
Abstract
Currently, off-label continuous administration of inhaled epoprostenol is used to manage hemodynamics during mitral valve surgery. A toxicology program was developed to support the use of inhaled epoprostenol during mechanical ventilation as well as pre- and postsurgery via nasal prongs. To support use in patients using nasal prongs, a Good Laboratory Practice (GLP), 14-day rat, nose-only inhalation study was performed. No adverse findings were observed at ∼50× the dose rate received by patient during off-label use. To simulate up to 48 hours continuous aerosol exposure during mechanical ventilation, a GLP toxicology study was performed using anesthetized, intubated, mechanically ventilated dogs. Dogs inhaled epoprostenol at approximately 6× and 13× the dose rate reported in off-label human studies. This novel animal model required establishment of a dog intensive care unit providing sedation, multisystem support, partial parenteral nutrition, and management of the intubated mechanically ventilated dogs for the 48-hour duration of study. Aerosol was generated by a vibrating mesh nebulizer with novel methods required to determine dose and particle size in-vitro. Continuous pH 10.5 epoprostenol was anticipated to be associated with lung injury; however, no adverse findings were observed. As no toxicity at pH 10.5 was observed with a formulation that required refrigeration, a room temperature stable formulation at pH 12 was evaluated in the same ventilated dog model. Again, there were no adverse findings. In conclusion, current toxicology findings support the evaluation of inhaled epoprostenol at pH 12 in surgical patients with pulmonary hypertension for up to 48 hours continuous exposure.
Introduction
Mitral valve (MV) disease (stenosis or regurgitation) often times results in pulmonary hypertension (PH). In addition to the preexisting PH in these patients, patients undergoing MV surgery and requiring cardiopulmonary bypass (CPB) are at risk to have exacerbations of preexisting PH, which, if not immediately treated, can lead to right heart dysfunction and/or failure. These underlying risks of PH frequently complicate the perioperative management of patients undergoing MV surgery 1 and may result in prolonged time in the intensive care unit (ICU) on ventilatory support, and other intensive therapies resulting in increased morbidity and mortality. 2
In patients undergoing MV surgery with CBP, there is a need to acutely and rapidly adjust pulmonary vascular resistance (PVR) during and after MV surgery without impacting systemic blood pressure. The use of general vasodilators and specific pulmonary vasodilators, administered intravenously, is normally contraindicated because of the propensity to decrease systemic arterial pressure and increase pulmonary edema. Mitral valve surgery patients would be expected to have a unique benefit from an inhaled vasodilator, such as epoprostenol.
Epoprostenol, is a naturally occurring prostaglandin molecule, also called prostacyclin (PGI2), which has 2 major pharmacological effects: (1) it is a potent vasodilator, and (2) it inhibits platelet aggregation, both effects through augmentation of cyclic adenosine monophosphate levels. Inhaled epoprostenol, in the approximate dose range 1 to 30 ng/kg/min, has shown dose-related efficacy in dogs and sheep by decreasing PVR and improving hemodynamic parameters in PH models of hypoxic pulmonary vasoconstriction and with infusion of a thromboxane A2 mimetic. 3,4 Furthermore, multiple reports have demonstrated that it is safe and beneficial to administer inhaled epoprostenol (or iloprost) to MV surgery patients with right heart dysfunction and/or failure. 5,6 When administered via inhalation, epoprostenol was shown to only impact lung hemodynamics without systemic hypotension.
To fill this need for an approved and well-characterized drug, VentaProst was developed as an integrated drug/device combination product consisting of a commercially available epoprostenol solution copackaged with a precision, single patient-use custom nebulizer compatible with ventilatory support equipment used in ICUs. Epoprostenol, the drug constituent part in VentaProst, has been approved in the United States for chronic intravenous administration in patients with pulmonary arterial hypertension under the proprietary names FLOLAN and Veletri. Food and Drug Administration (FDA) has also approved a generic epoprostenol. As such, VentaProst is amenable to receiving approval under the 505(b)(2) regulatory pathway. Relying on previous determinations of systemic safety from the abovementioned products, the primary goal of the toxicology program, to allow initiation of clinical trials of VentaProst, was to demonstrate acceptable safety/tolerance of VentaProst to the respiratory system. However, this resulted in several challenges leading to a unique nonclinical toxicology package. First, epoprostenol has a short half-life (3-6 minutes), potentially requiring the drug to be administered continuously for up to 48 hours to control PH, before, during, and after MV surgery. Additionally, as the intention was to use the intravenous formulations already approved, there was concern that these formulations marketed at pH 10.5 and 12 would be toxic to the lung when inhaled.
Based on ICH-M3(R2) guidance, 14-day inhalation toxicity studies in a rodent and nonrodent species would generally be recommended at doses up to 50-fold the clinical dose. Safety pharmacology (cardiovascular and respiratory) should also be demonstrated by the new route of administration. Rodent species can generally only tolerate nose-only inhalation for up to 4 to 6 h/d, while most nonrodent species can only be oronasally exposed for up to 2 h/d. 7 Epoprostenol administration for up to 14 days in unanesthetized animals didn’t seem to mimic the patient experience of up to 48 hours continuous inhalation exposure in anesthetized, intubated, and ventilated patients. Thus, these differences in physiological state, which affect both hemodynamic measurements and aerosol particle deposition, were not well modeled by the standard inhalation toxicology testing paradigm.
Based on these important distinctions from the traditional toxicology testing strategy, the following studies were proposed and accepted by regulatory authorities. First, a standard 14-day rodent inhalation toxicology study at doses up to 50-fold over the clinical dose was proposed to allow the evaluation of high-dose inhalation exposure to both the nasal passages and the lung. This evaluation was important as patients may receive aerosolized VentaProst both pre- and postoperatively via a nasal cannula, as well as directly to the lung through a ventilator during and immediately after surgery. More unique was the proposal to evaluate anesthetized, intubated, and ventilated dogs during a 48-hour continuous inhalation exposure to VentaProst. Eventually, 3 studies were performed with this method, a feasibility study (not discussed) and 2 Good Laboratory Practice (GLP) studies using different formulations. Additionally, GLP compliant safety pharmacology studies of respiratory and cardiovascular function were included by integrating such measurements into the dog inhalation toxicology studies.
