
Abstract
This study investigated whether the mechanism underlying the neurotoxic effects of cadmium chloride (CdCl2) in rats involves p66Shc. This study comprised an initial in vivo experiment followed by an in vitro experiment. For the in vivo experiment, male rats were orally administered saline (vehicle) or CdCl2 (0.05 mg/kg) for 30 days. Thereafter, spatial and retention memory of rats were tested and their hippocampi were used for biochemical and molecular analyses. For the in vitro experiment, control or p66Shc-deficient hippocampal cells were treated with CdCl2 (25 µM) in the presence or absence of SP600125, a c-Jun N-terminal kinase (JNK) inhibitor. Cadmium chloride impaired the spatial learning and retention memory of rats; depleted levels of glutathione and manganese superoxide dismutase; increased reactive oxygen species (ROS), tumor necrosis factor α, and interleukin 6; and induced nuclear factor kappa B activation. Cadmium chloride also decreased the number of pyramidal cells in the CA1 region and induced severe damage to the mitochondria and endoplasmic reticulum of cells in the hippocampi of rats. Moreover, CdCl2 increased the total unphosphorylated p66Shc, phosphorylated (Ser36) p66Shc, phosphorylated JNK, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, cytochrome c, and cleaved caspase-3. A dose–response increase in cell death, ROS, DNA damage, p66Shc, and NADPH oxidase was also observed in cultured hippocampal cells treated with CdCl2. Of note, all of these biochemical changes were attenuated by silencing p66Shc or inhibiting JNK with SP600125. In conclusion, CdCl2 induces hippocampal ROS generation and apoptosis by promoting the JNK-mediated activation of p66Shc.
Introduction
Environmental heavy metals are known to adversely affect mental health. 1 Cadmium (Cd) is one of the most common heavy metals on Earth. 1,2 It is associated with neurotoxicity, cognitive decline, and neurodegeneration and is a true risk factor for the development of depression, psychological disorders, and neurodegenerative disorders such as dementia, Parkinson disease, and Alzheimer disease (AD). 1 Humans are at high risk of repetitive exposure to Cd through the respiratory system, gastrointestinal system, and skin. 1,2 Nonoccupational Cd exposure primarily occurs via food, air particles, cigarette smoke, drinking water, and cosmetics. 2 However, occupational exposure to Cd ions occurs in individuals who work in refining, battery and pigment manufacturing, smelting, and welding. 2 In smokers, the blood levels of Cd are 3 times higher than nonsmokers (1.58 vs 0.47 µg/L), whereas it ranged between 5 and 50 µg/L in occupationally exposed individuals. 2
Evidence collected thus far has shown that Cd exposure can induce neurodegeneration in the cerebral cortices and hippocampi of neonatal, young, and adult rodents and promote cell death in cultured neurons. 1 -6 Cadmium ions can increase the permeability of blood–brain barrier (BBB) and pass through to cause neural damage, primarily through the generation of reactive oxygen species (ROS) and induction of oxidative stress. 4,7 Cadmium ions exert a prooxidant effect in neural and non-neural cells by suppressing the endogenous antioxidants such as glutathione (GSH), inhibiting the mitochondrial electron transport chain (ETC) and oxidative phosphorylation, and elevating intracellular levels of Ca2+. 7 -10 Moreover, Cd ions induce intrinsic (mitochondria-mediated) and extrinsic (Fas-mediated) apoptosis in the brains of rodents and in cultured neural cells. 5 -7,10,11 The main apoptotic pathways activated by Cd ions include the c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase 1/2, and mammalian target of rapamycin (mTOR) pathways. 5 Despite these findings, the precise mechanisms underlying the prooxidant and apoptotic effects of Cd ions remain largely unknown and require further investigation.
p66Shc is an adaptor protein with potent prooxidant and apoptotic properties. 12 -16 Studies on the cellular localization of p66Shc revealed that the majority of this protein (44%) is localized in the mitochondria, whereas the rest is localized in the cytoplasm and endoplasmic reticulum (32% and 24%, respectively). 15,16 Under normal physiological conditions, p66Shc levels are kept low. 12 Upon stimulation by an oxidant stimulus such as H2O2 or ultraviolet (UV) light, p66Shc is rapidly activated by phosphorylation at Ser 36 , Following this, p66Shc translocates to the mitochondria, where it impairs the ETC and oxidative phosphorylation, promotes the production of ROS, and stimulates the release of cytochrome c. 12,13,16 -18 Additionally, p66Shc induces ROS generation in mitochondria and other cellular compartments by activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase via Ras-related C3 botulinum toxin substrate 1 activation and reducing antioxidant levels via forkhead box O3a phosphorylation. 12,16,17 However, p66Sch is insensible for p53-induced mitochondria-mediated cell apoptosis. 4,19 -21 Notably, activation of JNK and protein kinase C beta (PKCβ) is known to mediate the phosphorylation and activation of p66Shc.
Interestingly, deleting p66Shc extends the cell life span of animals. 14 An accumulating body of evidence has demonstrated the role of p66Shc in mediating memory deficits and neurodegeneration in animal models of cerebral ischemia, diabetes-induced brain damage, and AD. 12,13,15,22 -25 In the majority of these studies, p66Shc deletion had neuroprotective effects. Indeed, genetic deletion or ablation of p66Shc reduced the generation of ROS, protected against oxidative stress-induced damage, induced sustained neurogenesis, improved spatial memory, and increased brain-derived neurotrophic factor levels. 25,26 In addition, p66Shc mediated ROS generation and cognitive impairment in diabetes mellitus and AD animal models, whereas the deletion of the p66Shc protected against brain oxidative stress and cognitive decline. 12,24 Moreover, β-amyloid-induced neurotoxicity and apoptosis were shown to be mediated by p66Shc activation and ROS overproduction. 13,22,23
Until now, the effect of Cd ions on the expression and activation of p66Shc in the central nervous system has not been investigated. Notably, Cd ion-induced neurotoxicity is mediated by the activation of JNK, a potent upstream activator of p66Shc. 10 In addition, Cd ions activate NADPH oxidase in numerous cells including hepatocytes. 26,27 These data support the hypothesis that Cd ions may induce ROS generation and impair apoptosis in neural cells by activating p66Shc. If proven to be correct, inhibition of p66Shc will provide a novel strategy for preventing Cd-induced neurodegeneration and memory deficits. Therefore, in this study, we investigated whether the neurotoxic effects of cadmium chloride (CdCl2) are mediated via p66Shc activation and explored the mechanisms underlying p66Shc activation in vitro.
Materials and Methods
Animals
Adult male Wistar rats (Charles River, Strain code: 003; 120 ± 5) were supplied from and maintained in the animal unit facility in King Khalid University, Abha, Kingdom of Saudi Arabia. During the experiment, the rats were maintained in a private room under controlled conditions (temperature of 23 °C ± 1 °C, humidity of 60%, and 12/12-hour light/dark cycle). The animals were fed normal rodent chow and had free access to drinking water. All procedures used in this study, including treatment, handling, and surgery, were approved by the Animal Ethics and Use Committee of King Khalid University, which follows the guidelines established by the US National Institutes of Health (NIH publication no. 85-23, revised 1996).
