
Abstract
Drug development is a term used to define the entire process of bringing a new drug or device to market. It is an integrated, multidisciplinary endeavor that includes drug discovery, chemistry and pharmacology, nonclinical safety testing, manufacturing, clinical trials, and regulatory submissions. This report summarizes presentations of a workshop entitled “Drug Development 101,” held at the 39th Annual Meeting of the American College of Toxicology in West Palm Beach, Florida. The workshop was designed to provide an introductory overview of drug development. Experienced scientists from industry and government provided overviews of each area, with a focus on safety assessment, and described some of the challenges that can arise. The role of chemistry and manufacturing was discussed in the context of early- and late-stage product development and approaches to assess, control, and limit impurities. The toxicologic assessment was emphasized in early-phase development, from the selection of a candidate drug through the determination of a first-in-human starting dose. Clinical trial development was discussed in the context of regulatory requirements and expectations. The final topic of issues and considerations in the review processes of different types of submissions to Food and Drug Administration included advice for best practices in authoring good Investigational New Drug and New Drug Application/Biologic License Application submissions and interacting effectively with regulatory reviewers.
Introduction (Hanan N. Ghantous)
Drug development is a complex process with an end goal of approval by the Food and Drug Administration (FDA) before the drug is marketed in the United States. (The term “drug” includes a myriad of small molecular entities and therapeutic biological products.) Approval occurs once data on the drug have been reviewed and the drug is determined to be safe and efficacious, with the benefits outweighing the known and potential risks for the intended use of the product. The process spans many years, from the discovery stage, to testing in animals, to human trials, culminating in marketing and postmarketing activities (Figure 1). The main parties involved in the process include the regulatory agencies (eg, FDA), sponsors (including pharmaceutical/biotechnology companies and academic clinical investigators), Contract Research Organizations, Contract Research, Development and Manufacturing Organizations, and clinical sites (eg, hospitals) where the principal investigators treat patients.
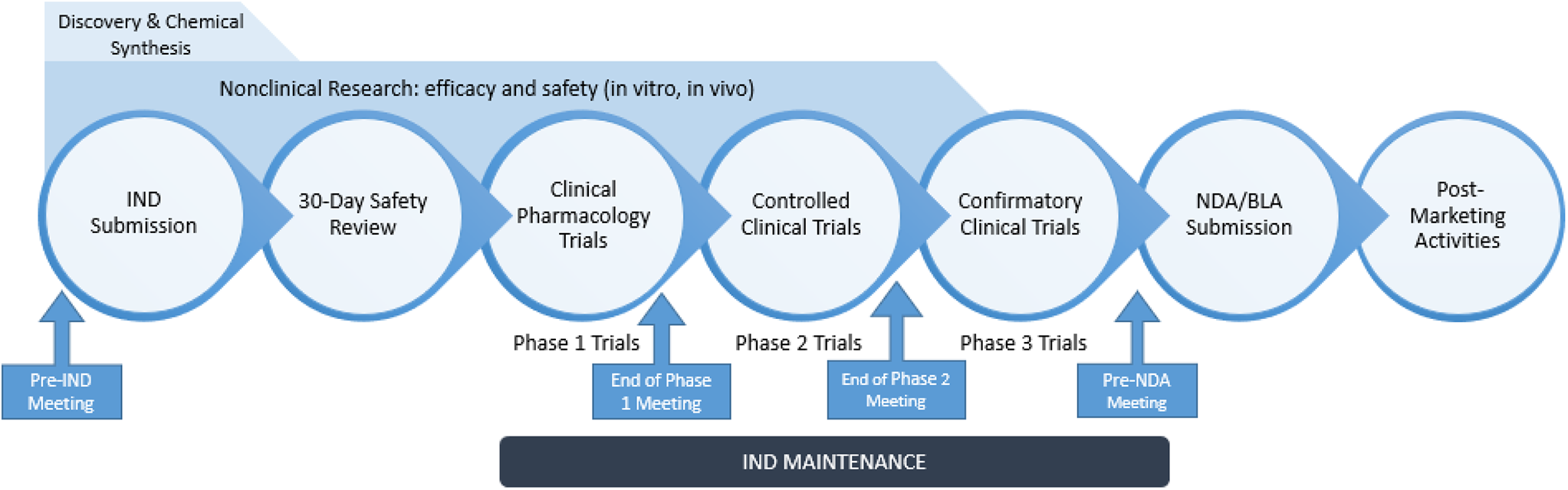
The stages of the drug development process.
The US Congress has passed dozens of laws giving regulatory authority to the FDA, only some of which are mentioned here. The Food and Drugs Act of 1906 prohibited the interstate transport of unlawful food and drugs under penalty of seizure of the questionable products and/or prosecution of the responsible parties. The basis of the law rested on the regulation of product labeling rather than premarket approval.
The 1938 Food, Drug, and Cosmetic (FD&C) Act, which replaced the 1906 law, was passed in the wake of a 1937 therapeutic disaster: Elixir Sulfanilamide, which contained an analog of antifreeze, was marketed to children for streptococcal infection and would ultimately result in the death of over 100 people including many children. The new law brought cosmetics and medical devices under control and required that drugs be labeled with adequate directions for safe use. Moreover, it mandated premarket approval of all new drugs, such that a manufacturer was required to prove to the FDA that a drug was safe before it could be sold. It irrefutably prohibited false therapeutic claims for drugs, although a separate law granted the Federal Trade Commission jurisdiction over drug advertising.
In 1962, after Congress passed the Kefauver-Harris Drug Amendments following the thalidomide tragedy, drug makers were required to prove that their products were efficacious before the FDA could approve them for sale. The FDA’s authority to regulate the drug development process through binding laws passed by Congress is described in the Code of Federal Regulations (CFR). In addition, there are regulatory guidance documents that contain nonbinding recommendations that summarize the Agency’s current thinking on a topic or issue. Guidances are published as required by law and are authored not only by the FDA and its staff but also by experts at the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, commonly referred to as the ICH. 1
The drug development process begins with discovery, including target identification and candidate molecule selection, followed by optimization of what is known as a lead compound or development candidate. Once a development candidate is selected (usually including a backup molecule), preliminary analytical tests to characterize physiochemical parameters as well as in vitro safety screens and animal studies are conducted to determine the pharmacological aspects of the compound and its potential mechanism of action. The process then moves into the development of the candidate compound and enters the regulatory process with first-in-human (FIH) studies, at which point the Sponsor submits an Investigational New Drug (IND) application to the FDA (Figure 1). At that time, the molecule changes in legal status under the Federal FD&C Act (in 1938) to become a new drug candidate, subject to specific requirements of the drug regulatory system, including the need to obtain an approved IND. The IND is necessary not only to initiate clinical studies but is also an exemption that allows shipment of the IND across state lines.
Before submission of an IND, the Sponsor may file a request for a pre-IND meeting with the Agency, which is optional. The pre-IND program allows Sponsors to receive guidance from the FDA regarding the data necessary to warrant IND submission. Once the IND is submitted, the Agency has 30 days to inform the Sponsor of any issues that may hinder the conduct of the proposed clinical study via a clinical hold (discussed later in the Regulatory section). Following initiation of the IND, the drug development process continues with phase 1, 2, and 3 clinical trials. Phase 1 clinical trials typically are small and focus on safety and pharmacokinetics (PKs), usually in healthy volunteers. Phase 2 clinical trials typically are larger, conducted in patients, and investigators continue to gather PK and safety data but also focus on data to support efficacy or proof of concept. Phase 3 clinical trials are confirmatory trials in patients and become the basis of and support for the New Drug Application (NDA) for small molecules or Biologic License Application (BLA) for large molecule/biological products. Throughout the regulatory drug development process outlined in Figure 1, FDA review staff participate in many meetings with Sponsors, who seek advice relating to the development of INDs and biologics.
The 21st Century Review describes the NDA and BLA review process in an organized and integrated manner as shown in Figure 2. 2 Similar to the IND, the NDA/BLA review is based on a defined time line. Considering the amount and complexity of the clinical and nonclinical data submitted in an NDA/BLA, the review process spans many months and typically is conducted by a larger group of regulatory scientists than that for an IND. The standard review time for a new molecular entity (NME) is 12 months from the date of application of submission, while review time for an application granted priority review is 8 months. For all other types of NDA/BLA applications, the standard review time is 10 months, while priority review is 6 months. 3
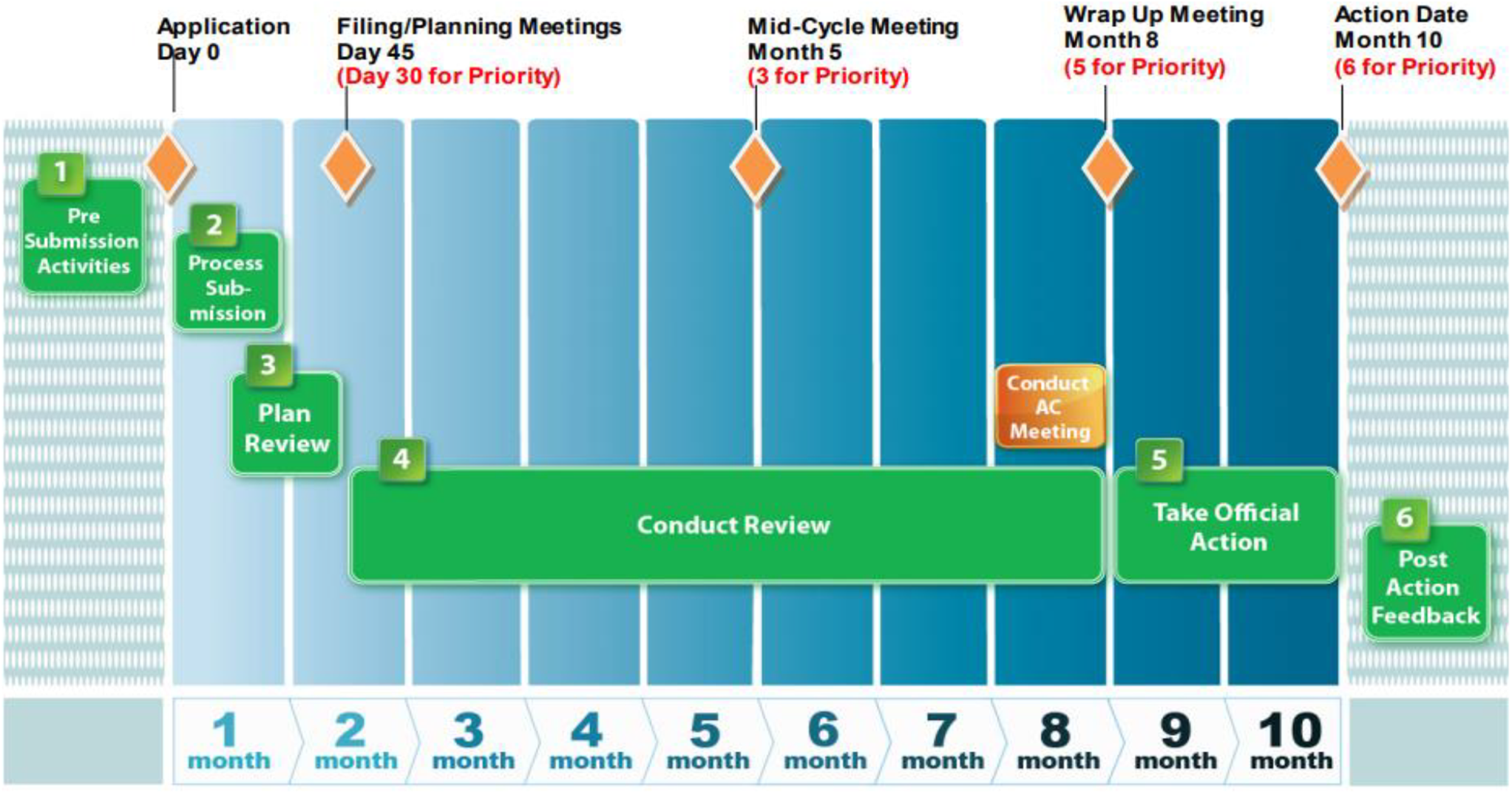
Time frame for Food and Drug Administration (FDA) evaluation of New Drug Applications (NDAs)/Biologic License Applications (BLAs).