Methods
Common Methods for Rat and Dog Studies
Vials of FLOLAN were purchased which were composed of 1.5 mg of lyophilized epoprostenol sodium, 3.76 mg glycine, 50 mg mannitol, and 2.93 mg sodium chloride. The vials were reconstituted with sterile diluent for FLOLAN (buffer/diluent), which consisted of 94 mg glycine, 73.3 mg sodium chloride, and water for injection and sodium hydroxide for pH adjustment (pH of the solution is 10.2-10.8). This pH 10.5 FLOLAN sterile buffer/diluent served as the control article for the rat and first ventilated dog study. For the second dog study, the same lyophilized FLOLAN powder was used as the active treatment but instead it was reconstituted with pH 12 sterile diluent for FLOLAN (pH 12 diluent) with the pH12 diluent used as the control article. The composition of the latter is identical to the former, save for a slight increase in the sodium hydroxide used for pH adjustment.
The pulmonary deposited dose rate was estimated using a modification of the following formula presented in Alexander et al
8
:
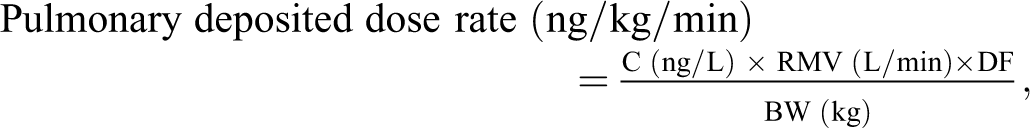
where
C = Concentration (ng/L) measured at: a) the breathing zone for rat b) from the air exiting the endotracheal tube (emitted dose) for dog.
RMV = Respiratory minute volume (L/min) a) allometric scaled, using RMV (L/min) = 0.608 × BW (kg)0.852* b) individual animal’s measured minute volume for dog
DF = Deposition Fraction, a) 10% of the emitted/delivered dose for rat b) 100% for pulmonary deposited dose rate in the dog
BW = Bodyweight (kg) a) pulmonary deposited dose rate calculated using each rat’s recorded body weight b) pulmonary deposited dose rate calculated using each dog’s recorded body weight
* equation parameters from Alexander et al. 8
Aerosol concentration measurements were made using open-faced filters at the breathing zone of the animal for rats, or at the end of the endotracheal tube for dogs. Similarly, filters from disassembled cascade impactors were analyzed to determine particle size distribution. The filters for concentration and particle size distribution were added to a conical tube containing 5 mL of diluent for epoprostenol sodium-borate buffer, pH 11.5: acetonitrile (4:1) and stored at 2 °C to 10 °C to stabilize the epoprostenol prior to chemical analysis.
No available chemical assay is sufficiently sensitive and specific to assess the in-vivo pharmacokinetics of epoprostenol. Epoprostenol is rapidly hydrolyzed at neutral pH in blood and is also subject to enzymatic degradation. Epoprostenol is metabolized to 2 primary metabolites: 6-keto prostaglandin F1α (PGF1α, formed by spontaneous degradation) and 6,15-diketo-13,14-dihydro-PGF1α (enzymatically formed), both of which have pharmacological activity orders of magnitude less than epoprostenol in animal test systems. Prostaglandin F1α was determined in plasma using a HPLC MS/MS validated method to demonstrate exposure.
All studies reported below were performed using GLP. All studies complied with all applicable sections of the Animal Welfare Act and were approved by the Institutional Animal Care and Use committees prior to start of study.
Rat Nose-Only Inhalation Study
A study was performed to determine the potential toxicity of inhaled epoprostenol when administered by nose-only inhalation exposure for 4 hours a day for 14 consecutive days, as well as to determine the reversibility of any potential toxic effects observed after a 14-day treatment-free period.
Male and female (65 per sex) Sprague Dawley derived rats [Crl: CD (CD) BR] were provided with Certified Global 18% Protein Rodent Diet. Water was supplied by means of an in-cage automatic watering system. During quarantine and nonexposure periods of the study, the animals were housed in polycarbonate “shoe-box” cages (10½″ × 19″ × 8″) lined with absorbent hardwood chip bedding. The animals were housed in pairs with animals of the same sex. The study design is displayed in Table 1.
Rat Study Design.
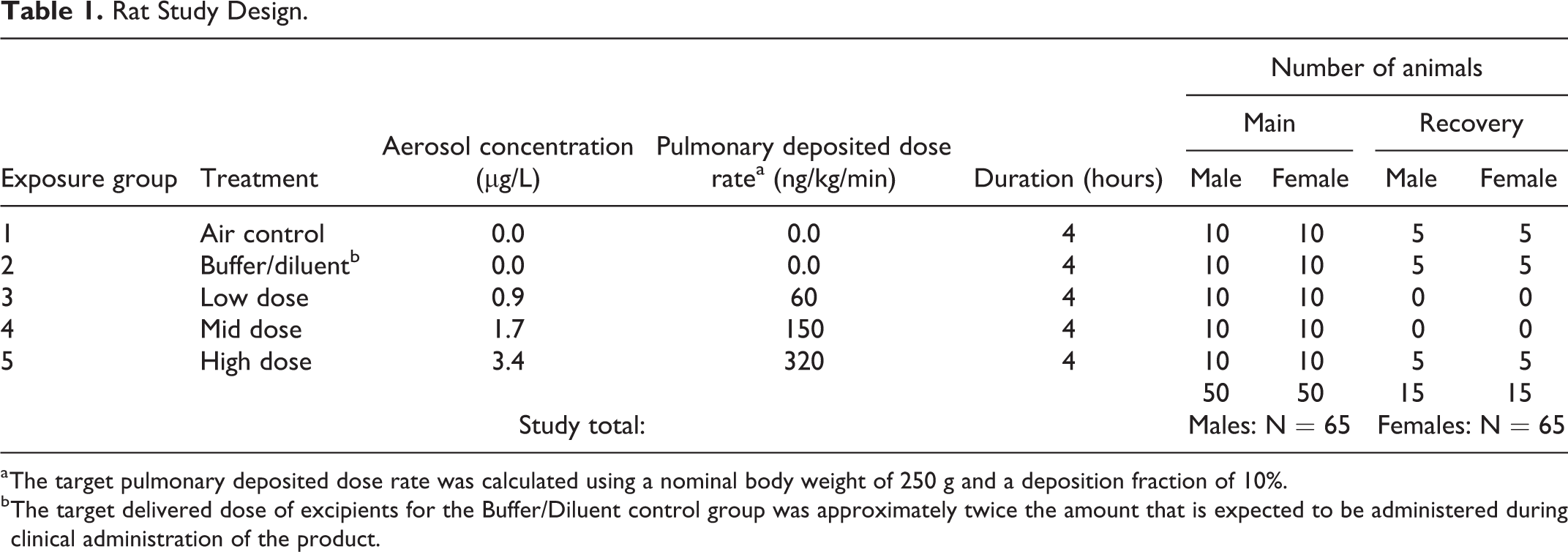
a The target pulmonary deposited dose rate was calculated using a nominal body weight of 250 g and a deposition fraction of 10%.
b The target delivered dose of excipients for the Buffer/Diluent control group was approximately twice the amount that is expected to be administered during clinical administration of the product.