Experimental Design
The animals were divided into 2 groups (n = 18/group). Those in the control group were administered 0.5 mL normal saline (vehicle), while those in the CdCl2-treated group were orally administered 0.5 mL CdCl2 solution (dissolved in normal saline to a final concentration of 0.5 mg/kg). The animals were administered saline or CdCl2 daily for 30 days. The dose of CdCl2 used in this study was adopted from a study by Agnihotri et al, 28 who showed that such a dose of CdCl2 is below the median lethal dose (LD50) over a treatment period of 30 days. Moreover, the same authors showed that CdCl2 at this dose affects the brain and not other organs and that CdCl2-induced hippocampal damage is mediated via oxidative stress and intrinsic cell apoptosis. 28 In this study, 4 rats died after CdCl2 treatment (days 5, 8, 10, and 12) and were replaced by other rats which underwent the same 30-day treatment of CdCl2.
Morris Water Maze Test
The spatial learning and reference memory function of all rats were tested using the Morris water maze (MWM) test. 29 The principle of this test is that the animal must learn to use distal cues to formulate a direct path and find the hidden platform when released from different and random locations around the perimeter of the tank. In the test, the spatial learning of each rat was assessed across repeated trials, whereas the reference memory of each rat was determined by assessing its preference for the previous location of the platform when it was removed during the probe trial. In brief, the maze consisted of a large circular swimming pool (diameter of 1.7 m and depth of 60 cm). Before the test, the pool was filled with water (temperature of 22 °C ± 1 °C) and divided hypothetically into 4 equal quadrants. A hidden escape platform (12 cm in diameter) was positioned in one quadrant below the surface of the water. Milk was added to the water to turn it opaque and obscure the platform. The test was carried out over 5 days (days 31-35), and each animal was given 4 daily trials each of 90 seconds (separated by 5 minutes) from different 4 locations (north, south, east, and west) to find the hidden platform. If the rat was unable to find the platform, it was directed to it by the investigator. Once the rat reached the platform, it was allowed to stay there for an additional 15 seconds. A probe trial was conducted 24 hours after the final training trial to test the reference memory of each rat. During this trial, the platform was removed and the total number of times that each rat crossed the previous location of the hidden platform was recorded. All training and probe sessions were carried out by an observer blinded to the experimental groups. All sessions were performed for n = 18 rats/group and data were presented as the mean ± SD of 4 different trails/day.
Collection of Serum and Preparation of Hippocampi
At the end of the MWM test, the rats were anesthetized with an intraperitoneal bolus of sodium pentobarbital (60-70 mg/kg). Then, 1 mL of blood was collected from each rats in a plain tube to collect the serum, which were stored at −20 °C for further analysis. Following this, the skull of each rat was opened and the brain was collected on ice. All hippocampi were isolated under a dissecting microscope, snap-frozen in liquid nitrogen, and stored at −80 °C for further use. Later, 25 mg samples of the hippocampi of 6 animals in each group were homogenized, individually, in 250 µL ice-cold phosphate-buffered saline (PBS; pH = 7.4) supplemented with 5 µL protease inhibitor (Cat. No. P8340; Sigma-Aldrich) and centrifuged at 1,000g for 10 minutes. The supernatants were then collected, stored at −20 °C, and used later for biochemical analysis. Another 25 mg of the hippocampus of each rat was used to isolate the nuclear and cytoplasmic fractions using a special kit (Cat No. 78835; Thermo Fisher Scientific). Another 6 hippocampi were homogenized in 0.5 mL 1× radioimmunoprecipitation assay buffer (RIPA) buffer (Cat. No. 156034; Abcam) containing 5 µL protease inhibitor cocktail and centrifuged at 10,000g at 4 °C. The supernatant was then collected, stored at −80 °C, and used later for Western blotting.
Biochemical Analysis
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (γ-GTT), urea, blood urea nitrogen (BUN), and creatinine (Cr) levels were measured using an automated analyzer (Cobas 8000 modular analyzer series; Roche Diagnostics). Hippocampal malondialdehyde (MDA), total levels of ROS and reactive nitrogen species (NRS), reduced GSH, manganese superoxide dismutase (MnSOD), tumor necrosis factor α (TNF-α), and interleukin 6 (IL-6) levels were measured using special rat colorimetric and enzyme-linked immunosorbent assay (ELISA) kits (Cat. No. ab118970, Abcam; Cat. No. STA-347, OxiSelect, Cell Biolabs, Inc; Cat. No. 7511-100-K, Trevigen; Cat. No. MBS2881838, MyBioSource; Cat. No. CSB-E11987r and CSB-E04640r, CUSABIO Technology LLC).
Isolation of Hippocampal Cells
The isolation and preparation of hippocampal cells from fetal rats were performed as previously described.
30
In brief, the brains of fetal rats (18 days old) were isolated from their pregnant females under CO2 sedation on ice. The whole hippocampi were digested with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer (pH = 7.4; Invitrogen) containing 1 mg/mL collagenase II, 1 mg/mL glucose, 0.2 mg/mL DNase (activity 500 U/mg), 4 mg/mL bovine serum albumin, 137 mM KCl, 0.7 mM Na2HPO4, 5 mM NaCl, and 100 μg/mL gentamicin (Sigma-Aldrich). The homogenates were then centrifuged at 200g for 2 minutes and the pellets were collected. The cells were resuspended and cultured (at a density of 4 × 105 cells/mL) in 6-well plates in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum and 5% horse serum (Sigma-Aldrich) for 24 hours at 37 °C in a humidified atmosphere containing 5% CO2. Then, the medium was changed and the cells were grown in Neurobasal medium supplemented with 2% B27, 0.5 mM
Small Interfering RNA–Mediated p66Shc Knockdown
To ensure complete p66Shc deletion, we knocked down both p66Shc-1 and p66Shc-2 according to the procedure established by Lone et al. 13 Control and p66Sch/12 targeted small interfering RNAs (siRNAs) were purchased from Thermo Fisher Scientific. The sequences of the p66Shc-1 and p66Shc-2 siRNAs used for this procedure have been described previously. 13 The transfection procedure was carried out using Lipofectamine RNAiMAX (Cat. No. 13778150; Thermo Fisher Scientific) and Opti-MEM (reduced serum medium; Gibco) according to the manufacturer’s instructions. A combination of the p66Shc-1 and p66Shc-2 siRNAs (1:1) was used. The final siRNA concentration was 75 pmol. Before transfection, cells were cultured in 24-well plates at 37 °C in a humidified atmosphere containing 5% CO2 for 24 hours until they reached 60% confluence. Following this, they were transfected with the siRNAs and incubated for 36 hours under similar conditions. Knockdown of p66Shc was confirmed by Western blotting.
