
Abstract
The main considerations for the development of a formulation for preclinical safety assessment testing are explored. Intravenous, inhalation, oral and dermal dosing are given focus and although different dose routes do present their own individual challenges there are common themes that emerge. In each case it is necessary to maximise exposure to achieve high doses to satisfy regulatory requirements for safety assessment testing. This often involves producing formulations that are at the limits of solubility and maximum volumes possible for administration to different test species by the chosen route. It is concluded that for all routes it is important to thoroughly explore the stability of the test item in the proposed formulation matrix well ahead of dosing any animals, giving careful consideration to which excipients are used and what their underlying toxicity profile may be for the relevant preclinical species. In addition, determining the maximum achievable concentrations and weighing that against the maximum volumes that can be given by the chosen route in all the test species at an early stage will also give a read on whether it would be theoretically possible to achieve suitably high enough doses to support clinical work. Not doing so can cause delays in the development programme and may have ethical repercussions.
Introduction
It is rarely possible to administer an active pharmaceutical ingredient (API) to any preclinical animal species by most dose routes. In the majority of cases, it is necessary to give the test item as part of a formulation which may vary between simple solutions or suspensions to complex drug delivery systems. This is usually because it is necessary to include an excipient to improve the test item’s shelf-life, stability, absorption characteristics, bioavailability or some other pharmaceutical function. The quality and consistency of the formulation need to be maintained during large scale manufacture. There are no excipients that are truly inert, and if required in high enough concentrations, they will exert some biological effect upon the test subject or patient. More importantly, some excipients, even if they are compatible with humans, may not be well tolerated in other species, and consequently, it may be necessary to use a different excipient in the preclinical species than is intended for use in the clinic. 1
For relatively small doses that are required for the clinic or in patients, the levels of the excipient may be low, but in order to achieve the high doses necessary to satisfy international regulatory safety testing requirements, it is often necessary to try to achieve high concentrations of the drug in solution. For example, depending on the class of drug and the relative level of systemic exposure that is to be targeted in patients, the International Congress on Harmonisation (ICH) M3 guidance document that covers small molecule development has specific guidance in regard to the highest dosage level that needs to be tested in both rodent and non-rodent species and this may be as much as 1 000 or 2000 mg/kg/day. 2 For all dose routes, there are maximum volumes that can be administered to a specific species per unit time, and as such, there may be a requirement to achieve a stable formulation for which the concentration is much higher than could ever be exposed to humans deliberately. 3 In addition, definitive preclinical safety assessment studies need to be conducted in accordance with Good Laboratory Practice (GLP).4,5 As such, it is necessary that any formulation is shown to be stable and homogeneous under the conditions of the preclinical study and will need to be demonstrated by subsampling of the formulations during every GLP study. This is not just for reasons of quality alone but is also for ethical reasons. If a study is conducted and the formulation is found to be unstable, for example, the test item has precipitated out of solution prior to or during the dosing of the animals, this will affect the dosage that the animal received and require a repeat of that study. Consequently, careful forward planning of formulation development is necessary before starting animal work.