During inhalation exposures, the animals were restrained in nose-only holding tubes in a rodent inhalation exposure system (CH Technologies). To condition the animals to placement and restraint in the exposure tubes, and to reduce stress during the aerosol exposure phase, each rat was acclimated prior to testing. Epoprostenol was aerosolized using Aerogen Solo nebulizers (Aerogen). Drug was fed to the nebulizer reservoir by a syringe pump, while filtered dry air, at 12 LPM, carried the nebulized test atmosphere to the exposure chamber. The infusion rate for the buffer/diluent group (group 2) was set at the nominal level of 0.12 mL/min to target twice the expected clinical dose rate. The infusion rates for the low, mid, and high groups were 0.18, 0.18, and 0.24 mL/min, at 120, 240, and 360 µg/mL epoprostenol, respectively. Aerosol concentration was measured at the breathing zone of the animal from open-faced particle filters. Particle size distribution was measured using a Mercer Impactor (In-Tox Products) and sampling filters were treated, stored, and chemically analyzed as described above.
Toxicological experimental end points consisted of morbidity/mortality and physical examinations/clinical observations, body weight, food consumption, bioanalysis, ophthalmic examinations, clinical pathology parameters (hematology, blood chemistry, coagulation, and urinalysis), organ weight, gross necropsy observations, and histopathology. Blood was obtained from the retro-orbital plexus of anesthetized animals on days 1 and 14 immediately postdose, using approximately half of the animals per sex per group at each time point for measurement of the biomarker PGF1α.
Anesthetized, Ventilated Dog Inhalation Toxicology at pH 10.5
Epoprostenol or epoprostenol diluent was administered once as a 48-hour continuous inhalation exposure. The solution was nebulized by an Aerogen Solo mesh nebulizer into the inhalation line of a ventilator circuit connected to the animal via an endotracheal tube. The study design is shown in Table 2.
Dog pH 10.5 Study Design.

a Group 1 animals received the diluent pH 10.5, administered with a volume equivalent to 1× the aerosol concentration applied clinically.
b Group 2 animals received the diluent pH 10.5, administered with a volume equivalent to 12× the aerosol concentration applied clinically.
For each animal, at the completion of dosing, one sample of aerosolized dosing formulation was collected on a filter to ascertain emitted dose (ED). The filter was attached to the end of the endotracheal tube which was connected to a ventilator operating at the same settings used to ventilate the dog. The duration of sampling was set to 20 minutes. A second set of samples of aerosolized dosing formulation were collected into a Mercer/Intox cascade impactor at completion of dosing for each animal from groups 3 and 4 for determination of particle size. The duration of sampling was set to 40 minutes, and the sample collection flow rate was 2 ± 0.05 L/min.
The inhalation exposure system, surgical preparation of the dogs, and measured end points have been described in detail. 9 Briefly, epoprostenol and pH 10.5 diluent were administered continuously, using a mesh nebulizer (Aerogen Solo, Aerogen Ltd. IRE), over 48 hours by inhalation. The aerosol passed through the ventilator (LTV Series 900 Ventilator, Carefusion) with an attached humidification chamber into the endotracheal tube. Dosing formulations were fed to the nebulizers at room temperature but replaced every 8 hours.
Eighteen Beagle dogs (9 males, 9 females), received from Marshall Bioresources were 11 months old at the start of the study. The body weights ranged from 7.2 to 9.1 kg. Animals were anesthetized throughout the dosing procedure with propofol and closely monitored. Morphine (0.2 mg/kg) was administered subcutaneously approximately every 4 hours following start of anesthesia. Animals were ventilated using pressure-controlled ventilation with synchronized intermittent mandatory ventilation (spontaneous breaths allowed between machine breaths). Ventilator airflow was adjusted during dosing, as necessary, to maintain appropriate ventilation. Any change in ventilation was also accounted for in the final determination of dose for each animal.
After animals were anesthetized and intubated, multiple venous and arterial cannula were inserted for analyte samplings, anesthesia administration, and physiological measurements (blood pressure and heart rate [HR]). An external telemetry transmitter (Data Science International, Model: D70-PCTP) with biopotential leads was also inserted in a Lead II configuration. To maintain the animals for over 48 hours under anesthesia, comfort care was provided that included ophthalmic ointment applied to both eyes, swabbing the mouth and the gums, and changing the position of the animal on a regularly scheduled basis. The animal was also placed on a heating pad set to maintain the animal’s body temperature at approximately 37 °C throughout the study. Enrofloxacin (10 mg/kg) was administered intramuscularly (IM) approximately every 24 hours following start of dosing. Cephazolin (0.90 mL of 334 mg, IM) was administered 3 times (approximately every 8 hours) in the last 24 hours of dosing. Partial parenteral nutrition started 4 hours after the start of dosing. A urinary catheter was inserted to empty the bladder, as necessary. Additional details of these procedures can be found in Bujold et al. 9
Clinical and vital signs were performed every hour. Clinical signs included reports of discharge, swelling, distension of the abdomen, tremors, and respiratory status. Vital signs included continuous measurements of SpO2 and HR by oximetry, mean blood pressure, body temperature, and regular assessments of response to stimuli. Pulmonary function was routinely assessed, recording tidal volume, respiratory rate, minute ventilation, peak inspiratory pressure, positive end-expiratory pressure, and inspiratory:expiratory ratio. These parameters were monitored continuously and recorded manually prior to dosing and every hour (±15 minutes) following the start of dosing. The electrocardiogram (ECG) was analyzed pretreatment and continuously monitored during treatment to assess potential wave configuration and rhythm abnormalities. Arterial blood pressure was recorded continuously for a period of at least 30 minutes before dosing and continuously throughout the 48-hour dosing period. The ECG and arterial blood pressure data were evaluated by a board-certified veterinary cardiologist.
Clinical pathology parameters (hematology, blood chemistry, coagulation) were measured before dosing and at 24 and 46 hours during dosing. Blood gas analysis was performed approximately every 3 hours while blood glucose was monitored hourly. Blood samples were collected for measurement of PGF1α from all animals at predose, 12, 24, 36, and 48 hours post start of dosing. Organ weight, gross necropsy observations, and histopathology were performed after the end of dosing.
Anesthetized, Ventilated Dog Inhalation Toxicology at pH 12
For the most part, the study was conducted similarly to the dog pH 10.5 study discussed above. Differences are discussed below. Epoprostenol (purchased as FLOLAN), FLOLAN-pH 12 diluent, and phosphate-buffered saline (PBS) control were administered once as a 48-hour continuous inhalation exposure. The study design is provided in Table 3.