Cell Culture and Treatment
The JNK inhibitor SP600125 (Cat. No. 120065; Abcam) was dissolved in dimethyl sulfoxide (DMSO) diluted in PBS (pH = 7.4). Hippocampal cells were cultured in prewarmed DMEM for 24 hours and treated with increasing concentrations of CdCl2 (5, 10, and 25 μM). 7 In some cases, cells were also treated with the highest dose of CdCl2 (25 µM) in the presence of 10 µM SP600125. Similarly, p66Shc-silenced cells were also grown for 24 hours in the presence or absence of 25 μM CdCl2. All experiments were performed in triplicate and repeated twice. Depending on the experimental setting, control cells were treated with DMSO, but others were not. Cell survival was assessed using a Cell Counting Kit-8 (Cat No. CK04-13; Dojindo). Levels of ROS/NRS were measured using the same kit used for the in vivo determination of ROS/RNS levels (Cat. No. STA-347, OxiSelect; Cell Biolabs, Inc). The levels of single-stream DNA (ssDNA), a marker of cell apoptosis, were measured using ELISA and colorimetric assays (Cat No. APT225; Millipore). Cell death was determined using an ELISA-based kit (Cat. No. 11 920 685 001; Roche Diagnostics GmbH). Total cell homogenates and the nuclear and cytoplasmic fractions were prepared from cultured cells as previously described in the in vivo section.
Western Blotting
Protein levels in the nuclear fraction, cytoplasmic fraction, and total homogenates of the hippocampi or cultured cells were determined using a Bradford assay (Cat. No 23300; Thermo Fisher Scientific). Moreover, 40 µg of proteins from each sample was separated on a sodium dodecyl sulfate polyacrylamide gel (8–12%), then transferred onto nitrocellulose membranes, and then blocked with skimmed milk (prepared in Tris-buffered saline with Tween 20 [TBST] buffer). Then, the membranes were incubated with primary antibodies against p-p66Sch (Cat. No. ab54518, 67 kDa, 1:500; Abcam), p66Sch (Cat. No. sc-967, 46/52/66 kDa, 1:500), JNK (Cat. No. sc-7345, 46/54 kDa, 1:1,000), p-JNK (Cat. No. sc-6254, 46/54 kDa, 1:1,000; Santa Cruz Biotechnology), p53 (Cat. No. 9282, 1:000, 53 kDa), cytochrome-c (Cat. No. 11940, 1:500, 14 kDa), cleaved caspase-3 (Cat. No. 9661, 1:500, 17/19 kDa), Bcl-2-associated X protein (Cat. No. 2772, 20 kDa, 1:1,000), p-nuclear factor-kappa B (NF-κB) (Ser536) (Cat. No. 3031, 65 kDa, 1:500), β-actin (Cat. No. 3700, 45 kDa, 1:1,000), α-tubulin (Cat. No. 2144, 50 kDa, 1:1,000), and lamin B1 (Cat. No. 15068, 68 kDa, 1:1,000; Cell Signaling Technology). The membranes were then incubated with the appropriate secondary antibody (prepared in TBST buffer). Antigen–antibody reactions were detected, photographed, and analyzed using a Pierce ECL kit (Thermo Fisher) and C-DiGit Blot Scanner (LI-COR). The membranes were stripped up to 5 times, and phosphorylated proteins were detected first. An internal known standard protein was run between gels and used for standardization.
Histological Analysis
Hippocampal specimens were fixed in 10% buffered formalin for 24 hours. Then, all tissues were dehydrated in ascending concentrations of ethanol (70% for 24 hours, 90% for 1 hour, and 100% for 1 hour), cleared with xylene, embedded in paraffin wax, and cut into small sections (5-µM-thick) using a Reichert Rotatory Microtome. All sections were routinely stained with Harris hematoxylin and eosin. For the electron microscopy study, hippocampal samples were cut into small pieces, fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer for 24 h, embedded in Spur’s resin, and cut into 1-µM-thick sections. Following this, the ultrathin sections were stained with uranyl acetate and lead citrate and examined under a JEM-1011 transmission electron microscope (Jeol Co) at 80 kV. All histology and electron microscopy slides were examined under a light microscope (Olympus PM-10 Photomicrographic System).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software (version 8; GraphPad Software Inc). Differences between the control and CdCl2-treated rats were analyzed using the Student t test. All other parameters were compared between multiple groups using one-way analysis of variance followed by Tukey post hoc test. Data are presented as mean ± SD, and a P value <0.05 was considered statistically significant.
Results
Cadmium Chloride Reduces the Body Weight of Rats, Impairs Renal and Hepatic Function, and Impairs Spatial Learning, Reference Memory, and Retention Memory
Administration of CdCl2 to rats for 30 days significantly reduced their final body weight and increased serum ALT, AST, γ-GTT, urea, BUN, and Cr levels, indicating that CdCl2 impaired renal and hepatic function (Table 1). In addition, 4 rats (22%) of the CdCl2-treated rats died on days 5, 8, 10, and 12 (data not shown). These rats were replaced with other rats that underwent a similar treatment with CdCl2 for 30 days to equalize the number of rats between the 2 study groups. The spatial and reference memory of the rats were tested by the MWM test, and the retention memory of the rats was tested using the passive avoidance learning test (PALT). The spatial memory of the rats was tested over the first 5 days of the MWM test, during which an escape platform was placed in one quadrant of the tank and submerged under milky water. One day later, the probe test was performed in which the platform was removed and the total number of times the rat crossed the previous location of the platform was counted. Between days 2 and 5 of the MWM test, CdCl2-treated rats showed prolonged escape latencies and longer swimming paths compared to control rats (Figure 1A and B). During the probe test, CdCl2-treated rats showed less number of times to cross the place where the hidden platform was initially placed (Figure 1C). These data indicate that CdCl2-treated rats exhibited impaired spatial and retention memory. During the PALT, rats with intact retention memory avoid entering the dark area as they previously received an electrical foot shock in this area. Cadmium chloride-treated rats entered the dark area faster than control rats, indicating that these animals exhibited impaired retention memory (Figure 1D).
Changes in Body Weights and Serum Levels of Liver and Kidney Function.a
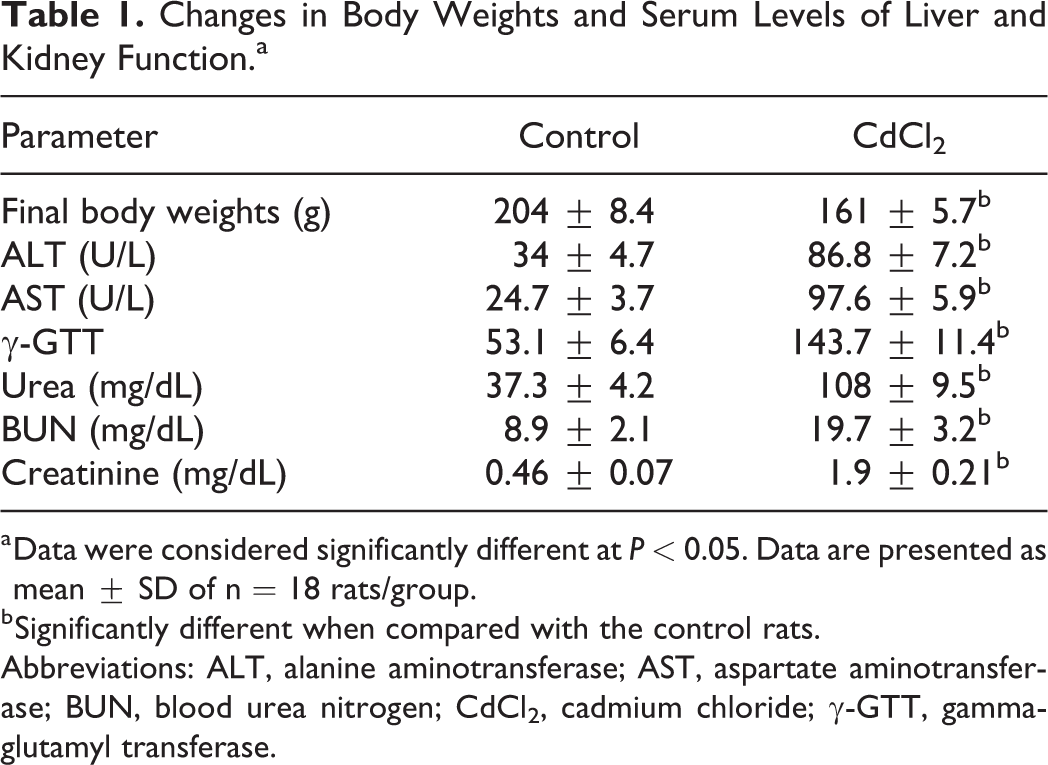
a Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 18 rats/group.