Choice of Excipients
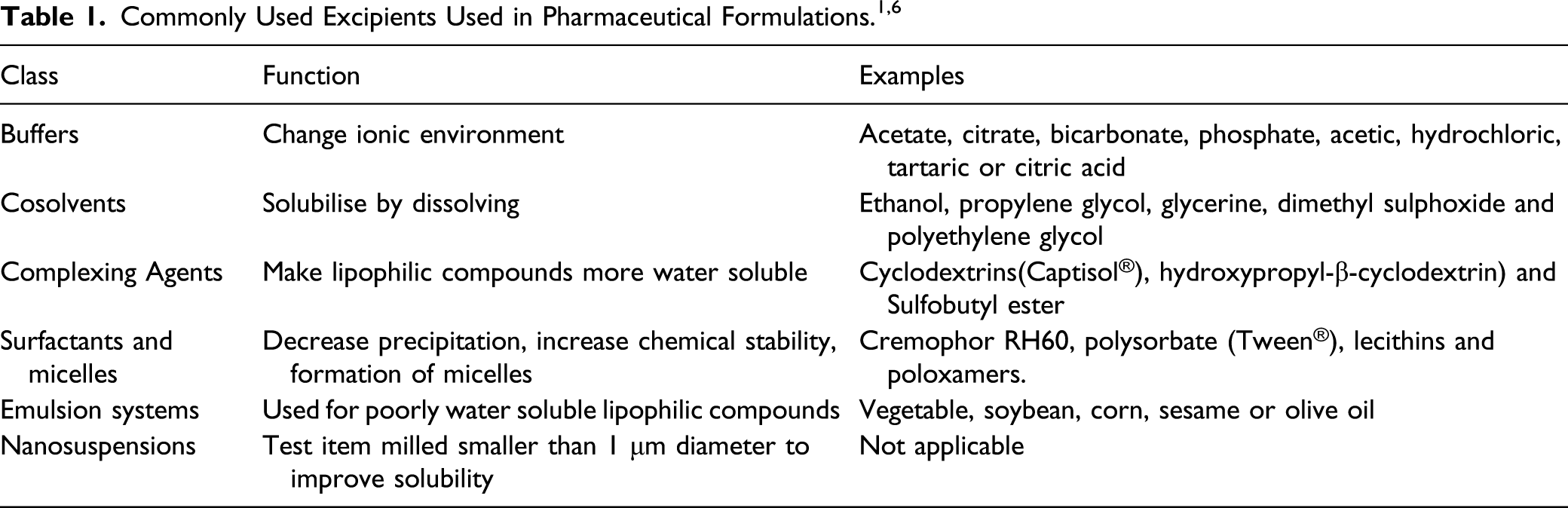
It is worth noting that if it becomes necessary to use a novel excipient or the test article or formulation contains impurities or breakdown products, then it may be necessary to quantify these in terms of their safety profile in isolation from the test item. In many cases, this may depend on the chemical class of the excipient or impurity and the quantity present in the administered product. If this situation is encountered, then it may be advisable to seek regulatory advice as the legislation that relates to this can be complex. Guidance relating to this is covered under various guidelines including ICH,7–9 Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). 10
Changing a formulation in any regard during a nonclinical development package is not advisable. It is very likely that this will change the exposure profile of the test item. For example, nanomilling a material is likely to enhance exposure significantly or changing an excipient will possibly change the safety profile in regard to background changes or may introduce new additive or synergistic effects such that additional bridging studies may become necessary. Consequently, time and care should be used in the selection of the most appropriate vehicle at the early stages of development as this is likely to save money, time and the unnecessary use of animals in the longer term. It should also be noted that some excipients that are relatively inert in isolation can be considerably more toxic in combination. For a more comprehensive list of excipients and their relative tolerability, the reader is referred to Gad et al. (2016). 6 When the use of biologically active vehicles cannot be avoided, it is normally advisable to include an additional dose group on the study with the simple base formulation in order to assess vehicle effects in isolation from the test item. The considerations relating to excipients will also vary in regard to the proposed dose route.
Routes of Exposure
Intravenous (IV) Delivery
For IV delivery and particularly long duration injection or infusion, it is particularly difficult to find a suitable vehicle. For certain indications such as oncology and biopharmaceutical therapies, the IV route is standard and most commonly used. Intravenous drugs are relatively more difficult to administer in comparison with oral or dermal drugs and not often particularly pleasant for the patient, and so in most cases, the decision to progress a test item for intravenous delivery will be out of necessity rather than by design. There are a number of factors that dictate this, and this will include the toxicity and kinetics of the active ingredient and the physical and chemical properties of the test formulation. Often, these types of drugs have stability problems at higher concentrations and therefore require the use of more unusual and less well characterised vehicles and need to be given at a higher dose volume in order to achieve high-dose levels which, in turn, will inevitably lead to a higher systemic dose of the excipients used. Another common reason is that the test item has a small therapeutic/toxicity window or exaggerated pharmacology at a high blood concentration (Cmax). In order to reduce this, the dose must be given more slowly to carefully control or lower peak exposure. As such, formulations either need to be given as a large volume or slowly in both cases; this will mean that the dose will need to be given over a long duration and will be in contact with the vein at the dose site for a relatively long duration. This dose route is, as a consequence, very susceptible to local irritation. This local irritation may be related to the properties of the test item itself but can also be related to the vehicle.