Dog pH 12 Study Design.
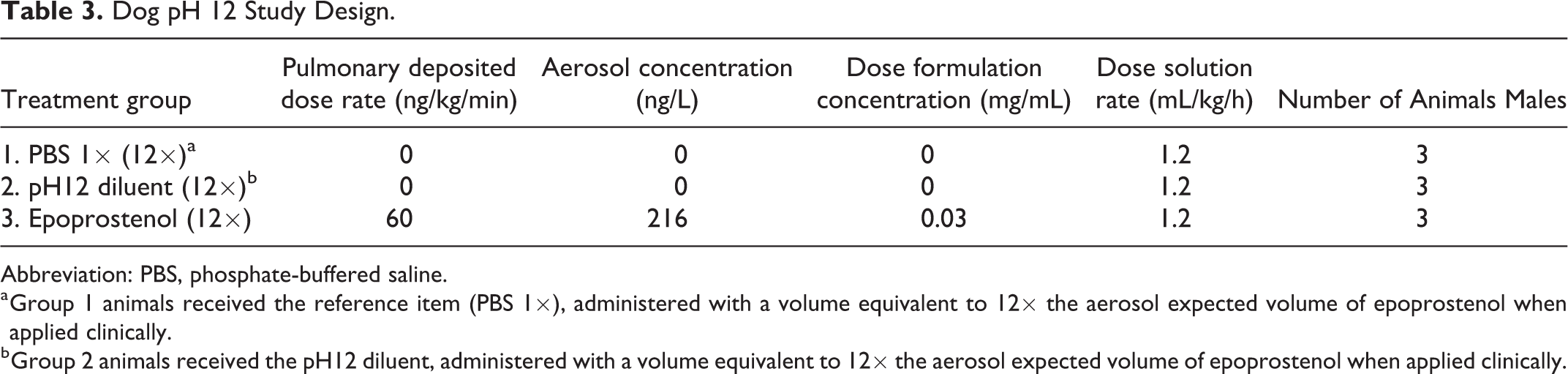
Abbreviation: PBS, phosphate-buffered saline.
a Group 1 animals received the reference item (PBS 1×), administered with a volume equivalent to 12× the aerosol expected volume of epoprostenol when applied clinically.
b Group 2 animals received the pH12 diluent, administered with a volume equivalent to 12× the aerosol expected volume of epoprostenol when applied clinically.
As no sex differences were noted in the previous study, only male Beagle dogs, received from Marshall Bioresources, were studied. Similarly, as there was no effect of the pH 10.5 diluent at 1× or 12× the clinical dose rate, PBS was used as a control for pH 12 diluent (12×). The methods used for dose formulation analysis, measurement of pulmonary deposited dose rate, and particle size distribution were similar to the pH 10.5 study. Surgical preparation, catheter and ECG placement, comfort care, thermoregulation, antibacterial treatment, and fluids and nutrition were the same. End points (clinical and vital signs, body weight, body temperature, pulmonary function, blood gas assessment, oximetry, ECG, and cardiovascular telemetry monitoring) were also the same. Clinical pathology, blood glucose, and bioanalysis time points were also the same as was the necropsy procedure, organ weight determination, and histopathology.
Results
Rat Nose-Only Inhalation Study
Formulation concentrations of epoprostenol were within the specified criteria of ±15% and stability over the 4-hour exposure period at room temperature was demonstrated. Mean measured epoprostenol aerosol concentrations were 0, 0, 0.772, 1.989, and 4.337 µg/L for the air control, pH 10.5 diluent, low, mid, and high epoprostenol dose groups, respectively. Overall, lung deposited dose rates of 0, 0, 60, 150, and 320 ng/kg/min were delivered after applying a 10% deposition factor to the delivered dose. 7 Mean mass median aerodynamic diameter (MMAD) values were 1.03, 1.27, and 1.38 µm for groups 3 to 5, respectively; corresponding geometric standard deviation (GSD) ranges were 1.66 to 1.85, 1.76 to 1.90, and 1.77 to 1.80, respectively. The mean MMAD value for the pH10.5 diluent was 1.34 µm; the GSD ranged from 1.28 to 2.70. The particle size data indicate that the aerosol was respirable in the rat. The metabolite 6-keto PGF1α, which is produced from endogenous PGI2, was detected at low levels (average <60 pg/mL) immediately post exposure in the blood of control animals as expected, while a dose-related increase was observed in treated animals (Figure 1).
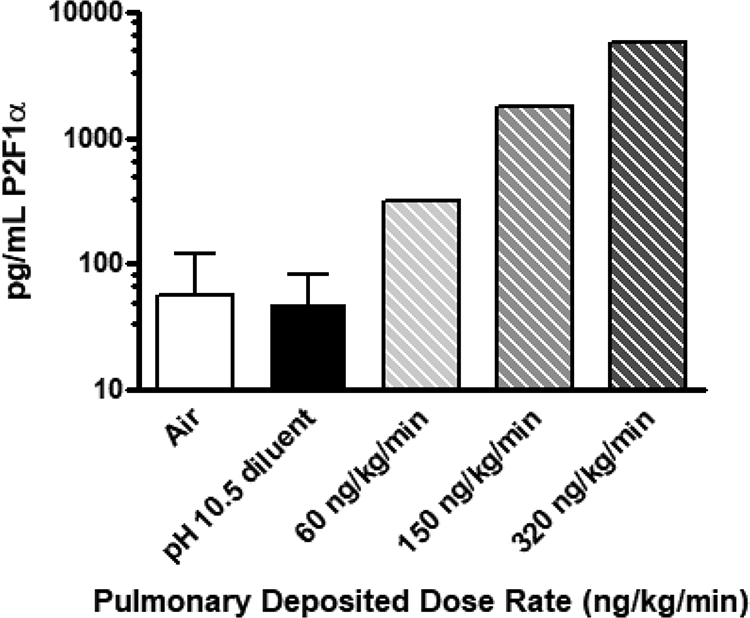
Dose–response for 6-keto prostaglandin F1α (PGF1α) after inhalation of epoprostenol. Note data are combined male and female rats on both days 1 and 14 as there were no statistical differences between day or sex (2-way analysis of variance). Error bars for the treatment groups are not visible on a log scale for the epoprostenol-treated groups.
No epoprostenol exposure–related mortality or clinical signs of toxicity were observed during the study. The most common observations noted after daily exposure were redness around the nose and eyes and wet inguinal fur; these were considered to be related to the restraint in the nose-only exposure tubes and were not present in subsequent pre-exposure observations. There were no exposure-related body weight changes, while sporadic reductions in body weight gains were noted in both males and females. However, based on the magnitude of change, the lack of dose–response, and that both male and female recovery animals gained as much as, or more than, the control animals during the recovery period, the changes were not considered adverse.