b Significantly different when compared with the control rats.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CdCl2, cadmium chloride; γ-GTT, gamma-glutamyl transferase.
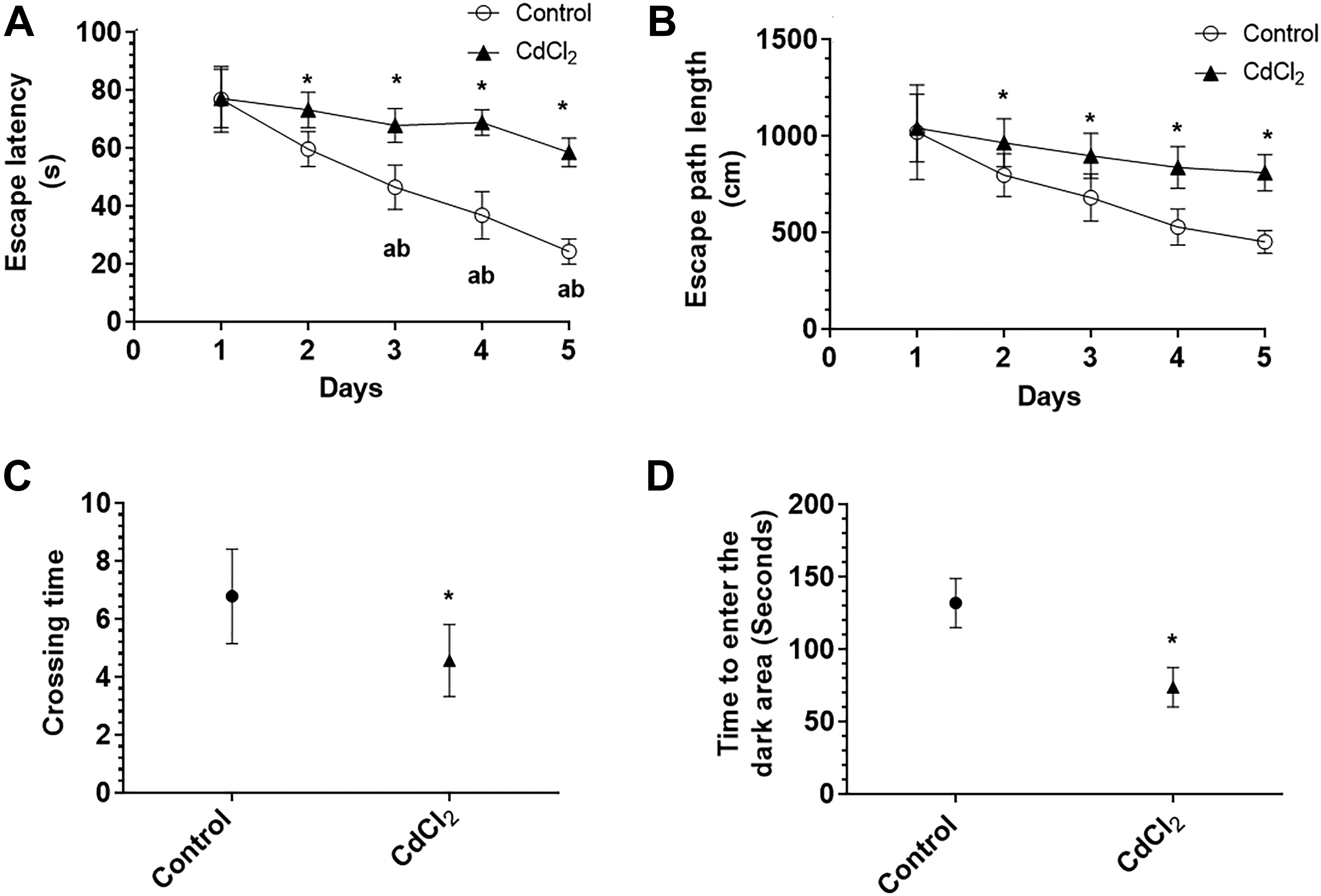
The escape latency (A) and length (B) of distance spent by the rats to find the hidden platform during the first 5 days and the number of crossing times (C) at the site of the removed hidden platform during the probe trial of the water Morris maze test, as well as time spent by the rats in the illuminated (lighted) area before entering the dark area during the retention memory test (D). Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 18 rats/group. *Significantly different when compared with the control rats.
Cadmium Chloride Induces Oxidative Stress and Inflammation and Alters the Structure of the Rat Hippocampus
As shown in Figure 2, treatment with CdCl2 for 30 days reduced the number of pyramidal cells in the CA1 region of the rat hippocampus and rendered these cells dark in color (Figure 2A and B). At the ultrastructural level, the hippocampal neurons of the CdCl2-treated rats showed pyknotic nuclei and severe damage to their mitochondria and rough endoplasmic reticulum (Figure 2C-F). In addition, overall, this sentence sounds unnatural—do you mean “In addition, significantly higher levels of ROS, MDA, TNF-α, and IL-6, significantly increased NF-κB p65 activity, and significantly lower levels of MnSOD and GSH were noted in the hippocampi of CdCl2-treated rats compared to the hippocampi of control rats (Figure 3A-G).
