical companies covering the relevant different specialities.
This article described the outcome of the eighth meeting of the European BioSafe members. The meeting covered a range of presentations, breakout session discussions on hot topics, and experiences in biologics safety assessment. The format of these meetings have evolved into a very productive open sharing of case examples and discussions of strategies, data, and health authority feedback. As for previous BioSafe Europe meetings, this resulted in a very engaged atmosphere with passionate discussions and exchange of ideas reflected in the descriptions above from the different sessions.
The positive and constructive outcome of the meeting confirmed the benefits of and need for continuing such annual meetings where scientists with a common passion for biologics safety assessment can network and exchange experiences and ideas (Figure 1).
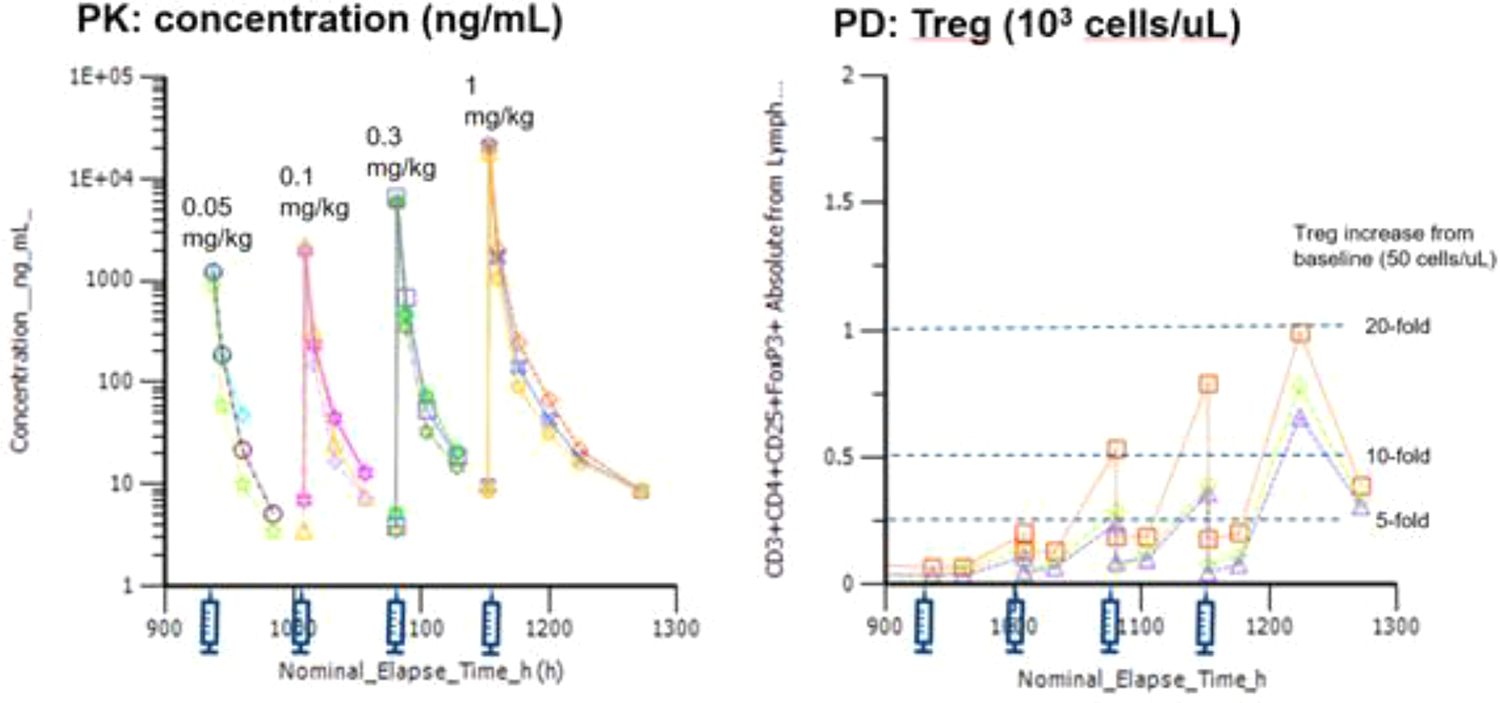
Applying an Espresso design: A clear PKPD (Treg increase) relationship was established in monkey (n = 3). Three male cynomolgus monkeys were administered IV doses of 0.05, 0.1, 0.3, and 1 mg/kg with 3 days interval. Matching daily sampling of PK and PD (regulatory T cells CD3+CD4+CD25+FoxP3+ by flow cytometry). Dose dependent exposure and associated increase in regulatory T cells demonstrating 5- to 20-fold increases relative to baseline values observed, demonstrating a clear PKPD relationship. IV indicates intravenous; PD, pharmacodynamic; PK, pharmacokinetic; Treg, regulatory T cells.
Footnotes
Author Contributions
Adam Hey contributed to conception and design and contributed to acquisition; Andreas Baumann contributed to conception and design and contributed to acquisition and interpretation; Sven Kronenberg contributed to conception and design and contributed to analysis and interpretation; Guenter Blaich contributed to conception and design and contributed to interpretation; Silke Mohl contributed to conception and design and contributed to acquisition, analysis, and interpretation; Rahni Fagg contributed to conception and design and contributed to interpretation; Peter Ulrich contributed to conception and design and contributed to analysis and interpretation; Benno Rattel contributed to conception and design and contributed to acquisition, analysis, and interpretation; Wolfgang F. Richter contributed to conception and design and contributed to acquisition, analysis, and interpretation; Andrea Kiessling contributed to conception and design and contributed to acquisition, analysis, and interpretation; Lucinda Weir contributed to conception and design and contributed to acquisition and interpretation. All authors drafted the manuscript, critically revised the manuscript, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Adam Hey and Peter Ulrich are employees of Novartis, Andreas Baumann is an employee of Bayer AG, Sven Kronenberg, Wolfgan Richter and Silke Mohl are employees of Roche, Guenter Blaich is an employee of AbbVie, Lucinda Weir and Rajni Fagg are employees of GSK, Benno Rattel is an employee of Amgen and Andrea Kiessling is an employee of UCB.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.