No changes in food consumption, ophthalmic findings, or clinical pathology effects were noted related to treatment. Changes were not observed in absolute organ weights at terminal and recovery necropsy for males and females. Organ-to-body weight ratios showed no change except increased lung weight to brain weight ratio in the mid-dose group; no other group had any significant difference. No epoprostenol-related gross findings were noted at terminal or recovery necropsies and microscopic examination of tissues from the high-dose group showed no epoprostenol-related findings.
Overall, based on lack of epoprostenol-related histopathology findings and any discernable differences in in-life observations (body weight, food consumption, clinical observations, and so on), the no-observed-adverse-effect level (NOAEL) in this study of inhaled pH 10.5 epoprostenol was considered to be greater than the pulmonary deposited dose rate of 320 ng/kg/min.
Anesthetized, Ventilated Dog Inhalation Toxicology at pH 10.5
Formulation samples containing epoprostenol (groups 3 and 4) were 30 µg/mL ± 10%, except for 1 sample. Evidence of epoprostenol in diluent samples was not found, with the exception of one group 2 samples showing a trace amount of epoprostenol. Dose formulation analysis was considered acceptable for the purpose of the study. Achieved pulmonary deposited dose rates for groups 3 and 4 were 31 and 67 ng/kg/min. As the dose was delivered through an endotracheal tube into the dog’s trachea, all of the aerosol was considered to be deposited in the lung. Particle size distribution was measured to demonstrate plausible distribution to all parts of the lung. However, problems were encountered using the novel aerosol particle sizing method developed for the study. Despite the issues encountered, where deposition across stages could be achieved sufficient to calculate a mass median diameter (3 of the 6 tests performed), the data indicated that the particle size was small enough (<2 µm) to be distributed to the alveoli. Further evidence that the dose reached the alveoli was indicated by a dose-related increase in PGF1α, peaking at 12 hours in group 3, while group 4 peaked at 24 hours. After peaking, concentration levels were stable for the remainder of the 48-hour exposure. No sex differences were noted.
Several clinical signs were observed throughout the observation period following the start of dosing including mucoid material discharge from the mouth and black/green/loose/liquid feces. Diffuse somatic swelling was observed which appeared to be related to the various procedures (eg, fluid administration). Occasional episodes of tremors, pedaling, and/or excessive repetitive movement were also noted for most animals; however, these clinical signs were considered related to the anesthetic wearing off. The clinical signs were observed in all treatment groups and thus did not appear to be specifically related to aerosolized epoprostenol or pH 10.5 diluent administration.
Body weight measurements were performed for dose–volume calculation purposes and to determine relative organ weights. As expected, no significant changes in body weight were noted over the 48-hour treatment period. When groups 2 to 4 were compared to epoprostenol diluent 1×-treated animals, there were no epoprostenol-related effects for the following parameters: body temperature, tidal volume, minute volume, peak inspiratory pressure, positive end-expiratory pressure, blood glucose, pH, PaCO2, PaO2, %SO2 and HCO3.
There were no differences in ECG (except HR) or arterial blood pressure parameters between groups, only normal variations in these parameters were observed. There was a trend toward a decrease in mean HR in the epoprostenol-treated groups for pooled sexes compared to pH 10.5 diluent 1×; however, this change was neither statistically nor hemodynamically significant (Figure 2).
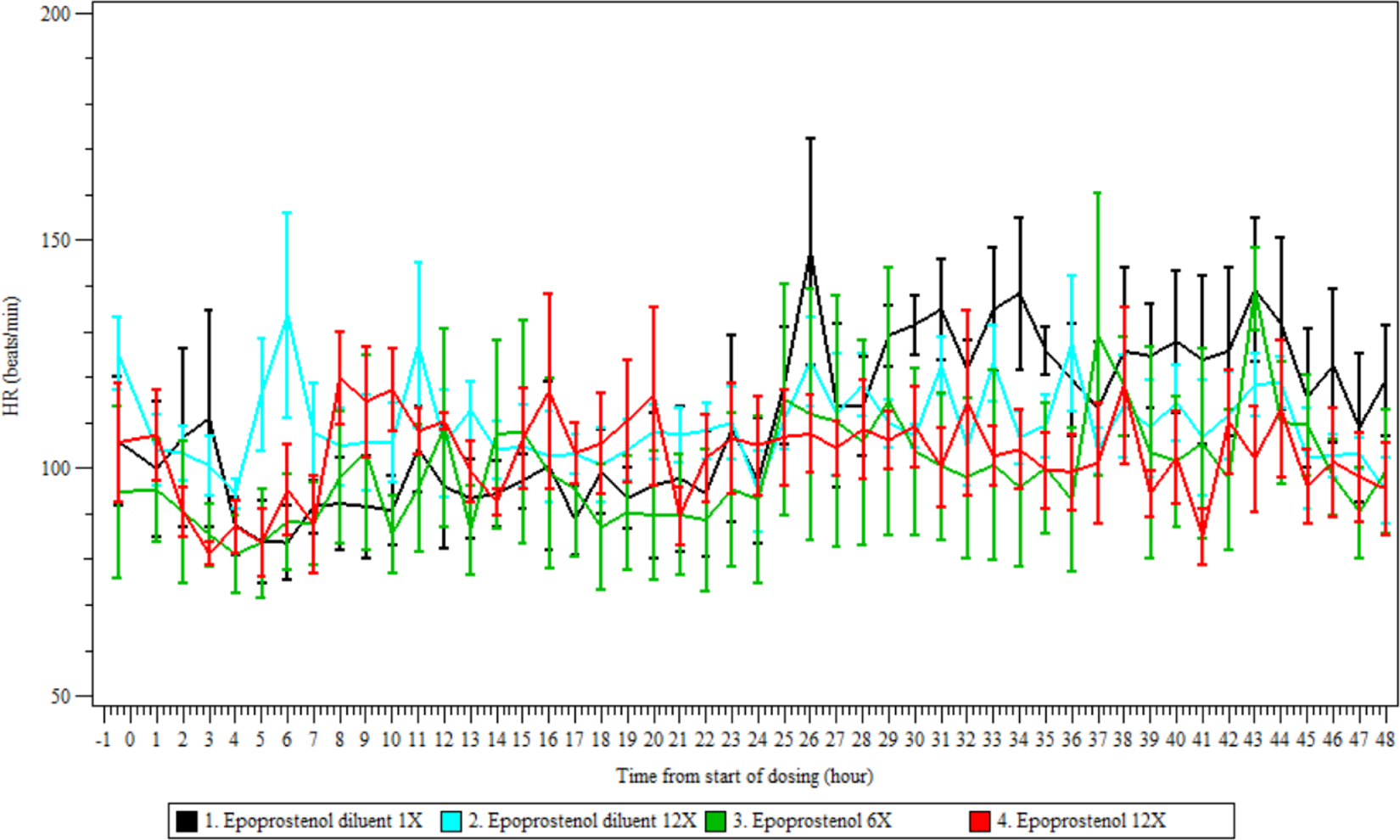
Heart rate in anesthetized ventilated dogs continuously exposed for 48 hours. Average heart rate (mean ± standard deviation) for 4 dogs per group (sexes combined) before and during a 48-hour inhalation exposure to pH 10.5 diluent or epoprostenol.