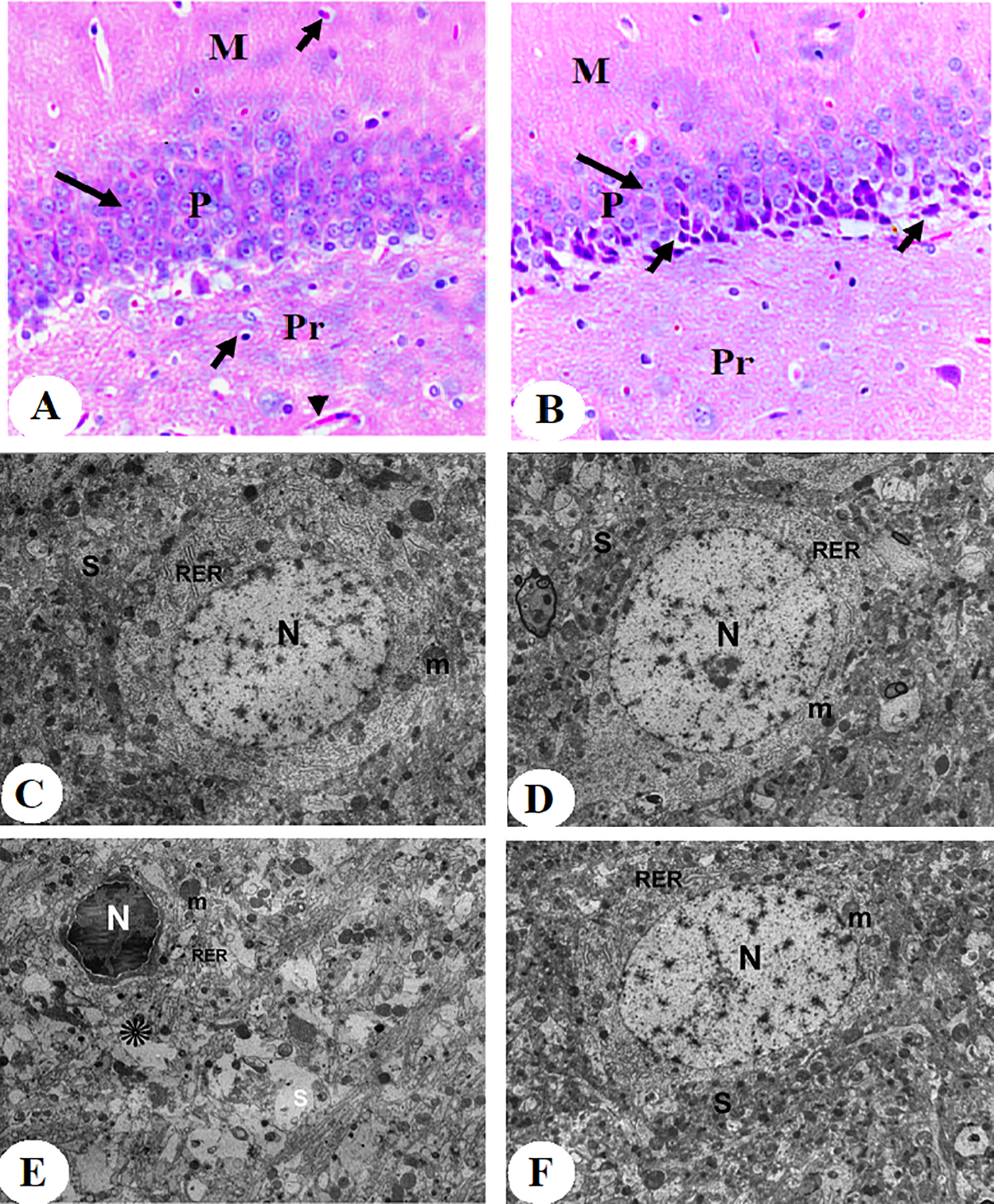
Histological images stained by hematoxylin and eosin (H&E) (A and B) and elector microscope images (C-F) of the hippocampi (CA1 region) of all groups of rats. Image A was taken from a control rat and showed the presence of the 3 normal layers including pyramidal layer (P), molecular layer (M), and polymorphic layer (Pr). The (P) layer clearly showed normal 4 to 6 intact layers of the pyramidal cells (long black arrow), most of which are with vesicular nuclei. Besides, normal glial cells (short arrow) and blood capillaries (arrowhead) were abundant in the M and Pr layers. Image B was taken from a CdCl2-treated rat and also showed all 3 layers. However, there is an obvious decrease in the number of layers and showed marked shrinkage in the size of the pyramidal cells (long arrow), most of which looked dark stained with the presence of many dead cells (short arrow). Images C and D were taken from control rats and showed a normal hippocampal neuron that has intact normal rounded nuclei (N), rough endoplasmic reticulum (RER), mitochondria (m), and synapses (S). The mitochondria look very healthy and are surrounded by a double membrane and contained mitochondrial cristae and matrix. Images E and F were from CdCl2-treated rats and pyknotic nuclei (N), damaged pleomorphic rough endoplasmic reticulum (RER), and mitochondria (m) which lost its normal cristae and matrix structures (×5,000).
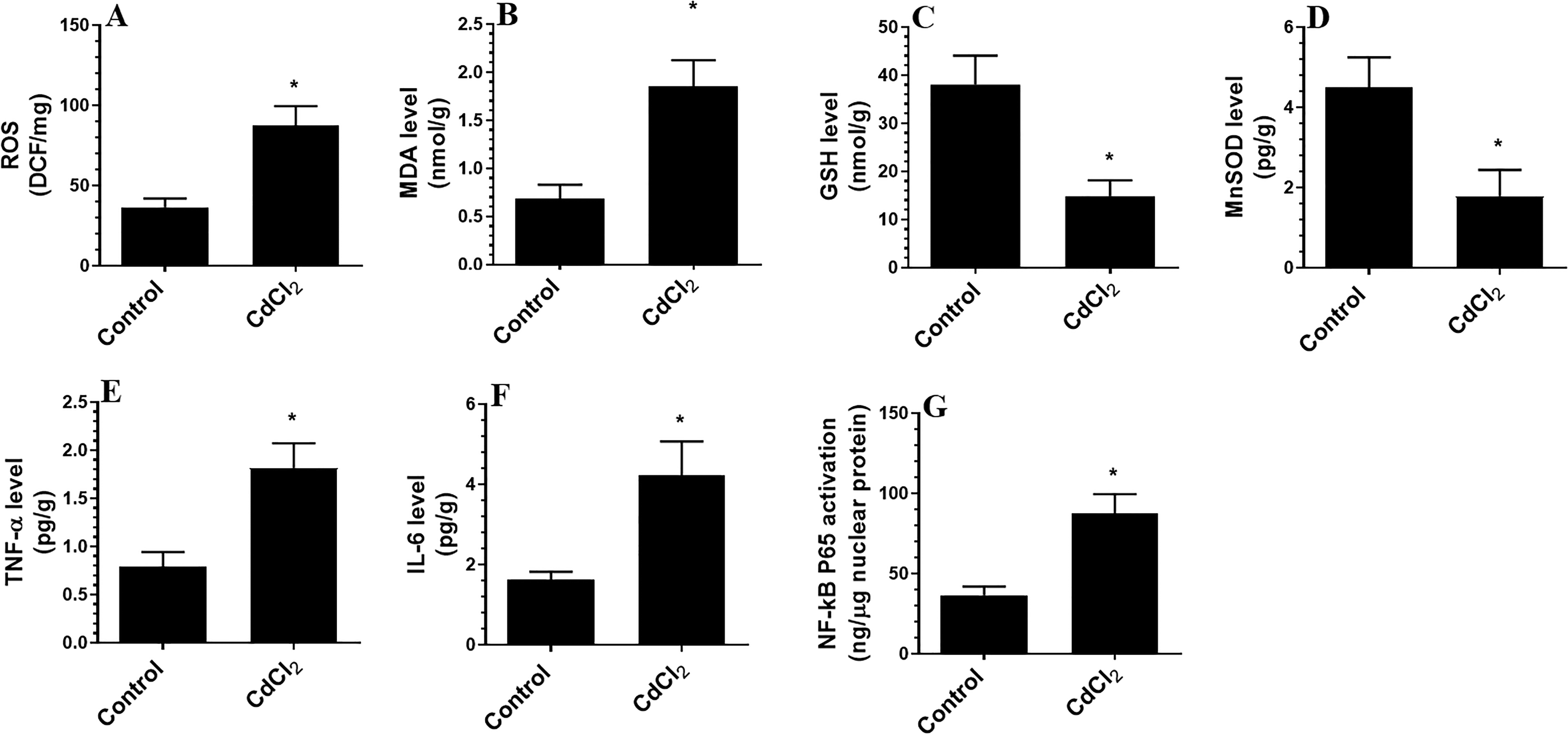
Levels of reactive oxygen species (ROS, A), malondialdehyde (MDA, B), glutathione (GSH, C), manganese superoxide dismutase (MnSOD, D), tumor necrosis factor (TNF-α, E), and interleukin 6 (IL-6, F) and activity of NF-κB in the hippocampi of all experimental groups. Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 12 rats/group. *Significantly different when compared with the control rats.