Scientists across different disciplines in both industry and FDA are involved in reviewing and analyzing data throughout the drug development process, from the discovery stage to the marketing phase. The main disciplines include chemistry, manufacturing, and controls (CMC), nonclinical (including pharmacology, PKs, and toxicology), and clinical (including human/patient pharmacology and medical). Oversight and supporting disciplines across CMC, nonclinical, and clinical include quality assurance, regulatory, statistics/biostatistics, legal, and project management. Industry teams also include commercialization, marketing, and sales staff.
Chemistry, Manufacturing, and Control Development (Eric C. Jensen)
Chemistry, manufacturing, and control encompasses a wide variety of technical disciplines that ultimately are responsible for the design, development, and validation of the processes and products associated with each new pharmaceutical product. It can be easy to get lost in the myriad technical details; however, the business of CMC development can be summarized in 2 high-level deliverables: The manufacture, packaging, and testing of materials to be used in all aspects of new drug development. These materials include the active pharmaceutical ingredient (API), material formulated for nonclinical testing, the formulated drug product, and appropriate material packaging for all clinical evaluations. Robust processes for installation in commercial manufacturing developed to ensure consistent product quality and uninterrupted commercial supply.
Beneath these 2 major deliverables, many detailed technical questions must be addressed to achieve a successful registration and launch. The CMC functions, which include process chemistry, formulation development, and analytical development, count on their development partners for timely answers to these questions so that decisions can be made for both product design and delivery. The medical team provides information concerning the desired dose strength(s) of the drug product; nonclinical teams are consulted concerning the amount of test article needed for safety assessment studies and qualification of the impurities which are part of the active ingredient. Both medical and marketing partners provide information concerning the patient population and disease state, which inform the selection of primary packaging platforms and, if applicable, delivery device features. Coming to final decisions on these important design criteria is an iterative process, as the entire drug development team learns increasingly more about the characteristics of the novel drug as it progresses through development.
The CMC in Early- and Late-Phase Development
The CMC becomes involved immediately after a firm decides on the identity (structure) of the new drug entity to be studied and remains involved throughout the drug’s life cycle, which could end either with a decision to terminate development or when the drug is taken off the market.
Early-phase CMC work is focused primarily on enabling the nonclinical and early clinical evaluations of the new drugs via the delivery of active ingredient and dosage forms for these studies (Figure 3). These materials typically are created using manufacturing processes that are not validated and have not undergone the full battery of evaluation expected for a commercial process. In fact, early-stage processes are designed with the sole purpose of creating the new drug without necessarily considering efficiency or reproducibility. Early drug materials are manufactured on a relatively small scale, compared to that for a commercial product. The drug product administered to subjects and patients in early-phase clinical trials is often a simple formulation not designed for scale-up. There is, however, the expectation that materials used in these studies will be produced under conditions very similar to those in a Good Manufacturing Practice–compliant operation. To ensure consistent quality, robust analytical testing is conducted in this phase of drug development. These test data serve as the initial data set for the characterization of the new product, as well as for the development of the control strategy ultimately implemented in the manufacturing of the commercial product.

Material supply sequence throughout drug development and commercialization.
In the early development phases of a new drug product, it is important that periodic feedback (eg, PK behavior) be brought to the CMC development team from the nonclinical and clinical teams concerning the growing body of knowledge about the product’s characteristics. Both API and drug product characteristics (eg, particle size for an oral synthetic drug) can often be modified to achieve a more desirable set of performance criteria once information about the “real-world” behavior of the product is available.
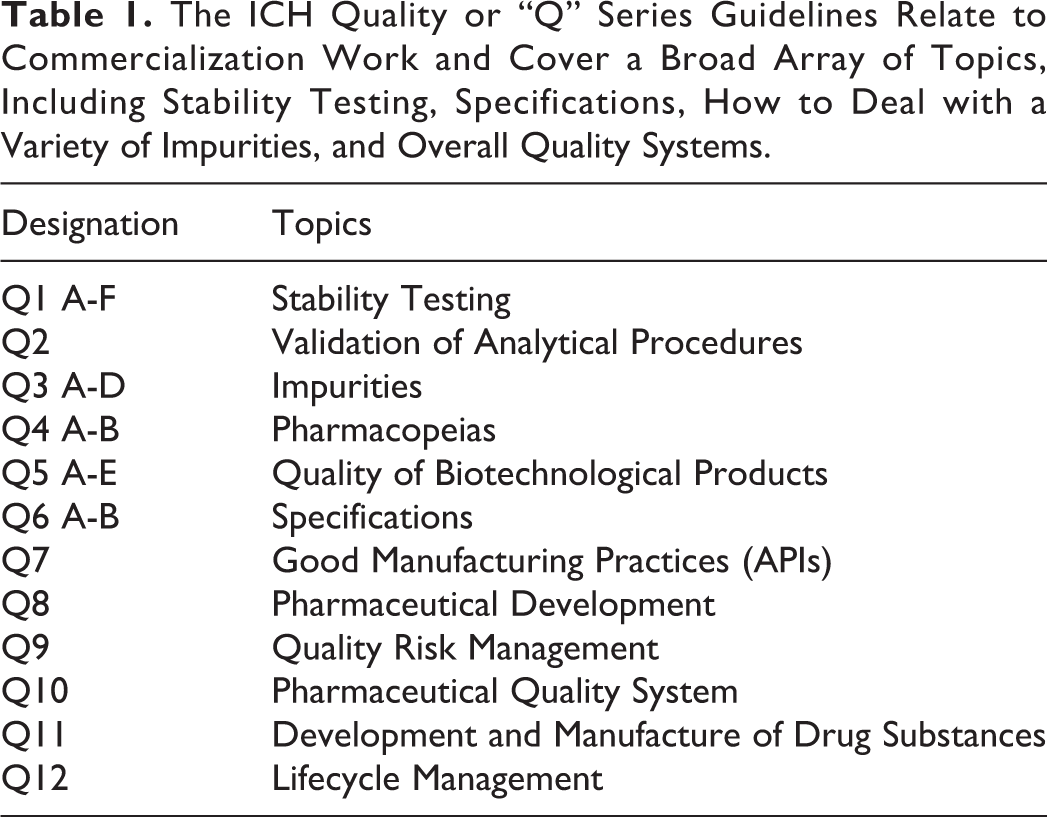
The CMC’s functions in late-phase development take on a new and different intensity once a company decides to commit to large-scale phase 3 studies and start commercialization work, a multi-year process during which the CMC time line is ideally aligned with the corresponding clinical development time line. The CMC team must reference the ICH Q series guidelines 1 (Table 1) to understand and deliver the information expected at the time of regulatory review; in particular, the elements expected as part of a Quality by Design (QbD) approach (see ICH Q7 through Q11) to development and commercialization.
The ICH Quality or “Q” Series Guidelines Relate to Commercialization Work and Cover a Broad Array of Topics, Including Stability Testing, Specifications, How to Deal with a Variety of Impurities, and Overall Quality Systems.
The work of providing support materials for the ongoing clinical evaluation of the new product (finishing Phase 2 clinical studies and performing phase 3 clinical studies) continues at an increasingly larger scale in proportion to the size and scope of phase 3 studies (Figure 3). The larger and more complex workload increases the burden on staff responsible for producing both the API and the drug products. In addition to the requirement for larger quantities of the new product, one or more comparator drug products may be added. These comparators typically are currently marketed products which must be purchased and possibly put into a “blinded” drug product format to prevent unblinding of the phase 3 trial. A blinded product has had the physical appearance of the product altered such that the study subjects and clinical investigators are “blinded”, for example, cannot distinguish between the investigational drug, comparator drug, and placebo.
The other major CMC deliverables during the commercialization phase are related to the ultimate goal of providing the firm’s manufacturing team with the information necessary for that group to begin commercial manufacture, packaging, and market distribution of the new product. This multiyear technology transfer process is driven primarily by an evolving set of design characteristics, often captured as specifications and quality attributes, for the API, its starting materials and intermediates, the drug product, primary and secondary packaging configurations, recommended storage conditions, stability profile, and expiry dating. Patient and disease profiles are used to inform the final selection of dosage form characteristics. Marketing also provides input on design criteria such as colors and logos depending on the firm’s intentions for brand identification features and differentiation from competitor products.
Beyond the design characteristic work and process development for each step in an API or drug product manufacturing sequence, CMC development team members, along with their colleagues in manufacturing, begin the important work of supply chain definition for all components of the manufacturing and packaging processes. This work typically is invisible to many other functions involved in new drug development; however, a strong supply chain is necessary to enable manufacturing to deliver an uninterrupted supply of quality medicine to patients upon the product’s approval. Numerous risk mitigation scenarios and redundancies can be built into a supply chain, so the organization has viable options should an unanticipated upset occur.
Regulatory and Manufacturing Deliverables
As part of demonstrating adequate safety and efficacy during phase 3, the CMC development team begins the task of summarizing their work to meet the needs of 2 major customer groups: the firm’s manufacturing operation and the global regulatory agencies who will review and assess the work of the CMC organization as part of the total regulatory review. To a great extent, the CMC team uses the same large body of data, information, and knowledge to create the documents needed by both groups.
For manufacturing, effective technical transfer of the manufacturing processes, specifications, and test methods will include sharing information, such that its members can successfully install and operate the manufacturing equipment and processes as intended, as well as provide knowledge to solve future technical issues and enable continued process improvements. Some of their reports (eg, detailed process development and optimization reports) have important future value both to themselves and associated technical service functions should they choose to take on additional process improvement work.
For the global regulatory agencies, an effective submission document (Common Technical Document, or CTD; see Regulatory section) will include details concerning API and product design choices; information about manufacturing methods, including a description of the multiple decisions that led to the final process selection; and evidence of technical understanding of material properties, product performance, manufacturing methods, packaging choices, and the product’s anticipated stability behavior. A clear description of the Sponsor’s technical decision-making process goes a long way toward helping the CMC reviewers understand the Sponsor’s choices. The Sponsor is responsible for providing evidence that the materials used in safety and efficacy studies are sufficiently comparable to those intended for the marketed product.