Changes in clinical pathology parameters (hematology, coagulation, clinical chemistry) were observed in all animals but were not considered epoprostenol-related, as the changes were comparable in pH 10.5 diluent and epoprostenol-treated groups. The changes were attributed to the experimental procedure (ie, surgery, 48-hour dosing period under continuous injectable anesthesia with partial parenteral nutrition).
No epoprostenol-related effects were identified from organ weight, gross pathology, or histopathology evaluations. Notable microscopic findings were observed without any treatment relationship in several tissues across all groups. Repeated findings in the larynx, pharynx, trachea, kidneys, gastrointestinal tract, and eyes were observed; however, the findings were believed to be related to the experimental procedures and not to drug administration as similar findings were observed in control animals.
Overall, administration of epoprostenol once as a 48-hour continuous inhalation in anesthetized Beagle dogs was well tolerated. All changes observed were considered procedure-related and no treatment-related effects were noted. As such, the NOAEL was considered to be equal to or greater than the highest dose pulmonary deposited dose rate of 67 ng/kg/min.
Anesthetized, Ventilated Dog Inhalation Toxicology at pH 12
Dose formulation solution samples demonstrated the absence of epoprostenol in PBS and FLOLAN diluent pH 12 samples (groups 1 and 2). Dose formulation samples from group 3 were within ±10% of the nominal concentration range and therefore, considered acceptable. The mean ED captured on a filter from the end of the endotracheal tube during a 20-minute sampling period for group 3 animals was 11.7 µg. On 1 of the 6 ED samples analyzed, there was no epoprostenol detected. Examination of the raw chromatogram revealed that all the deposited epoprostenol had been converted to its primary degradant, 6-keto PGF1α. Similarly, epoprostenol degradation was encountered for most of the aerosol particle size distribution measurements carried out over a 40-minute period. For the 5 particle size distribution samples judged as representative, the aerosol particle size ranged from 2.3 to 2.6 µm, indicating a respirable aerosol that should distribute across all lung regions. This was confirmed by an increase in plasma PGF1α only in the epoprostenol pH 12–treated group (group 3), which peaked at 24 hours. After peaking, concentration levels slightly decreased for the remainder of the 48-hour exposure. The disappearance of PGF1α postexposure from plasma was not followed.
Several clinical signs were observed throughout the observation period following the start of dosing including mucoid discharge from the mouth and green/loose/liquid feces. Diffuse, soft swelling in the limbs and around various parts of the body was observed that appeared to be related to the various procedures such as parenteral administration of fluids. Occasional episodes of tremors, pedaling, and/or twitches were also noted for some animals. However, these clinical signs were considered related to the experimental procedures (eg, anesthesia) as they were observed in all treatment groups and thus did not appear to be specifically related to epoprostenol administration.
Body weight measurements were performed for dose volume calculation, to compute dose rate and to determine relative organ weights. No effects on body weight were observed. Compared to PBS-treated animals, there were no pH 12-diluent or epoprostenol-related differences for the following parameters: body temperature, tidal volume, minute volume, peak inspiratory pressure, peak positive end-expiratory pressure, blood glucose, pH, PaCO2, PaO2, %SO2, and HCO3.
There were no differences in ECG or arterial blood pressure parameters between groups, only normal variations in these parameters were observed. There was a decrease in systolic, diastolic, mean arterial blood pressure following the start of dosing in all animals from all dose groups. This appeared to be related to the experimental procedure, as the effects in epoprostenol-treated animals were no different than those observed in the other 2 control groups.
Changes in clinical pathology parameters (hematology, coagulation, clinical chemistry) were observed in all animals but were not considered epoprostenol-related as the changes were comparable in PBS, pH 12 diluent and epoprostenol-treated groups. For example, triglyceride and cholesterol individual levels were increased by 3.2- to 25.4-fold and 1.8- to 3.1-fold, respectively, in all animals compared to pretreatment values. Urea, creatinine, albumin, and total protein levels decreased in all animals at completion of the 48-hour dosing period. The changes were all attributed to the experimental procedure (ie, surgery, 48-hour dosing period under continuous anesthesia with partial parenteral nutrition). No epoprostenol-related effects on organ weight or gross pathology were identified.
Within the larynx, pharynx, and/or trachea, minimal to severe erosion/ulceration, minimal to marked hemorrhage and/or minimal to moderate inflammation of the mucosa/submucosa, muscularis and/or the adventitia were noted in various animals with no relationship to the treatment group, similar to what was observed in the previous pH 10.5 dog inhalation toxicology study. Similarly, in the lungs and bronchi, minimal to moderate bronchioloalveolar and/or interstitial inflammation and minimal to mild hemorrhage were noted with no relationship to the treatment group. These changes in the larynx, pharynx, trachea, and lungs were considered due to experimental procedures and are expected with the presence of a cuffed endotracheal tube and anesthesia for the 48-hour administration period. Data demonstrating no significant treatment effect of pH 12 diluent or epoprostenol on lung histopathology compared to the PBS control group are shown in Table 4.
Lung Histopathology Incidence in Anesthetized and Ventilated Dogs Exposed for 48 Hours to Epoprostenol Aerosol.