Cadmium Chloride Upregulates p66Shc and NADPH Oxidase; Activates JNK, p66Shc, and NF-κB; and Increases Cytochrome c and Caspase-3 Levels in the Rat Hippocampus
The hippocampi of CdCl2-treated rats contained significantly higher protein levels of p66Shc, p-p66Shc (Ser 36 ), and NADPH oxidase than the hippocampi of control rats (Figure 4A and C). While the total protein levels of JNK and NF-κB were not significantly different between the groups, CdCl2-treated rats exhibited higher levels of p-JNK (Figure 4B) and p-NF-κB p65 (Ser356) (Figure 5A) compared to control rats. The CdCl2-treated rats also exhibited higher cytoplasmic levels of cleaved caspase-3 and cytochrome-c (Figure 5B and C).
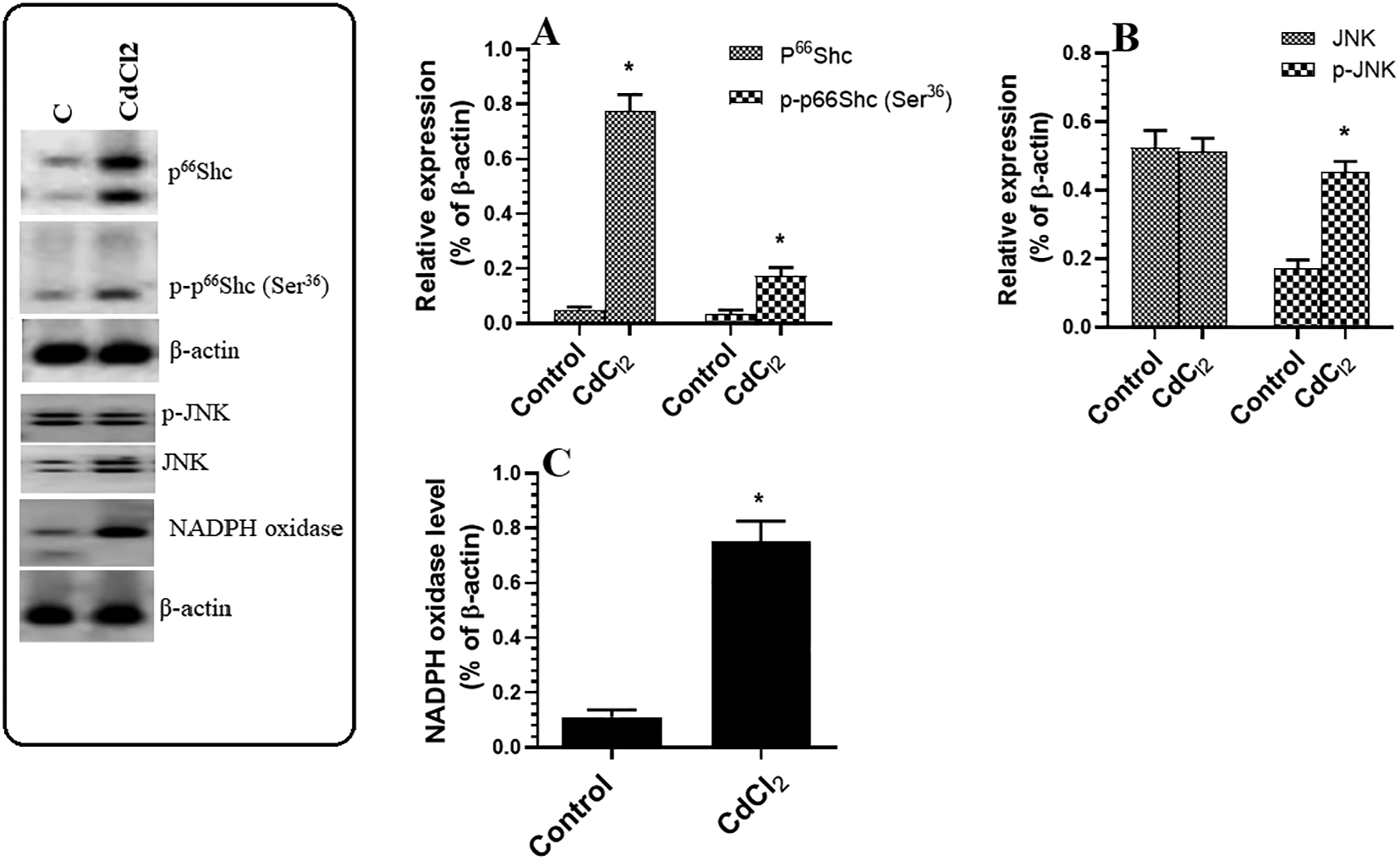
Protein levels of p66Shc/p-p66Shc (A), JNK/p-JNK (Thr183/Tyr185) (B), and NADPH oxidase (C) in the hippocampi of rats of all experimental groups. Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 12 rats/group. *Significantly different when compared with the control rats.