Extremes of pH and osmolality alone can also cause local irritancy and there is general concordance between the local tolerability of human blood and tissues for these factors in comparison to what is seen in preclinical animal models. In the relevant mammalian species, the pH of blood is approximately 7.5 and the osmolality 280–310 Osm mmol/kg, and in humans, a pH within the range of 5–9 and an osmolality between 100 and 600 Osm mmol/kg is generally considered acceptable for administration by most medical bodies. 11 Studies have shown that IV formulations outside of this range may cause haemolysis and/or phlebitis type pathologies and other inflammatory responses at the injection site. If use of formulations outside of this pH range is needed, modification to study designs can be made with guidance from a specialized preclinical toxicologist. 12 Some commonly administered IV drugs range in pH from ∼3.0 (dopamine HCl) to pH of ∼11–12 for ganciclovir sodium and phenytoin sodium.11,13
Drugs delivered by the IV route, as previously discussed, can be chemically unstable and may precipitate out of solution and cause coagulation or flocculation on contact with blood. This can also lead to the formation of crystal or other plaques that may result in the formation of emboli in the more vascular organs such as the lung. 14 If the drug needs to be delivered using a surgically catheterised model, it is likely to involve the slow movement of formulation through a large surface area of tubing, made of various materials and at a variety of temperatures. Under these conditions, it is possible that the test item (potentially at a low concentration) will adhere to the tubing or precipitate out of solution during administration and therefore reduce exposure or form plaques. For these reasons, it is advisable that formulations are tested in-vitro to assess haemocompatibility and for the possibility of test item adherence prior to their first administration into an animal. In-vitro haemocompatibility models are widely used and commonly available, and it is usually possible to include an assessment of test item adherence as part of the formulation stability and validation studies that need to be conducted at an early stages of a drug development programme. 15
Inhalation Delivery
Progressively, more classes of test item are being developed for inhalation delivery. The most common physical forms for delivery of a pharmaceutical by this route are as an aerosolised powder or liquid, nebulised vapour, or as a gas or pressurised spray. In development of formulations by this route, careful consideration needs to be given to the particle size of the test item that is delivered. Particle size will affect the region of the lung to which the test item is delivered and can therefore affect not just its efficacy but also its absorption characteristics and toxicity. As with all formulations, it is necessary to confirm the achieved concentration of the formulation, but with such doses, assessing particle size is also therefore necessary, as well as sampling of the test atmosphere is also required. There are various methods for the generation of test atmospheres which can involve a variety of different pieces of equipment and methodologies and can take some time to develop even by an experienced team. For more detailed information on this area, the reader is referred to in Teppler et al 16 and Bujold et al. 17 Gases may be released from a pressurised cylinder via a regulator value and flow metre where it will be diluted by air to provide the required atmosphere. Vapours may be formed by evaporating a liquid in a stream of air driven by a syringe pump. The resultant vapour may require atomisation to produce a mist which is accomplished by pumping in air at a high pressure into a nebuliser with the test vapour. 18