The necessary CMC data and information must be ready for regulatory submission in roughly the same time frame as the final clinical and toxicology sections. Thoughtful planning by experienced scientists coupled with robust communication with the partner organizations is the key to bringing all parts of a drug development program into alignment.
Practical Realities
The CMC organizations rarely have as much experience with materials and processes as they would like. Sponsors want to compress time lines, carefully manage scarce financial resources, and balance priorities across multiple drug development projects. The CMC development teams must therefore engage in creative, innovative experimental design and conduct data-rich experiments to gain as much knowledge as they can in a compressed time frame, given Sponsors’ constraints. Manufacturing benefits from earlier studies with predictable outcomes, since their operations are not designed to enable continued experimentation on the factory floor. While CMC development strives to give manufacturing as complete a package as possible, given the inability to achieve a full-scale experience for each process, some gaps will be evident.
Biologics (Large Molecules) Versus Synthetics (Small Molecules)
Historically, the term “CMC” implied primarily synthetic molecules. In recent decades, “chemistry” has expanded to include biological and biotechnical platforms and manufacturing processes. The same basic principles apply across all types of molecule platforms. However, biologics have a higher degree of molecular diversity and structural heterogeneity requiring additional tests for better quality control, as described in ICH Q6. 1 Characterization of large molecules may include an assay for biologic activity, an understanding of immunological properties, and a description of tertiary structure. Process-related impurities including host cell proteins, host cell DNA, and endotoxin and viral contamination may complicate the scenario. As with small molecules, product-related impurities include precursor molecules and degradation products. For biologics, characterization of the molecule is largely defined by characterization of the process, and close alignment of tests and specifications with the manufacturing process is required. Creation of the master cell bank and working cell banks to support product supply throughout the product life cycle is central to the development of a successful biologic drug.
Control Strategy
Using the relevant ICH and FDA guidance documents, Sponsor companies have been encouraged to take a QbD approach to new drug development. The QbD was first described by Joseph M. Juran as a methodical approach to product development.
4
These principles are embodied in a series of ICH Q series guidelines (Table 1) which describe useful concepts and approaches for the development of new pharmaceutical products. As stated in ICH Q10,
1
the control strategy is one of the major deliverables for the CMC functions during commercial development: a planned set of controls, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.
Typical Areas of Major Focus for Impurity Control Strategy Development for Commercial Products.

As part of a holistic impurity control strategy, the CMC team must undertake a series of studies to thoroughly understand both the chemical and physical stability profiles of the drug substance (API) and the drug product once they are in final form. Commercial manufacturing operations often maintain safety supplies of API to ensure a supply buffer should an interruption in production occur, so it is possible that batches of API will be stored as a contingency for some time. The finished drug product is assigned an expiry date to provide assurance to patients and medical professionals that it is stable and safe to use for an assigned time period after production.
The CMC team uses stress studies early in the development life cycle to begin to build the stability knowledge base. Stress studies serve an important role in this process, as they are designed to reveal in a much shorter time frame the stability behavior of the product under stress conditions intentionally chosen to cause degradation (eg, thermal, water/humidity, oxidation, photosensitivity). Once potential degradation products are identified and matched with the conditions which cause them to appear, the team has the initial information necessary to inform their plans as to compensating controls (eg, lower storage temperatures, protective packaging) which can be implemented.
Overall, the CMC team will adopt a thorough understanding of impurity origins and their fate during processing. Ideally, the team will identify those impurities which are important, usually for safety considerations, early on and do what they can to minimize their formation. The team will also look for ways to remove or minimize impurities in the API so that they will not be included in the drug product. At the conclusion of this work, the team will describe their control parameters and specifications for these impurities, which will be applied to future batches. For specified impurities in the drug substance and drug product, a toxicologist must qualify each impurity according to ICH Q3A and B, respectively. Likewise, residual solvents, elemental impurities, extractables and leachables (where applicable), and genotoxic impurities (GTIs) must be qualified for safety according to ICH Q3C, D, and M7 (see Role of Toxicology section below).
In conclusion, CMC development activities encompass both material delivery and knowledge creation. Effective control strategy development requires investigation of multiple avenues and approaches. A QbD approach enables the delivery of robust processes and analytical controls. Finally, toxicologic assessment of risk due to impurities and other elements is critical to the control strategy development.
The Role of Toxicology in Drug Development (Paul D. Cornwell)
In the simplest sense, toxicology is the study of the nature, effects, and detection of poisons and the treatment of poisoning. While “poisons” may not be the most appropriate term in the context of drug development, the goal of toxicology in drug development is to characterize potential hazards of new drug candidates and devices and to contextualize those hazards in terms of potential human risk.
Toxicology Support of Early Drug Development
Toxicology plays a critical role in every stage of drug development (Figure 4). At the very earliest stages, published literature, in silico tools, and any existing data commonly are used:
Evaluation of safety based on assessment of a target: Published literature can be useful for predicting the types of toxicity that may be associated with the target and thus are useful for subsequent study designs.
Predictive Quantitative Structure Activity Relationship (QSAR) modeling: It may be possible to compare the structure of an NME to a database of other substances for which there are toxicology data, as substances with similar structures commonly have similar effects.
Data mining: Drug development organizations often have a significant database of in vivo toxicology data on chemical structures that commonly are used by that entity; mining those data may yield useful toxicologic information due to structural or pharmacodynamic (PD) similarities. These data may derive from single or repeat-dose toxicology studies at multiple dose levels in one or more animal species, safety pharmacology studies, genetic toxicology studies, developmental and reproductive toxicology (DART) studies, phototoxicity studies, and so on. End points might include clinical observations, clinical pathology, microscopic pathology, functional end points (eg, cardiovascular parameters), or novel biomarkers of toxicity.
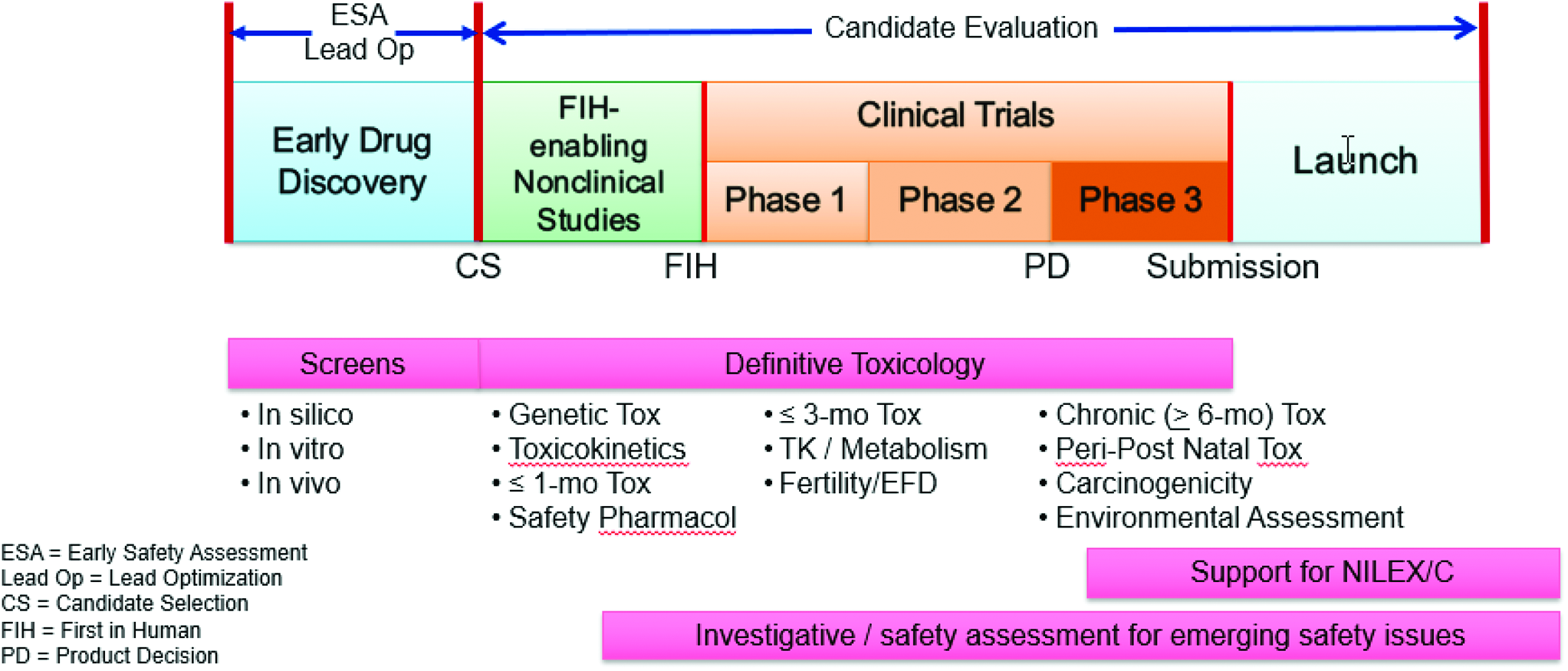
Toxicology supports all phases of the drug development paradigm. NILEX/C indicates new indication or line extension/commercialization.
In vitro studies, which typically use very small amounts of a test article, can be useful for identifying toxicology risks or discriminating between multiple compounds during early drug development. In studies with cells in culture, biomarkers, or surrogates, of toxicity or techniques, such as genomics, transcriptomics, or proteomics, can be leveraged to understand potential toxicities and their mechanisms. At these early stages, nonclinical animal models may also be employed, although typically very little test article is available for these studies. Strategies to reduce the amount of test article needed in this situation include the use of smaller animals, shorter study durations, and other alternative study designs to identify or characterize toxicity. 5
As early drug development programs advance, the number of chemical scaffolds or structural classes under consideration is narrowed based on properties relating to safety, efficacy, and chemical drugability. The toxicology goals of this phase may include: Understanding the mechanism of toxicity to guide the design of subsequent studies. Selection of lead compounds with fewer safety liabilities. Identification of target organs and therapeutic or safety margins (Figure 5). Identifying safety issues that must be managed during development.