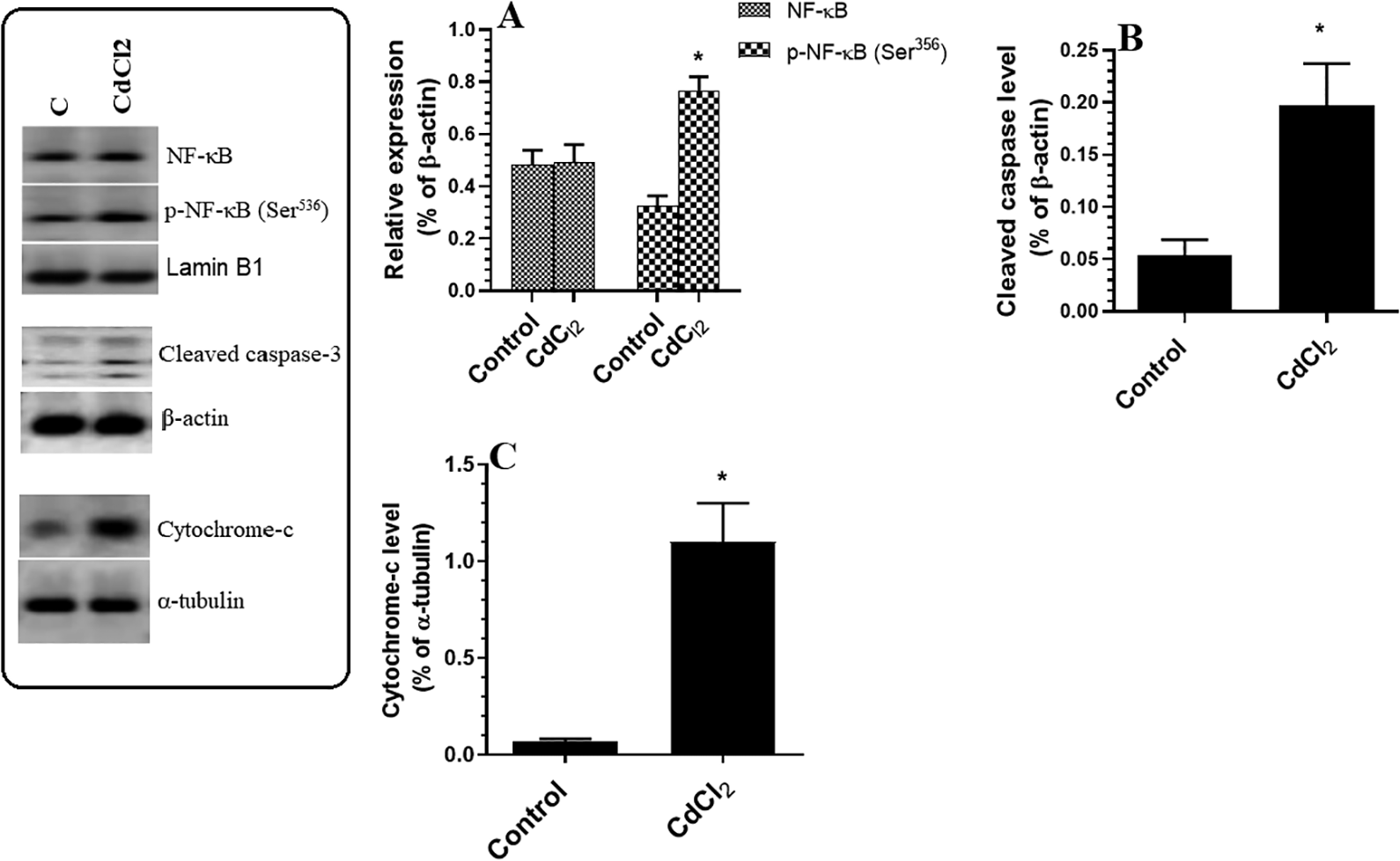
Protein levels of NF-κB p65/p-NF-κB p65 (Ser356) (A), cleaved caspase-3 (B), and cytoplasmic cytochrome-c (C) in the hippocampi of rats of all experimental groups. Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 12 rats/group. *Significantly different when compared with the control rats.
Cadmium Chloride Promotes DNA Damage and Apoptosis and Upregulates p66Shc and NADPH in Cultured Hippocampal Cells in a Dose-Dependent Manner
Treating cultured hippocampal cells with increasing doses of CdCl2 (0, 5, 10, and 25 µM) significantly reduced cell survival, promoted cell death, and increased the levels of ROS and cleaved ssDNA in a dose-dependent manner (Figure 6A-D). Similarly, CdCl2 treatment significantly increased the total protein levels of p66Shc, p-p66Shc (Ser36), NADPH oxidase, and cleaved caspase-3 (Figure 7A-C). On the other hand, pretreating CdCl2-treated cells with SP600125, a specific JNK inhibitor, significantly prevented all of the aforementioned CdCl2-induced changes (Figure 6A-D and Figure 7A-C).
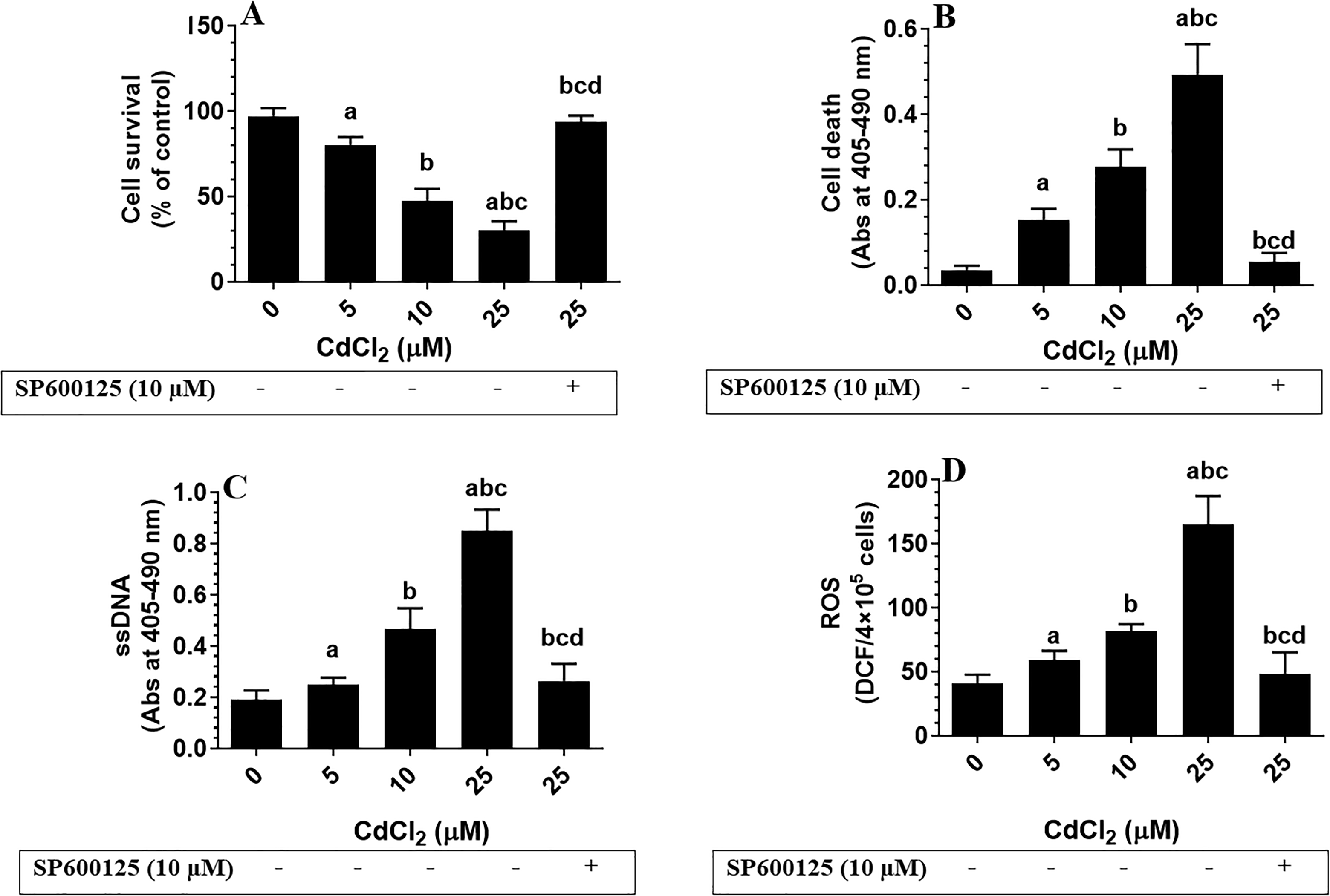
Cell survival (A) and levels of cell death (B), single-stranded DNA (ssDNA) (C), and reactive oxygen species (ROS, D) in the cultured fetal hippocampal cells. Cells were cultured at a density of 4 × 105 in B27 Neurobasal medium for 10 days and then transferred to Dulbecco’s modified Eagle’s medium for 24 hours and treated with increasing concentration of Cadmium chloride (CdCl2; 5, 10, 25 μM). Also, some cells were pretreated with 10 µM SP600125, a specific c-Jun N-terminal kinase inhibitor, and then incubated with the highest dose of CdCl2 (25 µM) for 24 hours. Control cells were treated only with dimethyl sulfoxide (0.1%) and received no CdCl2 (0.0 µM). Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of 3 trials each of triplicate. aSignificantly different when compared with the 0.0 µM CdCl2-treated cells. bSignificantly different when compared with the 5 µM CdCl2-treated cells. cSignificantly different when compared with 10 µM CdCl2-treated cells. dSignificantly different when compared with 25 µM CdCl2-treated cells.