Pharmaceutical powders can be delivered simply as the pure active ingredient. To improve aerodynamic properties, the powder may be mixed with an excipient such as lactose. Other modifications may include spray drying, which can be used to give more defined particle size, and density adjustment which is a good method for insoluble or fragile materials. The most common method for administering a pressurised spray is through a metred dose inhaler. Given the equipment used in many of these devices, it is necessary to account for any leachates, for example, propellant cans or cylinders may leach plasticisers out of the rubber valve. 19
Non-rodent preclinical species such as non-human primates (NHP), dogs and minipigs typically receive the test formulation via purpose made facemasks while rodents may be restrained in tubes with their snouts directed into a cylinder containing the test atmosphere. By using respiration rate known as conversion ratios and the test atmosphere concentration, it is possible to estimate dose level but the actual dose level each group of animals is exposed to can only really be determined accurately once the blood exposure data becomes available at the end of the study. 20
Oral Delivery
Development of formulations for oral administrations is generally far simpler than those developed for IV or inhalation delivery. For test items that are administered by gavage, it is usually appropriate to administer a suspension rather than a solution. It is usually possible to dissolve most drugs in simple aqueous vehicles such as water, methycellulose or corn oil, and so excipient-related problems are less commonly encountered. Where the test item is intended to be given to the patient in a capsule or as a tablet, this can normally be given in the same form to most preclinical species including dogs, rats, NHPs and minipigs. It is worth mentioning that dogs do have a sensitive vomit reflex, are more prone to loose or liquid faeces and their gastrointestinal (GI) tract is, in general, less similar to humans than that of the minipig. Consequently, the minipig is rapidly becoming the more favourable non-rodent species for the preclinical testing of oral drugs. 21 There are also several excipients such as Solutal®, Vitamin E TPGS and several oils that should be avoided in dogs orally. For novel excipients or where the excipients are at particularly high concentrations or where doses are given at a high volume, it is advisable to trial the vehicle in a small number of dogs before committing to a formulation to develop further. Similar to all routes of administration, consideration should be made for the kinetics and absorption characteristics of the drug and particularly the region of the GI tract where the drug is absorbed. For example, aqueous formulations are likely to be absorbed through the stomach when capsules can often survive breakdown in the stomach and therefore the test item is absorbed further along the GI tract, typically in the small intestine. 22 Absorption characteristics can also be affected by the excipients used in the vehicle. Shah et al 22 also serve as a good source of general guidance for the development of oral formulations in preclinical species.
Admixture of the drug to the diet of the animals is a relatively uncommon dose route. As with all preclinical testing, it is necessary to dose in terms of mg/kg, and as such, dose levels need to be predicted for each dose based on the most recently recorded body weight and food consumption data prior to formulation. The analytical chemistry that supports dietary administration is usually quite challenging as the test item can bind preferentially to the various components of the diet. Dietary administration is not practical in NHPs as they are predisposed to playing with and throwing food, and therefore, food consumption and achieved intake of test item is difficult to measure accurately.
Dermal Delivery
Comparison of Minipig With Human Skin Thickness. 23
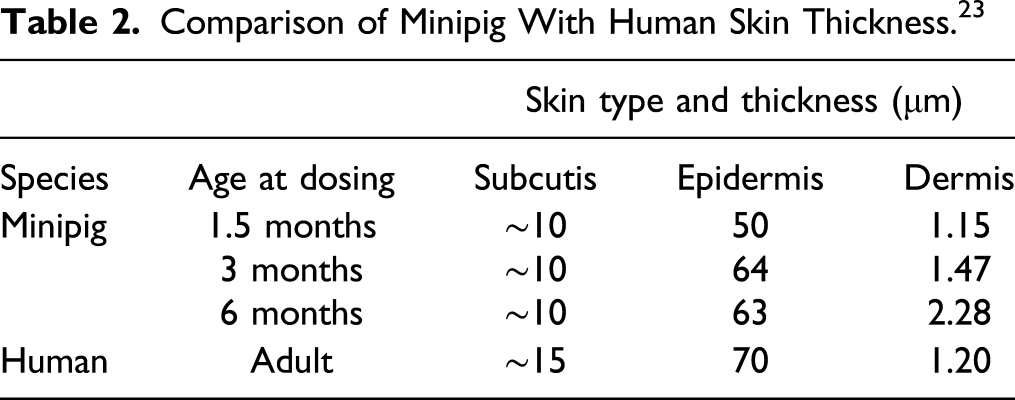
For further information on the relative properties of minipig vs human skin, the reader is encouraged to consult Wojcinski et al 24 and Stricker-Krongrad et al 25 for further guidance on the preclinical development of drugs for dermal delivery.