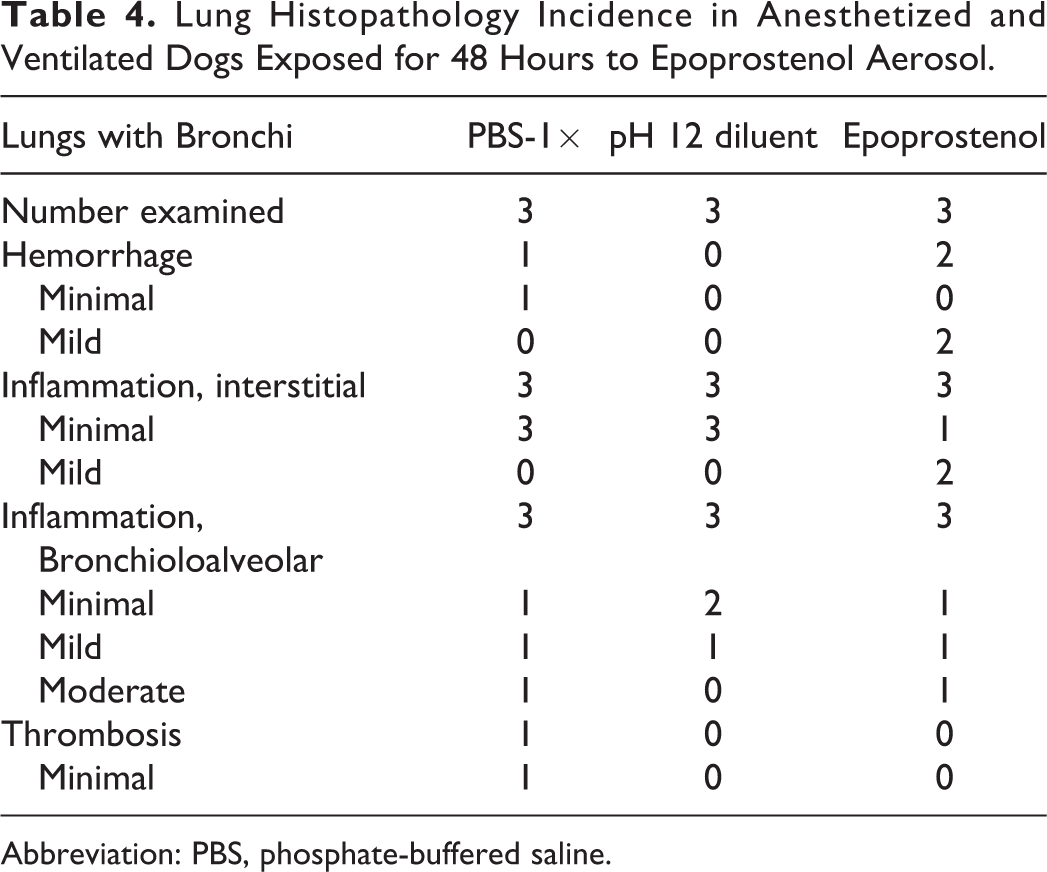
Abbreviation: PBS, phosphate-buffered saline.
Overall, administration of epoprostenol, once as a 48-hour continuous inhalation in anesthetized Beagle dogs reconstituted with pH 12 diluent, was well tolerated. All changes observed were considered procedure-related and no epoprostenol-related effects were noted. As such, the NOAEL was considered to be a pulmonary deposited dose rate of 50.4 ng/kg/min.
Summary/Conclusion
As an intravenous vasodilator, epoprostenol is approved by the FDA for the treatment of pulmonary arterial hypertension (World Health Organization group 1) to improve exercise capacity. However, the systemic side effects and lack of pulmonary selectivity of intravenous prostacyclins (eg, epoprostenol and iloprost) have significantly limited their use in the ICU 10 and has led to their off-label use in inhaled forms. 11 Inhalation delivery of epoprostenol offers an attractive alternative to intravenous delivery because it minimizes systemic exposure associated with clinically hazardous effects such as hypotension and exacerbation of pulmonary and cardiac shunts. The rationale for administering epoprostenol continuously as an aerosol through ventilatory support equipment stems from its extremely short half-life of approximately 2 to 6 minutes in vivo. The inhalation route enables delivery of high local concentrations of vasodilator into the lung vasculature, reducing PVR, and right heart afterload. As a result, the use and effectiveness of inhaled epoprostenol is widely reported in a variety of surgical applications 12,13 to mitigate the effects of PH. Nonetheless, such practices remain off-label, as inhaled epoprostenol is not FDA-approved. However, there is extensive published peer-reviewed literature discussing the need for perioperative administration of inhaled vasodilators in cardiothoracic surgery patients with PH that could lead to right heart failure. 14,1 5
Thus, facing the lack of a well-characterized and approved method for using inhaled epoprostenol, clinical centers have developed protocols to deliver the intravenous formulation through ventilators using commercially available nebulizers. Despite the existence of site-specific treatment protocols, the nominal dose rate (often 50 ng/kg/min, ranging 10-100 ng/kg/min) is administered without regard for lung delivery efficiencies. This results in little to no consistency in the dose that is delivered to the lungs. Thus, there is a critical need to develop standardized equipment and procedures so that the dose to the lung is consistent, controlled so that dose–response relationships can be established.
The literature typically describes administration of ∼50 ng/kg/min of epoprostenol via aerosol 12 with no differentiation between the ED (dose exiting the nebulizer) and the dose delivered and deposited into the lung after aerosol loss in the ventilator circuit and endotracheal tube. Typically, administration of epoprostenol aerosol by ventilator is highly variable and inefficient as a result of many factors including the type of nebulizer, continuous nebulization of aerosol during exhalation, the location of the nebulizer in the ventilatory circuit, the amount of humidification in the circuit, and the particular ventilator parameters being used. In adults, MacIntyre et al 15 reported 3% of the nominal dose (dose added to the nebulizer) deposits in the lungs with a jet nebulizer during mechanical ventilation. This estimate is in agreement with the later findings of Fuller et al 16 also using jet nebulizers. In vitro models using either a pressurized metered dose inhaler with spacer or the Aerogen Solo Vibrating Mesh Nebulizer suggest ∼17% lung deposition is possible; however, Fink et al 17 demonstrated that in vitro models, tend to overestimate delivered doses as patients exhaled about 5% more aerosol than the filter model would suggest. Consequently, the 17% deposition in vitro, is likely closer to 12% in vivo. The literature data indicate that current off-label use results in a range of 2% to 12% of the nominal dose rate delivered into the ventilator circuit actually reaches and deposits in the lung, that is, approximately 5 ng/kg/min from a nominal dose rate of 50 ng/kg/min.
Typically, inhalation toxicology studies report the delivered dose to the animal in units of mg/kg/d. Delivered dose is a measurement of the concentration of aerosol at the breathing zone of the animal corrected for respiration rate, the duration of inhalation exposure, and the animal’s body weight. 8 The delivered dose does not reflect the amount of drug deposited in the lungs of the animal. Unfortunately, there is no simple method for determining the pulmonary deposited dose, which is dependent on a host of variables of which some of the more important ones are the breathing pattern and lung anatomy of the animal, and the particle size, among others. By convention, based on particle deposition data of different particle sizes in different species, 18 a deposition factor is applied to the delivered dose of 10% in rodents and 25% in nonrodents, to estimate the pulmonary deposited dose. 7 In the case of epoprostenol, as the half-life is so short and because surgeons are used to delivering it intravenously at a known rate, we have expressed the pulmonary deposited dose as a rate per minute for easy comparison to the clinical literature.