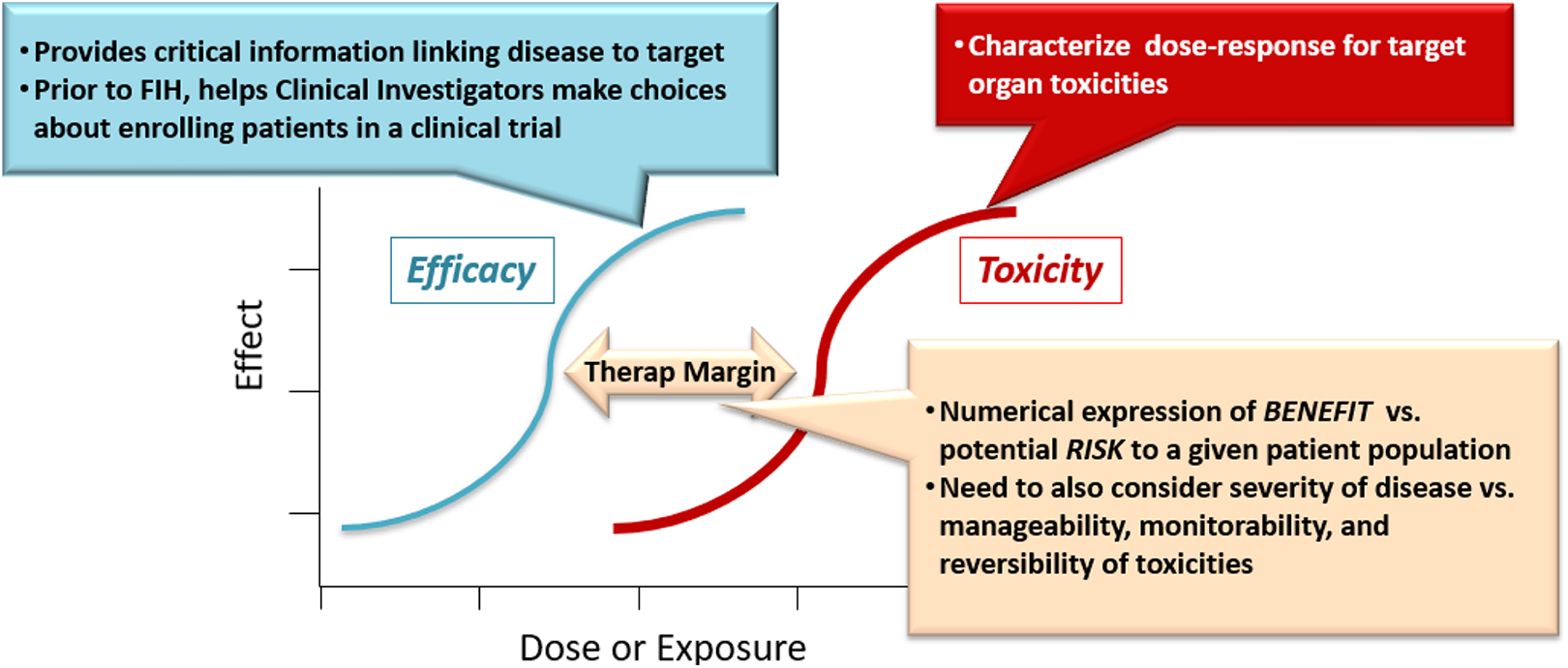
The therapeutic margin is defined as the “space” between efficacious doses or exposures and those that are toxic.
Toxicology studies commonly conducted during the early phases of development include:
Screening and dose-range finding studies in animal models (1-7 days typically): These studies are conducted to identify toxicities that would prevent development and to better design subsequent toxicology studies.
Pilot rodent/nonrodent studies (7-14 days typically): The main goals of these studies are to better understand toxicity following repeated dosing, to estimate the therapeutic margin relative to that toxicity, and to enable the design of subsequent Good Laboratory Practices (GLP) studies.
Genetic toxicity: Because genetic damage is unacceptable for most disease indications, performing an initial empirical assessment of genetic toxicity is common. Common study types include the Ames assay, in vitro and in vivo micronucleus tests, and a study of chromosome aberration.
Safety pharmacology: When identified as a risk in other assessments, it may be advisable to conduct safety pharmacology assessments at this stage of development. Safety pharmacology studies typically include the in vitro hERG assay and in vivo single-dose studies of the central nervous system, cardiovascular, and respiratory function.
Toxicology Support for Initial and Early-Phase Clinical Trials
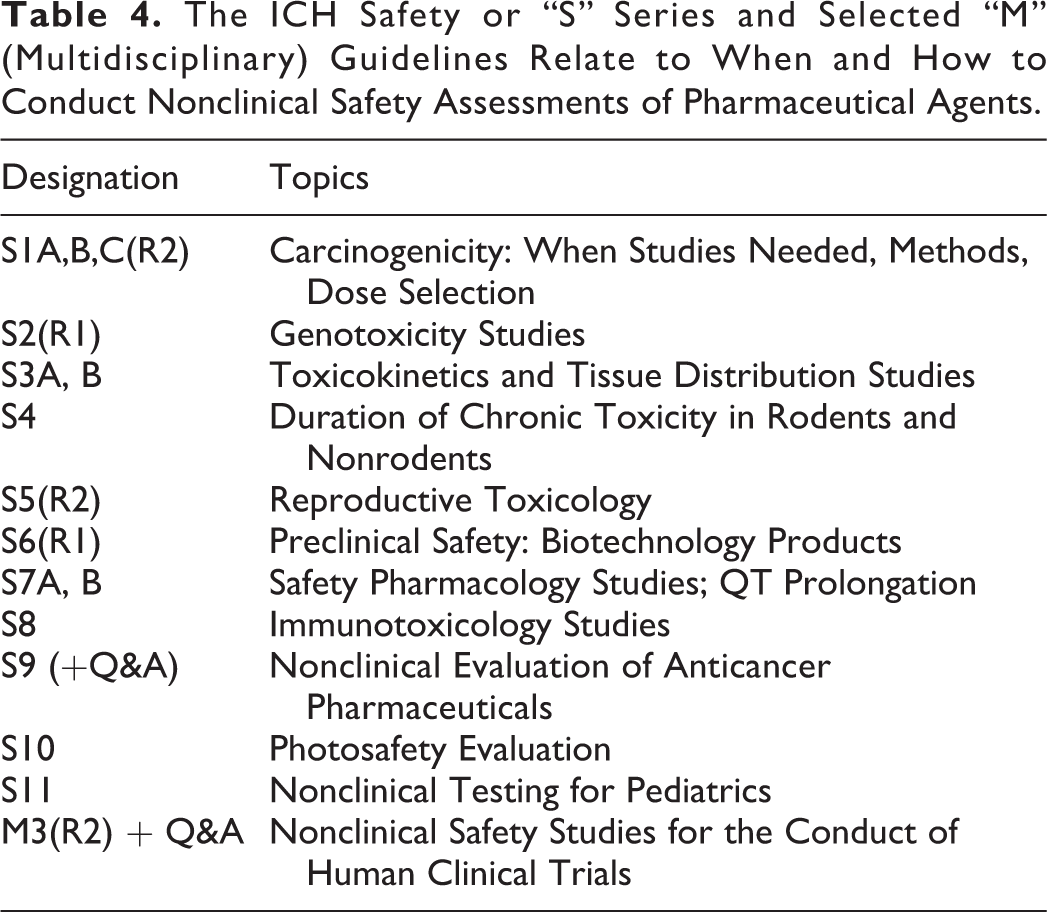
For a drug candidate to advance into clinical testing, the required toxicology studies vary by candidate type (eg, small molecule, biologic, vaccine) and indication (Table 3). In addition to ICH regulatory guidance (Table 4), it is important to note that a Sponsor has many opportunities to consult with regulators throughout the course of drug development. Figure 1 shows some typical opportunities for meeting with the US FDA. Such interactions are extremely valuable in reaching common ground regarding appropriate nonclinical safety packages, study designs, and data interpretation.
Typical Nonclinical Safety Assessment Studies Conducted to Support FIH Clinical Trials.

The ICH Safety or “S” Series and Selected “M” (Multidisciplinary) Guidelines Relate to When and How to Conduct Nonclinical Safety Assessments of Pharmaceutical Agents.
For small molecule, nononcology drugs, the relevant overarching regulatory guideline is ICH M3R2. 1 Repeat-dose studies, with reversibility, as appropriate, should be conducted in 2 nonclinical species, at least one of which should express the pharmacological target. In general, the duration of repeat-dose toxicology studies needs to equal or exceed that of the planned clinical trial (ICH M3R2). Dose selection in these studies is guided by the goal of exploring a dose associated with toxicity (a maximum tolerated dose [MTD]) and identifying doses/exposures associated with effects or a lack of effect, for example, a no observed adverse effect level (NOAEL). Studies of drug disposition and metabolism are necessary to understand dose–exposure–toxicity relationships (Figure 6). The timing of genetic toxicity assessment is guided by ICH M3R2, while the content of the genetox package is governed by ICH S2. Safety pharmacology studies assess the acute effects of a drug candidate on systems that are vital for life, for example, cardiovascular, central nervous, and respiratory systems; ICH S7A, B. Studies on these systems may be conducted as stand-alone studies; however, these end points increasingly are being incorporated into the repeat-dose toxicology studies.
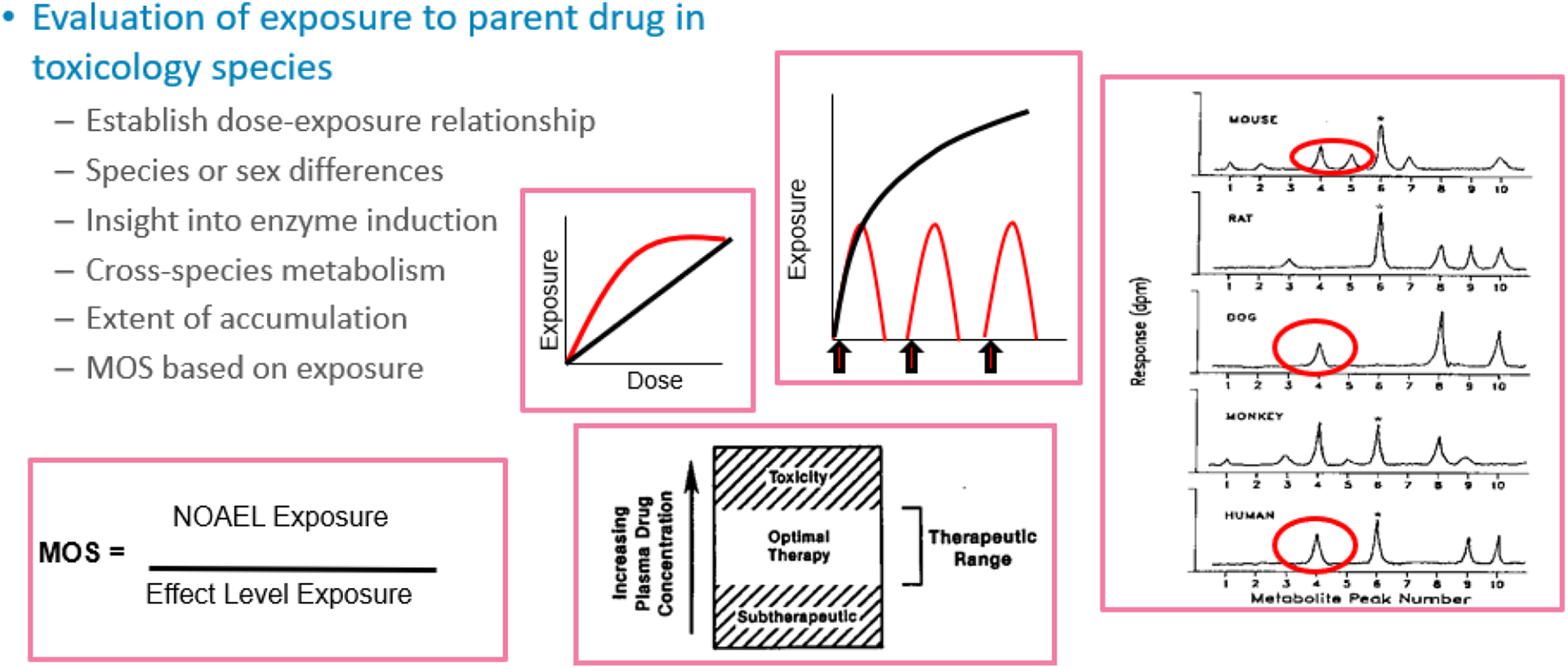
Characterization of drug dispositional properties informs interpretation of toxicology studies. Subfigures represent various relationships of dose–exposure response and comparative metabolism. MOS indicates margin of safety.