Other Delivery Routes
There are few if any routes of drug delivery in humans that cannot be mimicked in preclinical species to a lesser or greater extent. We have already discussed the most common routes which hold particular challenges, and Emami et al 26 provide further general guidance in this area. There are a variety of other injectable routes including, but not limited to, subcutaneous, intraperitoneal, intrathecal, intramuscular, intradermal, intraarticular, intraarterial and intraocular administration. Other routes in which the test article comes into direct contact with mucous membranes include intravaginal, buccal cavity or rectal administration. Using these routes will require care in the choice of excipients used in the formulation as these routes are particularly prone to local irritation.
Solutions to Overcome the Limitations of Dosing Clinical Formulations in Preclinical Species
Often, the exposure of the preclinical species to the test formulation is considerably lower than that observed in humans, and it is therefore necessary to give as high a dose as possible by the test route in order to achieve a sufficient safety margin to permit first-in-man clinical trials. For example, intramuscular administration in humans, where large muscle blocks are available, is difficult to perform in rodents where the muscle blocks are smaller relative to the size of the equipment and dose. Therefore, it may become necessary to administer high concentrations, volumes and/or increase the number of dose sites. Care should also be taken to ensure that the volume administered can be tolerated and is practical by the chosen route and preclinical species.3,27
It may sometimes be necessary to administer the test item by multiple dose routes at the same time. The assessment of a maximum practical dose is all that is necessary to understand the local toxicity of the test item, but in order to fully assess a sufficiently high systemic exposure/toxicity, it may be necessary to administer the test item, additionally, by a second dose route. This additional route may not be an intended route for use in the clinic. The implication here is that it will likely be necessary to have two different formulations of your test material one for each route. Dose routes that are particularly prone to this type of issue are inhalation and dermal and typically the additional routes that would be used are intravenous, subcutaneous or oral administration. It will also be necessary to confirm local toxicity by this additional route. In all likelihood, this additional dose is relatively modest and would be given at the same level to all treated groups. As such, only a small confirmatory study would be necessary to make this assessment. An example of this might occur in the treatment of psoriasis in humans where the dermal dose is intended for administration to the broken human skin where a relatively high systemic exposure is seen compared with an animal model with intact skin. An alternative strategy here might be to cause long-term abrasion of the skin to mimic long-term chronic exposure at higher levels. This, however, is not desirable from an ethical perspective where an additional subcutaneous or intramuscular injection might be considered more favourable to mimic the human situation.
Biopharmaceutical products generally do not require complex vehicles and usually do not need to be administered in high volumes by any route, as a safety margin of 10×, the likely human dose is often adequate to satisfy regulatory requirements. 28 A further concern for such products when they are administered intravenously is total protein load which can lead to toxicities relating to clearance and immune complex formation, particularly following repeat dosing. This is somewhat related to the size and total dose of the protein administered and not easy to predict but higher doses given repeatedly should be avoided and this should be considered in addition to those previously mentioned for other classes of drug. 29
There are considerations that need to be made when redeveloping a drug for a dose route for which it is not currently licenced. Even if it is not necessary to change the formulation, there will likely be an expectation for bridging studies in order to assess local irritancy by the new dose route. The bridging studies required to support regulatory submissions following a change in dose route will depend on the routes in question based on the relative risks. There is some very useful guidance issued by the US FDA which provides their recommended approach. 30 In general, if you are transitioning from a route that would give a relatively low level of systemic exposure, dermal or intraocular, for example, to a dose route that would provide a greater level of exposure, such as intravenous or oral, then the extent of bridging work required would likely be greater.
Conclusion
For most, if not all dose routes, preclinical toxicity studies often involve administering doses far higher than in the clinical setting. In terms of dosing volume and duration, it is not always possible to administer test formulations as high in preclinical species as is possible in humans and this means that higher test item concentrations in formulations are typically required. Intravenous and inhalation delivery more commonly present more of these challenges than with other dose routes. For any dose route, it is important to consider the physicochemical properties of the formation as a whole when assessing potential toxicity. In doing this, it may be necessary to use different excipients which need to be qualified in the preclinical species. Determining the maximum achievable concentrations and weighing that against the maximum volumes that can be given by the chosen route in all the test species at the earliest stages is considered essential for the smooth progress of any drug development plan. Not doing so can cause delays in the development programme and may have ethical repercussions.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.