In our inhalation toxicology studies in the rat, a pulmonary deposited dose rate of 320 ng/kg/min was achieved. This dose rate, for 4 hours a day for 14 days, was without adverse findings (NOAEL), suggesting an approximately 64-fold margin over the expected clinical pulmonary deposited dose rate of 5 ng/kg/min. In the first dog study at pH 10.5, the pulmonary deposited dose rate of epoprostenol was 67 ng/kg/min at the NOAEL. This dose rate would provide about a 13-fold safety margin for up to 48 hours continuous exposure relative to an estimated efficacious clinical dose. In the second dog study, which examined epoprostenol at pH 12 for a 48 hours continuous exposure, the NOAEL was 50.4 ng/kg/min, which is approximately 10-fold the estimated dose rate currently administered during off-label clinical inhalation of epoprostenol. However, several caveats must be considered in the interpretation of these data and the assessment of risk.
There was concern that the pH of the diluent alone, at a sufficient exposure volume, would result in lung pathology. The initial studies in rats and dogs used epoprostenol with pH 10.5 diluent. The pH 10.5 diluent was used for the control animal exposure. The results of these studies were somewhat surprising. The rat study was completed first and showed no effect of the pH 10.5 diluent compared to air control exposure. For the first dog study, 2 levels of diluent exposure were evaluated (1× and 12× the expected volume to be administered clinically). As the inhaled volume expected to enter the lung was relatively small at 1× clinical volume (∼0.9 mL/h), and because the diluent was part of an approved product, it was believed that a pH effect of the diluent would be evident if it occurred at 12× the inhaled volume with 48 hours of continuous aerosol exposure. As there were no obvious effects of the diluent at pH 10.5, a bridging study was run using pH 12 diluent. The reason for trying the pH 12 diluent was because the higher pH formulation has better room temperature stability (8 vs 24 hours). Again, no obvious lung injury occurred, and no blood acid/base changes occurred after 48 hours of dosing with the pH 12 diluent or with epoprostenol mixed in the pH 12 diluent compared to PBS control. Although the lack of findings was somewhat surprising, it has been hypothesized that the buffering capacity of the lung was sufficiently large to quickly neutralize the small amounts of liquid deposited across the large surface area of the lung. 19 However, pulmonary injury was observed in both dog studies but was believed to be the result of endotracheal intubation, ventilator-induced lung injury, and prolonged anesthesia as the findings were similar to the findings observed in PBS control dogs. Therefore, it is possible that more subtle lung injury, associated with pH 10.5 or 12 diluent or epoprostenol, may have been obscured by the procedure-related lung lesions.
Currently, there is no method available to detect the appearance and rapid hydrolysis and enzymatic metabolism without resorting to radioactive labeling of the drug product. Studies have demonstrated that the inactive 6-keto-PGF1α hydrolysis product serves as a stable surrogate measure with a linear dose relationship to intravenously infused epoprostenol. 20,21 However, the blood level of 6-keto-PGF1α attributable to hydrolysis of endogenously produced prostacyclin, or exogenously administered epoprostenol, cannot be differentiated. The metabolite, 6-keto PGF1α, was detected at low levels (average <60 pg/mL) immediately post exposure in the blood of control animals as expected, while a dose-related increase was observed in animals inhaling epoprostenol.
Due to short room temperature stability, rapid hydrolysis and the very low concentration of epoprostenol in the aerosol, measurements of concentration and particle size were technically challenging and required nonstandard aerosol sampling methods for the dog studies. Because the ventilator supplied all the animal’s breathing volume, sampling from the aerosol stream was not feasible as occurred in the rat study. As the aerosol concentration was very low, gravimetric measurements of concentration and particle size would have been very time-consuming (several hours of sampling) and thus not practical. To remedy these 2 issues, an in vitro method was developed using the same aerosol exposure equipment used to dose the dogs, except that an artificial lung was attached to the end of the endotracheal tube. However, to collect aerosol on the filter, the method required the ventilator settings to be switched from pressure control to volume control mode because in pressure mode, the human artificial lung compliance was too low (ie, too stiff). Once the samples were collected on the filters (20 minutes for aerosol concentration, 40 minutes for impactor samples) they were immediately immersed in a stabilizing solution. However, even with this procedure, sufficient drug had been hydrolyzed to the metabolite such that combining the peaks for epoprostenol and the PGF1α metabolite was sometimes necessary to obtain realistic concentration and particle size data. This method was deemed acceptable as there was a clear and proportional increase in degradant with loss of main peak, although there is inherent uncertainty in the aerosol size data.
Overall, a novel toxicology testing strategy was devised to better estimate the risk and determine the safety associated with aerosolized epoprostenol. Rat inhalation data provided useful information related to potential nasal injury from the product, as aerosolized epoprostenol may be delivered via nasal prongs before and after surgery to stabilize and control PH. Both rat and dog data were useful in demonstrating the lack of local lung injury after 14 days of exposure in the rat and 48 hours of continuous exposure in dogs. Concerns surrounding aerosol administration of the diluent and diluent plus epoprostenol at high pH values (10.5 or 12) were favorably resolved as no lung injury or acid/base blood abnormalities were observed. Systemic safety of the product had previously been demonstrated and clinical use has not provided data to reveal any new cause for concern. In summary, the data suggest that inhaled epoprostenol should be safe for administration to patients undergoing MV surgery with CPB to control perioperatively PH.
Footnotes
Acknowledgments
The authors thank the technical staff at CiToxlab (now CRL) for their help in developing the model and remaining awake during many sleepless nights. Similar thanks to the technical staff at IITRI for their diligence in overseeing these studies.
Author Contributions
J.T. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, drafted the manuscript, and critically revised the manuscript for important intellectual content. J.P. substantially contributed to conception or design and contributed to acquisition, analysis, or interpretation of data. K.B. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, and drafted the manuscript. J.B.F. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, and critically revised the manuscript for important intellectual content. R.M. contributed to acquisition, analysis, or interpretation of data, and critically revised the manuscript for important intellectual content. D.S. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, and drafted the manuscript. S.A. substantially contributed to conception or design and contributed to acquisition, analysis, or interpretation of data. P.E-D. contributed to acquisition, analysis, or interpretation of data, and drafted the manuscript. A.C. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, and critically revised the manuscript for important intellectual content. All authors gave final approval and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Drs. Fink, Malcomson, Entcheva-Dimitrov and Clark disclose being employees of Aerogen Pharma Corp. The remaining authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by Aerogen Pharma Ltd.