The development of drugs (both small molecules and biologics) intended to treat advanced cancer is guided by ICH S9. 1 Analysis of multiple development packages supporting oncology programs has demonstrated that 1-month repeat-dose studies in 2 species are generally adequate to support clinical development of oncology products through phase 2. Doses are selected to identify the highest nonseverely toxic dose in large animals and to approximate a severely toxic dose in 10% of small animals (STD10); these doses, in turn, guide the selection of a safe clinical starting dose. Demonstration of an NOAEL is not required, and a recovery phase often is not necessary. Genetic toxicology studies are not required for early-phase clinical development because most participants in oncology clinical trials already have been exposed to drugs whose mechanism of action involves genetic toxicity. Finally, safety pharmacology end points typically are included in repeat-dose studies rather than as stand-alone studies.
The development of biologic drug candidates, for example, antibodies, peptides, is governed primarily by ICH S6R1. 1 While 2 species also are desirable for toxicology studies with large molecules, repeat-dose studies supporting biologics may be conducted in a single species only, as most biologics are highly specific for a (human) pharmacological target and the availability of pharmacologically relevant nonclinical species may be limited. Further, toxicity is almost always attributable to intended pharmacology, with a low risk of off-target toxicity; thus, the use of a pharmacologically relevant species is essential. Biologics containing non-naturally occurring amino acids, nucleic acids, or inorganic linkers may require some genotoxicity testing; however, most biologics do not, as these molecules usually are too large to gain entry into the cell/nucleus. As with oncology drugs, safety pharmacology risks typically are assessed in the repeat-dose toxicology studies.
First-in-human dose selection and escalation
The selection of a starting dose can be one of the most difficult and critical decisions made for an FIH clinical trial. In these trials, dose levels should not cause harm to trial participants, who usually are healthy volunteers; however, a dose should not be so low that an excessive number of cohorts/participants are required to reach an appropriate dose range for phase 2 trials. The FIH dose normally is defined by nonclinical studies of efficacy, which establish doses needed to achieve the intended efficacious effect, and toxicology, which identify what doses are “safe,” for example, not associated with adverse effects. Methods and guidance for FIH dose selection vary by indication, for example, oncology versus nononcology, and drug type, for example, small molecule versus biologic. The most common methods are described in this section.
For most nononcology molecules, a maximum recommended starting dose typically is based on an NOAEL (expressed as a human equivalent dose [HED]) in the most sensitive nonclinical species to which a safety factor (SF) is applied.
6
A 10-fold SF usually is applied to determine an appropriate maximum clinical starting dose; however, a number of factors could suggest the need for a larger or smaller SF, including: nonmonitorable versus monitorable toxicity in repeat-dose studies, nonreversible versus reversible toxicity in repeat-dose studies, slope of dose/response curve in repeat-dose studies (steep vs shallow), uncertain human bioavailability due to variable bioavailability in nonclinical species, and novel therapeutic versus well-characterized target/class.
Toxicities that are severe, nonmonitorable, or nonreversible could result in the need for a more cautious approach. For cases in which there are significant uncertainties or concerns regarding safety, the NOAEL-based method of choosing a starting dose may not be appropriate. Such concerns warranting a more conservative approach may derive from particular knowledge or uncertainties on the mode of action, the nature of the target, and/or the relevance of animal models (eg, as may be the case with immune-modulating therapeutics and some novel biologics). Tragic incidents which occurred in 2 clinical trials 7,8 drove the need to explicitly consider if a drug candidate is high risk and, in these cases, to define more cautious approaches to safeguard clinical trial subjects. The European Medicines Agency issued guidance on factors that should be considered in determining if a new drug candidate is considered high risk and how to conduct clinical trials accordingly. 9,10 For example, a Sponsor should consider at what dose/exposure pharmacological effects are expected, for example, minimal anticipated biological effect level, or MABEL, 11 as well as the ramifications of exaggerated pharmacology in setting clinical doses. High-risk factors such as those described above drive a cautionary approach for selection of the initial FIH dose and can also drive recommendations for the rapidity and magnitude of dose escalation practices.
Nonclinical data also are included in documents that support early-phase clinical trials, such as the regulatory application to conduct clinical trials (IND), clinical trial protocols, the investigator’s brochure, and informed consent document (see Clinical Trial and Regulatory sections). The goal of the nonclinical package is to provide adequate data to allow safe dosing to human volunteers, for example, in phase 1 trials, 6 and to balance the potential benefit from exposure to a new drug candidate with the risks from that exposure in clinical trials with patient subjects.
Toxicology Support for Late-Phase Drug Development Activities
As drug development progresses, the toxicology package may expand to include studies of chronic toxicity, DART, and carcinogenicity (Figure 4). The safety of human metabolites and drug substance impurities must be qualified in toxicology studies. Clinical safety data often supersede nonclinical data. However, some types of risks, such as DART, including fertility, embryo–fetal development, and pre- and postnatal toxicity, as well as carcinogenicity, cannot be ethically tested in humans; thus, these risks must be informed by other data, including nonclinical toxicology data.
Submissions for New Drug or BLAs, that is, NDA, BLA, are formatted according to CTD (see Regulatory section). The CTD includes tabular and textual summaries of the nonclinical pharmacology, PK, and toxicology data (module 2, section 2.6), an integrated nonclinical summary (section 2.4), and the nonclinical study reports (module 4). Nonclinical data also are utilized in the clinical and CMC sections of the CTD. In drug labels, the US Package Insert, for example, nonclinical data, feature prominently in several sections. These include Use in Specific Populations, such as pregnant or nursing mothers and pediatric patients (section 8), and Nonclinical Toxicology (Carcinogenesis, Mutagenesis and Fertility; Animal Toxicology and/or Pharmacology; section 13).
Other Toxicology Considerations
The NOAEL
One of the most contentious issues in the interpretation of nonclinical toxicology studies can be the identification of the NOAEL. The NOAEL commonly has been defined as the highest dose/exposure that does not cause biologically important increases in the frequency or severity of adverse effects between the exposed population and the appropriate control. 12 Many of the differences in assessment of the NOAEL are driven by differences in opinion regarding whether observation or changes are adverse or not. 13,14 Minimal toxicity can occur at the NOAEL if those effects are not considered to affect the welfare of the animal or to be precursors of serious adverse events. Further, the NOAEL can vary from study to study and is influenced by study design. For example, it must be one of the experimental doses, it does not consider slope of the dose–response curve or nature of effects at higher doses, and it is sensitive to sample size (ie, higher with fewer animals).
Factors driving compound requirements for nonclinical toxicology studies
As discussed earlier in this section, limited availability of test article often limits the extent of toxicologic evaluation in early drug development. The need for large amounts of a compound is driven by several factors, including the need to demonstrate “system failure,” for example, toxicity, the nonclinical species employed, for example, larger species require more test article, the duration of the study, and other aspects of study design.
The goal of most toxicology studies is to identify toxicity, and very high doses may be required to achieve this end. Identification of a MTD in early, shorter studies, for example, 2 to 4 weeks, is important so that appropriate dose levels can be justified for longer duration studies, for example, up to 1 year for general toxicology studies and up to 2 years for carcinogenicity studies.
Toxicology support for CMC
In partnership with CMC, toxicology data often are used to qualify the presence and acceptable levels of impurities in the drug substance [1; ICH Q3]. In this context, qualification means a demonstration of the safety of the given level of impurity. Types of impurities that can be qualified by nonclinical data include:
Total and individual impurities related to the manufacturing/synthesis process: Typically, novel impurities (having no published toxicity data) that are generated during the synthesis process must be qualified [1; ICH Q3A, B].
Residual solvents: Acceptable levels of residual solvents normally are qualified based on published toxicity data (1; ICH Q3C].
Inorganics, including metals and elemental impurities: Acceptable levels of inorganic impurities are normally qualified based on published toxicity data [1; ICH Q3D].
Genotoxic impurities: GTIs are qualified using a threshold of toxicologic concern based on a large body of knowledge about genetic toxicity [1; ICH M7].
ICH Q3A and Q3B outline methods for determining if an impurity needs to be qualified, how it should be qualified (a toxicology study may be needed), and how limits for process-related (novel) impurities should be set for marketed products. ICH Q3 guidance, it should be noted, does not pertain to allowable limits for investigational substances. ICH S9 (development of oncology treatments) allows for alternative impurity limit-setting options in order to increase the speed at which oncology products intended to treat advanced cancer can reach the market.
In conclusion, the nonclinical safety assessment of a drug commences at the earliest stages of target identification and candidate selection through early- and late-phase clinical trials, regulatory submission, and beyond, that is, for the entire life cycle of the drug. Effective partnerships with colleagues from CMC, drug disposition, medical, and regulatory are critical to the ultimate goal of delivering safe and effective medicines to patients.
Introduction to Clinical Trials (Aimee Hodowanec)
The overall objectives of drug development are to determine that a drug is safe and effective for its proposed use, that a drug’s proposed labeling is appropriate to allow for its intended use, and that the methods used in manufacturing a drug are adequate to preserve its identity, strength, quality, and purity. Clinical trials are an integral part of achieving these objectives. Specifically, the effect of a treatment is confirmed, and the risk–benefit profile of a drug is established through the clinical development program. Additionally, data from clinical trials are vital for determining how a drug will be used after approval, as reflected in the package insert, also referred to as the label or prescribing information. Throughout the clinical stages of drug development, scientific rigor and close safety monitoring must be maintained. The protection of the rights and safety of research subjects is of paramount importance.
Drug development consists of a stepwise series of events occurring across several distinct phases of development (Figure 1). Not all important questions about a drug can be expected to be answered in a single study. Therefore, many sequential studies, generally with progressively increasing complexity, are needed. Each new study builds on the results of the previous study, using the data gained to inform the trial design. In general, phases are conducted sequentially, but there may be overlap between the phases and reason to conduct additional phase 1 trials later in development (eg, to assess drug–drug interactions [DDIs]). To successfully move a drug through the clinical stages of development to approval, development programs and study designs must be efficient, yet thorough.
In the United States, phase 1 through 3 clinical trials are conducted under INDs. The FDA oversees clinical trials under IND to help assure subject safety, interpretability of study results, and compliance with established regulations. An IND usually is opened with a phase 1 study, though in some instances it may be opened with a phase 2 or 3 trial if early studies have already been conducted outside the United States. See the regulatory section for details regarding the regulatory aspects of opening an initial IND.
Nonclinical information provides a basis for determining if conducting the proposed clinical investigation(s) will be reasonably safe [§21CFR312.23(a)(8)]. Specifically, nonclinical information is needed to inform drug dosing in early clinical trials. The initial duration of drug exposure in humans should not exceed the duration of exposure in animals, and the initial dose used in humans should not exceed the HED for the NOAEL dose in animals (see Toxicology section). As previously described, an SF is typically applied to the HED. 6 In addition, nonclinical information may inform subject selection and safety monitoring in clinical studies. For example, ocular toxicity in animal studies may prompt pre- and posttreatment ophthalmologic assessments in clinical trials, and testicular toxicity in animals may lead to the exclusion of nonvasectomized males from clinical trials.
Phase 1 of development consists of trials aimed at assessing the safety of a drug and how the drug interacts with the human body. These trials generally include only a small number of subjects, for example, 20 to 100 subjects, and usually involve only short-term dosing, for example, single-dose to 2 weeks in duration. Most often, phase 1 trials are conducted in healthy volunteers. However, in some cases, it may not be appropriate or feasible to study the drug in healthy volunteers. In such instances, phase 1 trials will enroll patients with the condition of interest. When patients with the disease are enrolled, early evidence of drug activity may be gleaned from a phase 1 trial. Examples of common phase 1 trial designs include single ascending dose PK/PD studies; multiple ascending dose PK/PD studies; food-effect studies; absorption, distribution, metabolism, and excretion studies; and DDI studies. Across the Phase 1 trials, the data needed to design well-controlled, scientifically valid phase 2 trials should be collected.
Phase 2 of development serves to better characterize the safety of a drug and to conduct dose exploration to identify the optimal safe and efficacious dose for phase 3 trials and registration. These trials are sometimes referred to as proof-of-concept trials, referring to their role in establishing the activity of the drug for a specific indication. They usually are controlled trials conducted in patients with the disease or condition for which the drug is being developed and remain relatively small, for example, usually no more than several hundred patients. An example of a common phase 2 trial design is a randomized, placebo-controlled, parallel-group, multiple-dose cohort safety and effectiveness trial. Phase 2 trials are crucial in informing the design and conduct of phase 3 trials. Specifically, phase 2 of development is the time to identify appropriate end points and diagnostic assays for use in phase 3 trials.
In general, if phase 2 trials provide preliminary evidence of effectiveness of an investigational drug, the drug may then enter phase 3 of development. (In some instances, trials may be conducted as combined phase 2/3 trials using a single protocol which allows for a seamless transition between trial phases to expedite a drug development program.) Phase 3 trials are large (eg, hundreds to thousands of subjects) studies conducted in the target population. Given the large, heterogeneous populations in phase 3 trials, these trials may reveal less common adverse effects not detected in earlier stages of development. The primary purpose of phase 3 trials is to provide evidence of the safety and effectiveness of a product to support a marketing application. The establishment of efficacy generally requires a controlled trial to distinguish the effect of a drug from other influences, such as a spontaneous change in the course of the disease, placebo effect, and biased observations.
Data collected from nonclinical and clinical studies ideally culminate in the submission of an NDA/BLA. In order to obtain marketing approval, there must be “substantial evidence” to support the claims of effectiveness for new drugs in “adequate and well-controlled” trials. The major elements of an adequate and well-controlled trial include a clear statement of objectives, a valid comparison with a control, adequate assurance that study subjects have the disease or condition being studied, a method of assigning patients to treatment and control groups that minimizes bias and assures comparability, methods of assessing response that are well-defined and reliable, and an analysis plan that is adequate to assess the drug’s effects. The method of assessing response is referred to as the end point.
An end point is an event or outcome that is documented in the course of a clinical trial and is analyzed according to a previously established analysis plan. It is used to determine whether the null hypothesis of the trial should be accepted or rejected. A trial will typically have primary, secondary, and exploratory end points. The primary end point is the single best indicator of effect and should be associated with a clinically meaningful benefit. Examples of clinically meaningful end points include survival, symptom improvement, and prevention of clinical events such as myocardial infarction. In some instances, a surrogate end point or marker that is reasonably likely to predict a clinically meaningful benefit may serve as the primary end point. The optimal primary end point for a specific indication may vary depending on the phase of development. Secondary end points are intended to support the primary end point. Often, several secondary end points are assessed in a trial, one or two of which may be included in labeling if considered particularly noteworthy.
According to the FD&C Act, 505(d), substantial evidence of effectiveness is defined as “Evidence consisting of adequate and well-controlled investigations, including clinical investigations, by experts qualified by scientific training and experience to evaluate the effectiveness of the drug involved, on the basis of which it could fairly and responsibly be concluded by such experts that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the labeling or proposed labeling thereof.” Typically, 2 adequate and well-controlled trials are needed to provide independent substantiation of the experimental results; however, in some instances, 1 trial may be considered adequate. Examples of scenarios in which a single trial may be acceptable include applications for pediatric uses in which data can be extrapolated from prior adult trials, a large trial with statistically persuasive results, and the demonstration of efficacy on 2 different clinically meaningful end points within a single trial. 15
Over the course of a drug’s development, questions and concerns may arise that prompt the convening of an advisory committee. An advisory committee hearing is convened by the FDA for matters of interest to the public. A panel of scientific and medical experts will be called to serve as independent sources of expertise and advice. New drugs frequently are presented to an advisory committee prior to an NDA or BLA decision. In this case, scientific data in the application will be presented and discussed. The FDA asks specific questions of the committee, usually related to areas in the application needing larger discussion or areas of uncertainty, for example, safety concerns, nontraditional study designs, or uncertainty of clinical benefit. At the conclusion of the meeting, the advisory committee will recommend a course of action which the FDA notably is not bound to follow. The committee is not intended to represent or advocate for a particular interest. Most advisory committee meetings are open to the public, with associated documents publicly available on the FDA website. Occasionally, members of the public participate as patient representatives. 16
For approved products, labeling is crucial for conveying the relevant safety and efficacy information needed to use a drug as intended. Drug labels have multiple components. A product will have either a medication guide or a patient package insert geared to patients. Medication guides are required for a prescribed drug or biological product if certain information is needed to prevent serious adverse effects, if a serious side effect should be factored into patient decision-making, or if patient adherence to directions for the use of a product are essential to its effectiveness. If a medication guide is not required, a patient package insert will be provided.
A package insert is written for health care providers. Major sections of the package insert include Indications and Usage, Dosage and Administration, Contraindications, Warnings and Precautions, Adverse Reactions, Drug Interactions, Use in Specific Populations, for example, in pregnant, elderly, and pediatric populations; nonclinical, for example, carcinogenicity, mutagenicity, fertility; and clinical studies. Most information in it is derived from phase 3 trials.
In conclusion, clinical drug development relies on a well-thought-out, stepwise series of studies establishing a drug’s safety and effectiveness. Many factors shape the design of each individual study, such as the phase of drug development, the intended population, and the disease or condition under study. The most important information garnered from clinical drug development will be reported in the package insert, allowing health care providers to prescribe drugs safely and effectively in the appropriate patient populations.
Regulatory Laws, Recommendations, Submissions, Agency Actions, and the Review Process (Ilona Bebenek)
Regulatory Laws and Recommendations
The Federal FD&C Act of 1938 is the basic food and drug law of the United States. With numerous amendments, it is the most extensive law of its kind in the world. It is intended to assure, among other things, that drugs and devices are safe and effective for their intended uses and that all labeling and packaging is truthful, informative, and not deceptive. The final regulations published in the Federal Register, a daily published record of proposed rules, final rules, meeting notices, and other information, are collected in the CFR. The CFR is divided into 50 titles which represent broad areas subject to Federal regulations. The FDA’s portion of the CFR interprets the FD&CA and related statutes. Title 21 of the CFR is reserved for rules of the FDA (subchapter D [Parts 300-399]). For toxicologists, the most relevant section of the FDA regulations is 21 CFR Subchapter D, Parts 312 and 314, and Subchapter F, Parts 601. These sections describe INDs, NDAs, and BLAs. In addition, GLP regulations are outlined in Subchapter A [Part 58]. These regulations direct all actions to be taken by drug Sponsors that are required under federal law. The laws passed by Congress are binding and are not subject to interpretation.
Guidance documents represent FDA’s current thinking on a topic and provide guidelines as to the processing, content, and evaluation/approval of drug applications and to the design, production, manufacturing, and testing of regulated products. While compliance with laws passed by Congress is mandatory, guidance documents provide nonbinding recommendations. Guidance documents also establish policies intended to achieve consistency in the Agency’s regulatory approach and establish inspection and enforcement procedures. It is important to note that guidance documents are not enforceable, and alternate approaches to those described in the guidance may be used with adequate justification, so long as the alternative approach satisfies the requirements of the applicable statute, regulations, or both.
Many guidelines are authored by The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), which is unique in bringing together regulatory authorities, including representatives from the FDA and the pharmaceutical industry, to discuss scientific and technical aspects of drug registration. 1 Harmonization is achieved through the development of ICH Guidelines via a process of scientific consensus with regulatory and industry experts working together. The key to the success of this process is the commitment of the ICH regulators, including the FDA, to implement the final guidelines. A unique aspect of the ICH is the effort it places on harmonization of drug development worldwide. Regulatory members include representatives from the European Union, the United States, Japan, Canada, Brazil, Korea, Singapore, and China. Observer members include those from South America, Africa, the Middle East, and Asia. Although FDA coauthors many of the ICH Guidelines, it also has its own guidance documents, as do health authorities in other countries, including Canada, Japan, and the countries within the European Union.
Manuals of Policies and Procedures (MAPPs) are federal directives and documentation of internal policies and procedures in the FDA Center for Drug Research and Evaluation (CDER). The MAPPs are required by law and are available to the public so that the Agency’s governance can be more transparent; they are the equivalent of Standard Operating Procedures (SOPs) used within organizations. Similarly, Standard Operating Procedures and Policies (SOPPs) are used by staff in FDA’s Center for Biologics Evaluation and Research (CBER) in the performance of their duties, and also are public.
Regulatory Submissions
Throughout drug development, Sponsors must provide evidence of the safety and efficacy of their products for human use. As a result, the process is an almost continuous string of communication and correspondence, mainly regulatory submissions, between the Agency and the regulatory affairs departments of the Sponsor. Regulatory submissions can be defined as a series of documents provided by a Sponsor to the FDA in order to demonstrate compliance with the laws, regulations, and guidance that encompass drug development.
Regulatory submissions are organized and submitted in a common format known as the CTD, which is an assembly of all the quality, safety, and efficacy information for a drug (Figure 7). The CTD is organized into 5 modules. Module 1 is region-specific and modules 2, 3, 4, and 5 are intended to be common to all regions and represent CMC, safety, quality, nonclinical study reports, and clinical study reports, respectively. The CTD guidelines are published in the ICH M4 document. 1
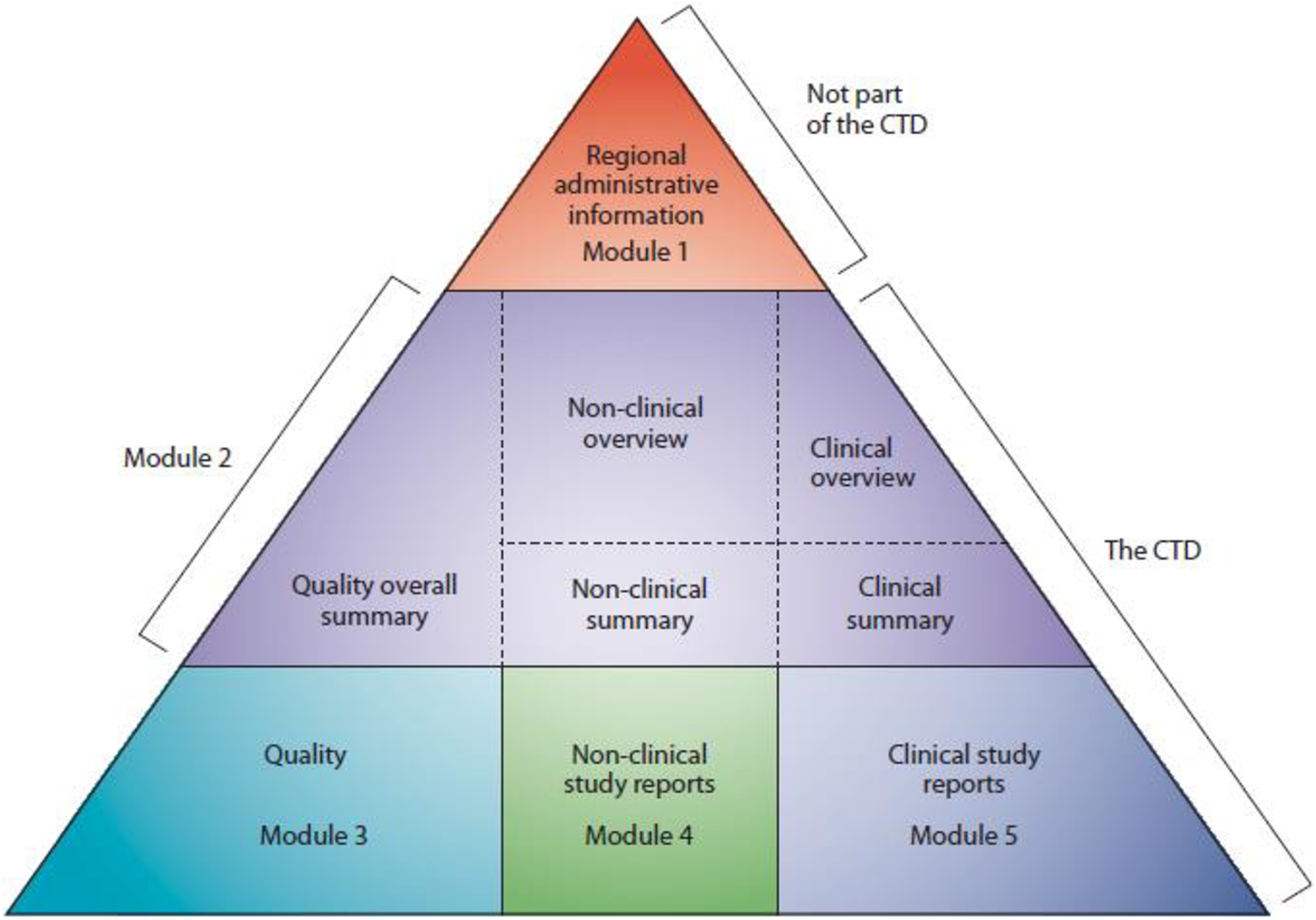
The Common Technical Document is the formatted assembly of all quality, safety, and efficacy information (ICH M4). Source: ICH M4 Guideline: https://www.ich.org/page/ctd
Pre-IND Application
Prior to submitting an IND, a Sponsor may take advantage of the Agency’s Pre-IND Consultation Program. This program is designed to foster early communication between Sponsors and new drug review divisions to provide guidance on the data necessary to warrant IND submission. In addition, through the Pre-IND, the Sponsor may gain an understanding of the Agency’s expectations, focus its strategy, and potentially reduce time to market. Pre-IND advice may be requested for many different issues including CMC, design of nonclinical pharmacology, toxicology, and proof-of-concept studies in animal models and clinical trial design; in addition, advice may be requested for data requirements for an IND application, initial drug development plans, and regulatory requirements for demonstrating safety and efficacy in human subjects. The Agency response may include answers to Sponsor questions, recommendations, or comments on planned or existing development, as well as other questions or information requests. A Pre-IND meeting is granted as a written response (most common), teleconference, or face-to-face meeting.
Investigational New Drug Application
In its simplest definition, an IND is a Sponsor’s request for FDA’s authorization to administer an investigational drug or a biological product to humans. The FDA’s role in the development of a new drug begins when the Sponsor, having screened the new molecule for pharmacological or therapeutic potential, would like to test its diagnostic or therapeutic potential in humans. At that point, the molecule changes in legal status under the FD&CA, and becomes a new drug, subject to requirements of the drug regulatory system under an IND. Current federal law requires that a drug be the subject of an approved marketing application before it can be transported or distributed across state lines. A Sponsor must seek an exemption from that legal requirement in order to ship the investigational drug to clinical investigators in many states. Technically, the IND is the means by which the Sponsor obtains this exemption from the FDA.
Several types of INDs are currently in use. The investigator IND, that is, research IND, or noncommercial IND, is submitted by a physician who both initiates and conducts an investigation and under whose immediate direction the investigational drug is administered or dispensed. A physician might submit a research IND to propose studying an unapproved drug or an approved product for a new indication or in a new patient population. The Emergency Use IND enables the FDA to authorize the use of an experimental drug in an emergency situation that does not allow time for submission of an IND, in accordance with 21CFR, Sec. 312.23 or Sec. 312.20. It is used also for patients who do not meet the criteria of an existing study protocol or if an approved study protocol does not exist. The treatment IND is submitted for experimental drugs showing promise in clinical testing for serious or immediately life-threatening conditions, while the final clinical work is conducted and the FDA review takes place. Lastly, the Commercial IND, the type most commonly submitted to the FDA, typically is submitted by a pharmaceutical company with the intent ultimately to pursue an NDA/BLA.
The IND submission, or “package,” must contain information in 3 broad areas, including animal pharmacology and toxicology (nonclinical studies), manufacturing (process, materials, and quality characteristics), and clinical (the protocol for the proposed clinical study and any previous human experience, if available). The package also includes commitments to obtain informed consent from the research subjects, to obtain review of the study by an institutional review board, and to adhere to the IND regulations. 17
New Drug Application or BLA
Since 1938, every new drug has been the subject of an approved NDA/BLA before being commercialized in the United States. The NDA/BLA application is the vehicle by which Sponsors formally propose that the FDA approve a new pharmaceutical for sale and marketing. Data gathered during the animal studies and human clinical trials which are submitted to an IND become part of the NDA/BLA. The documentation required for submission should include complete data on the drug, including pharmacology, the results of all animal studies conducted, clinical trial results, and a list of ingredients, as well as other CMC information. The goals of the NDA are to provide enough information to enable the FDA reviewer to reach decisions regarding safety and efficacy for the proposed use(s), whether the benefits of the drug outweigh the risks, whether the proposed labeling is appropriate and what it should contain, and whether the manufacturing methods and controls used to maintain the drug’s quality are adequate to preserve the drug’s identity, strength, quality, and purity.
While there are several types of NDAs, the most frequently encountered is the type 1 NME NDA (505b1). An NME is an active ingredient that contains no active moiety that has been approved previously by the Agency in an application submitted under Section 505 of the 1938 FD&C Act or that has been marketed as a drug in the United States.
The 21st Century Review process describes the activities conducted during the review of NDA and BLA applications under the Prescription Drug User Fee Act V program (Figure 2). This process has enabled FDA review staff to engage in greater deliberation during the review phase, complete reviews earlier, and initiate earlier discussion of labeling and postmarketing requirements/commitments with industry—all resulting in more consistent decision-making, as well as in enhanced communications and transparency with industry. New molecular entities being reviewed on a standard time line have a 12-month review period, and those undergoing a priority review have an 8-month review period. Non-NMEs have 10- and 6-month review periods for standard and priority reviews, respectively.
A priority review designation will direct overall attention and resources to the evaluation of applications for drugs that, if approved, would constitute significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions in comparison to standard applications. Significant improvement may be demonstrated by evidence of increased effectiveness in treatment, prevention, or diagnosis of a condition, elimination or substantial reduction of a treatment-limiting drug reaction, documented enhancement of patient compliance that is expected to lead to an improvement in serious outcomes, or evidence of safety and effectiveness in a new subpopulation.
The FDA decides on the review designation for every application. However, an applicant may expressly request a priority review. The designation of a drug as “priority” does not alter the scientific/medical standard for approval or the quality of evidence necessary. Breakthrough therapy, accelerated approval, and fast track are additional approaches for expedited NDA review.
An NDA supplement is an application to enable a Sponsor to make changes in such areas as formulation, manufacturing, patient population, and indication of an already-approved product. The CDER must approve all important changes to NDAs and BLAs to ensure the conditions originally set for the product are still met. A major change requires the submission of a supplement and FDA approval prior to distribution of the drug product made with the change.
The FDA Meeting Requests
Throughout the regulatory development process, and often at critical points such as end of phases (Figure 1), Sponsors may seek advice relating to the development and review of their drugs under an IND or NDA. This type of communication may be conducted by convening an FDA meeting, of which there are 3 types. Type A meetings address stalled product development or critical safety issues and often include discussions of clinical holds. Type B meetings usually occur at defined stages of drug development, such as pre-IND, as mentioned above, or at end of phase. They also include discussion of the overall development program for products granted breakthrough therapy designation status. Type C meetings are any meetings other than type A or type B meetings. These meetings may address the development and review of a product, including early consultations on the use of a biomarker as a new surrogate end point, for a novel use. The meetings may be conducted in person, as teleconferences, or using written responses only.
Agency Actions
Investigational New Drug
Following the submission of an initial IND, the FDA has 30 days to review the submission and determine if proceeding with the clinical trial is safe. During this period, the review team will determine if study participation could expose research subjects to unreasonable risk, if the investigator(s) are qualified to conduct the proposed investigations, and if the proposed investigations are likely to yield data capable of meeting statutory standards for marketing approval.
If deficiencies are identified, the IND may be placed on clinical hold. A clinical hold is an order issued by the FDA to the Sponsor of an IND application in order to prevent a proposed clinical investigation or to suspend an ongoing investigation. All or part of the clinical trial under an IND application may be placed on clinical hold. A clinical hold may be imposed within the 30-day review time or at any time during the IND phase. It may be designated either a complete clinical hold or a partial clinical hold. The Agency will notify the Sponsor of study deficiencies and identify requirements that must be satisfied in order to lift the hold.
A common reason for imposing a clinical hold is concern that subjects would be exposed to an unreasonable and significant risk of illness or injury, and the IND does not contain sufficient information required to assess the risks to subjects. Frequently, the basis for a clinical hold hinges on inadequate toxicology data or a signal in toxicology studies which needs to be addressed further. Additional toxicology studies may be requested to resolve the hold deficiencies. An applicant may submit a complete response to the clinical hold, and the Agency must evaluate the response and decide whether to lift it within 30 days. The Federal Code of Regulations [§312.42(b)(1)] contains a complete listing of indications for clinical hold.
New Drug Application/BLA
Briefly, once an NDA or BLA is submitted to the Agency, it is processed and assigned to the appropriate review staff. Upon the initial submission, the Agency will determine whether or not the application is fileable. The Sponsor also may be notified that filing issues were identified, and asked to resolve them, or a refuse-to-file letter may be sent within 60 days of the application receipt date. A refuse-to-file letter will list details of the deficiencies and discuss reasons that the NDA/BLA cannot be filed. Assuming an application is fileable, the review team reviews, analyzes, and summarizes all of the submitted data. During this time, team members participate in several meetings, which aid in the communication and discussion of data and any review issues (Figure 2). Labeling comments and postmarketing requirements and commitments are sent to the applicant, typically toward the end of the review process. Advisory committee meetings and pediatric review committee meetings are also conducted before the end of the review process, as are discussions about Risk Evaluation and Mitigation Strategies. An approval letter will be issued if the review process is complete and the application was deemed acceptable—containing sufficient evidence of safety and efficacy—by the review team and the FDA Division. Alternatively, a complete response letter may be issued, indicating that the review cycle is complete and that the application is not ready for approval. This letter will again describe the deficiencies and outline recommendations to address them.
The Review Process for INDs and NDAs/BLAs
During the early stages of an IND, and especially during the 30-day IND review (commercial), the data review is heavily slanted toward the nonclinical data. Although the review time line has a 30-day clock, typically the review must be completed within 14 to 21 calendar days, as review by supervisors and the safety meeting all must occur before the 30-day period as well (Figure 8). During the review process, results are discussed with the pharmacology/toxicology supervisor and colleagues from other disciplines, and additional input may be requested from Agency expert panels or groups. In addition, information requests may be sent to the Sponsor with questions or requests for additional or supplemental data. Before the end of the 30 days, the review team will meet with the division director and formulate a decision, as described above. As drug development continues through phase 1, 2, and 3 studies, nonclinical toxicology studies to support each phase are submitted and reviewed (Figure 4).
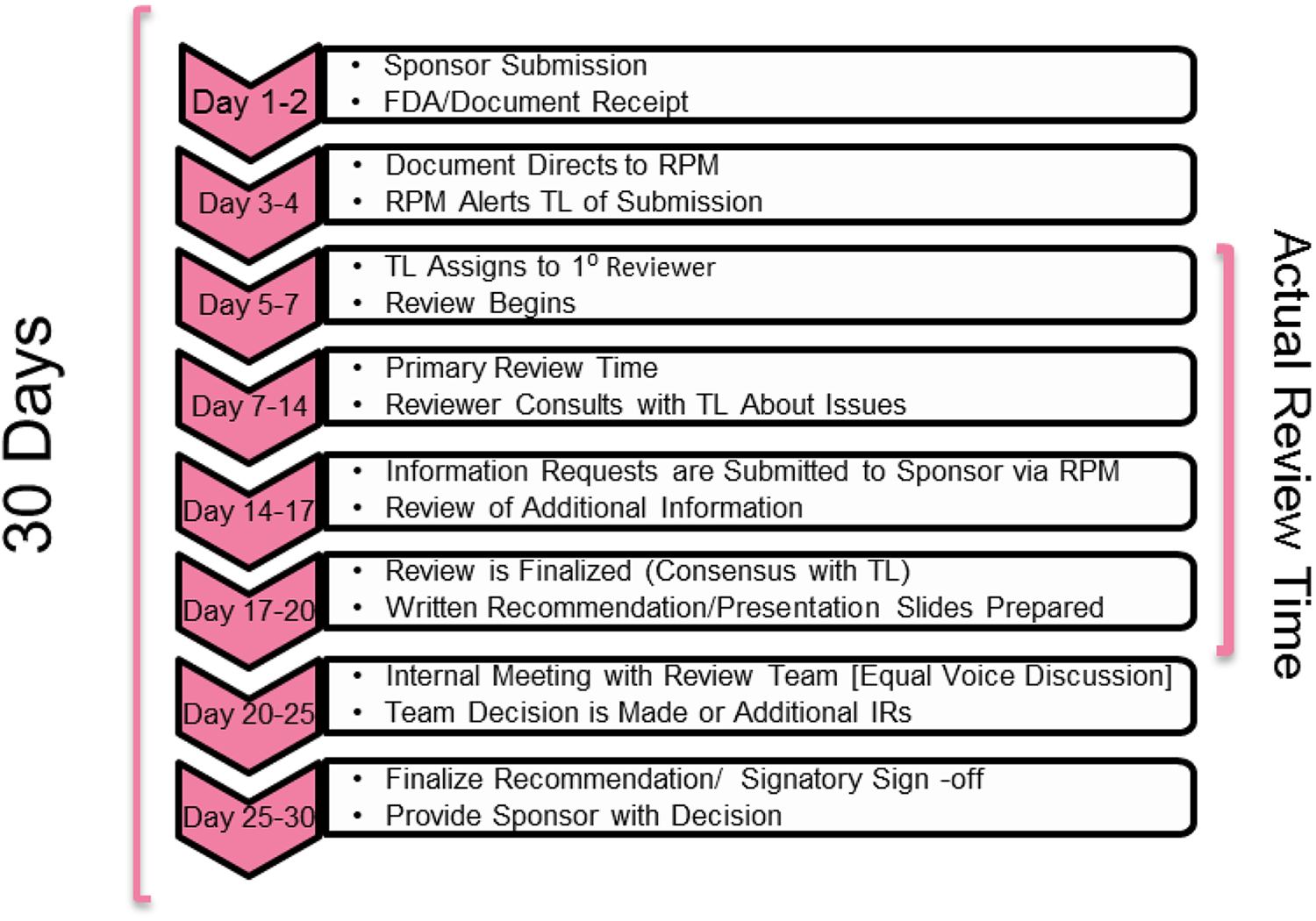
The Food and Drug Administration (FDA) regulatory review of an Investigational New Drug (IND) occurs according to a detailed process and time line.
Typically, at the time of an NDA submission, a large portion of nonclinical studies have been conducted and reviewed, along with any outstanding studies. In general, IND-enabling studies, as well as studies needed for further drug development to support an NDA/BLA, are described in ICH M3(R2). 1 When reviewing data for studies submitted in support of an IND or NDA/BLA, reviewers consider many factors, including compliance with recommended guidance documents, adequacy of supporting safety data prior to initiation of clinical trials or to support marketing, and consideration of nonclinical study issues that warrant follow-up, including toxicities that are unmonitorable in clinical trials. Also under consideration are the proposed dose, adequate margins of safety based on HED 6 or clinical pharmacology data (human exposure), and conduct of appropriate studies to support the duration, stage, and dose of the proposed clinical trial or marketed doses. In addition, a detailed review of the excipients and impurities/degradants in the drug substance and product is conducted (more relevant at the time of NDA/BLA submission). A final, integrated multidisciplinary review document will be submitted into the Document Archiving, Reporting, and Regulatory Tracking System. As part of the NDA review process, results from nonclinical studies are also communicated on the NDA label, and proposed labeling is reviewed and edited based on these results.
Best Practices Suggestions
The presentation in the Continuing Education course included a number of best practice suggestions for the submission process, correspondence with FDA, and submission content. Submissions should be complete, appropriately organized, and easily searchable. Study reports and summaries should be complete, error-free, for example, correct grammar and spelling; corresponding values in tables and text, accurate, and follow common editing and formatting best practices, for example, bookmarks in PDF files, hyperlinked pages in tables of contents. Figures and tables should be used where appropriate, since often it is easier to understand and evaluate data when it is tabulated and presented in an organized fashion. Limitations of a study, as well as opinions and explanations of unusual findings, should be provided. This is especially important in cases of mortality (unexplained death). Discrepancies between the opinion stated in the study report and that of the pathology report should be addressed. When possible, all available information from different studies should be integrated; historical control data should be provided if it is used as a reference in the study report or any summaries. Enough information, for example, sufficient detail, unit consistency, should be provided to enable someone not familiar with the study to understand any calculations. Regarding correspondence, it is recommended that Sponsors engage the Agency early with any questions (Pre-IND and other correspondence), that the questions be specific, and that the correspondence be as direct and as brief as possible. Allowing sufficient time for an Agency response to protocols or proposals is especially important in order to enable incorporation of any necessary changes. Also, upon receipt of Agency questions or information requests, the provision of complete responses in a timely manner is also recommended.
In conclusion, a regulatory submission should have a logical flow and tell a story, include adequate details, and support regulatory expectations with regard to science and content; in other words, provide the means by which regulators can make informed decisions in the interest of patient safety and public health.
Summary
Drug development—it’s not “rocket science”…or is it? Developing drugs is a risky enterprise characterized by a high rate of attrition. 18 Bringing a drug from discovery through testing in nonclinical models and ultimately in human trials, then through regulatory review and, hopefully, commercialization, is an extremely complex and multifunctional process. The contributions and expertise of many are integral: chemists, toxicologists, drug disposition scientists, clinicians, and regulatory scientists. More importantly, drug development is a “team sport” which requires thoughtful planning and integration of the many interdependent parts. Program teams of scientists work together in close collaboration—a good team member understands the deliverables and challenges that other functions face and contributes to the team effort to anticipate and address them. Toxicologists must understand new drug product characteristics in order to qualify impurities, dose–exposure–effect relationships, and strategies for successful regulatory interactions and submissions. The need for nonclinical to clinical translation is obvious; which effects observed in animal models might occur in humans, and at what doses or exposures? How might those effects be monitored in the clinic? Certain nonclinical end points, such as carcinogenicity, reproduction, and development, will never truly be characterized in humans, but how should potential risks be interpreted, communicated, mitigated, or managed? A thoughtful, evidence-based, and appropriately conservative approach is often applied to nonclinical-to-clinical translation, and potential risks in humans are identified and managed in the context of the proposed human trial or NDA approval, involving such factors as indication, patient population, and existing treatments.
Transparent and effective communication with regulatory counterparts is imperative. The best practice suggestions offered in the last section of this work were based on significant regulatory experience and serve as excellent advice for Sponsors and regulators alike.
Footnotes
Authors’ Note
The views expressed are those of the authors. The content and opinions in this article are those of the authors and do not represent Eli Lilly and Co or Inotiv. No official support or endorsement by the US Food and Drug Administration was provided.
Acknowledgment
The authors thank Joanne Berger, FDA Library, for manuscript editing assistance.
Author Contributions
L. Buckley contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. I. Bebenek contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. P. Cornwell contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. A. Hodowanec contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. E. Jensen contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. C. Murphy contributed to analysis and drafted the manuscript. H. Ghantous contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.