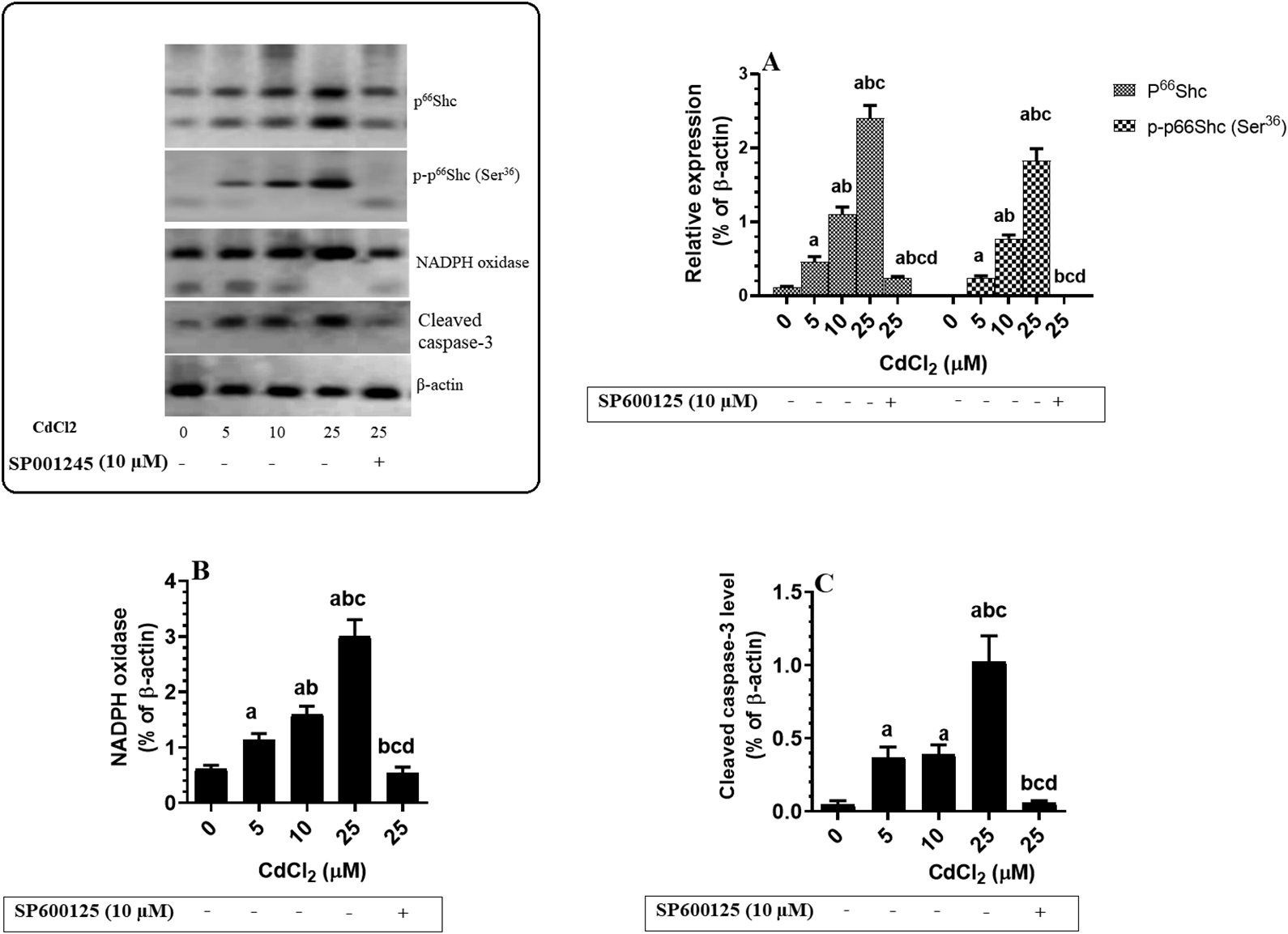
Protein levels of p66Shc/p-p66Shc (A), nicotinamide adenine dinucleotide phosphate oxidase (B), and cytochrome-c in the hippocampi of rats of all experimental groups. Cells were cultured at a density of 4 × 105 in B27 Neurobasal medium for 10 days and then transferred to Dulbecco’s modified Eagle’s medium for 24 hours and treated with increasing concentration of CdCl2 (5, 10, 25 μM). Besides, some cells were also pretreated with 10 µM SP600125, a specific c-Jun N-terminal kinase inhibitor, and then were incubated with a higher dose of CdCl2 (25 µM) for 24 hours. Control cells were treated only with DMSO (0.1%) and received no CdCl2 (0.0 µM). Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 12 rats/group. aSignificantly different when compared with the 0.0 µM CdCl2-treated cells. bSignificantly different when compared with the 5 µM CdCl2-treated cells. cSignifcantly different when compared with 10 µM CdCl2-treated cells. dSignificantly different when compared with 25 µM CdCl2-treated cells.
Small Interfering RNA–Mediated Silencing of p66Shc Protects Cultured Hippocampal Cells From CdCl2-Induced Cell Death, ROS Generation, and NADPH Oxidase Activation
To confirm that p66Shc activation is involved in CdCl2-induced neurotoxicity and hippocampal damage, we silenced p66Shc using specific siRNAs against p66Shc-1 and -2 and treated the cells with the highest effective dose of CdCl2. Silencing p66Shc did not affect any of the measured parameters but significantly prevented CdCl2-induced cell death and promoted cell survival (Figure 8A and B). Moreover, silencing p66Shc completely prevented the CdCl2-induced increase in the levels of ROS, ssDNA, and cytochrome-c and CdCl2-induced NADPH oxidase activation (Figure 8C-H).
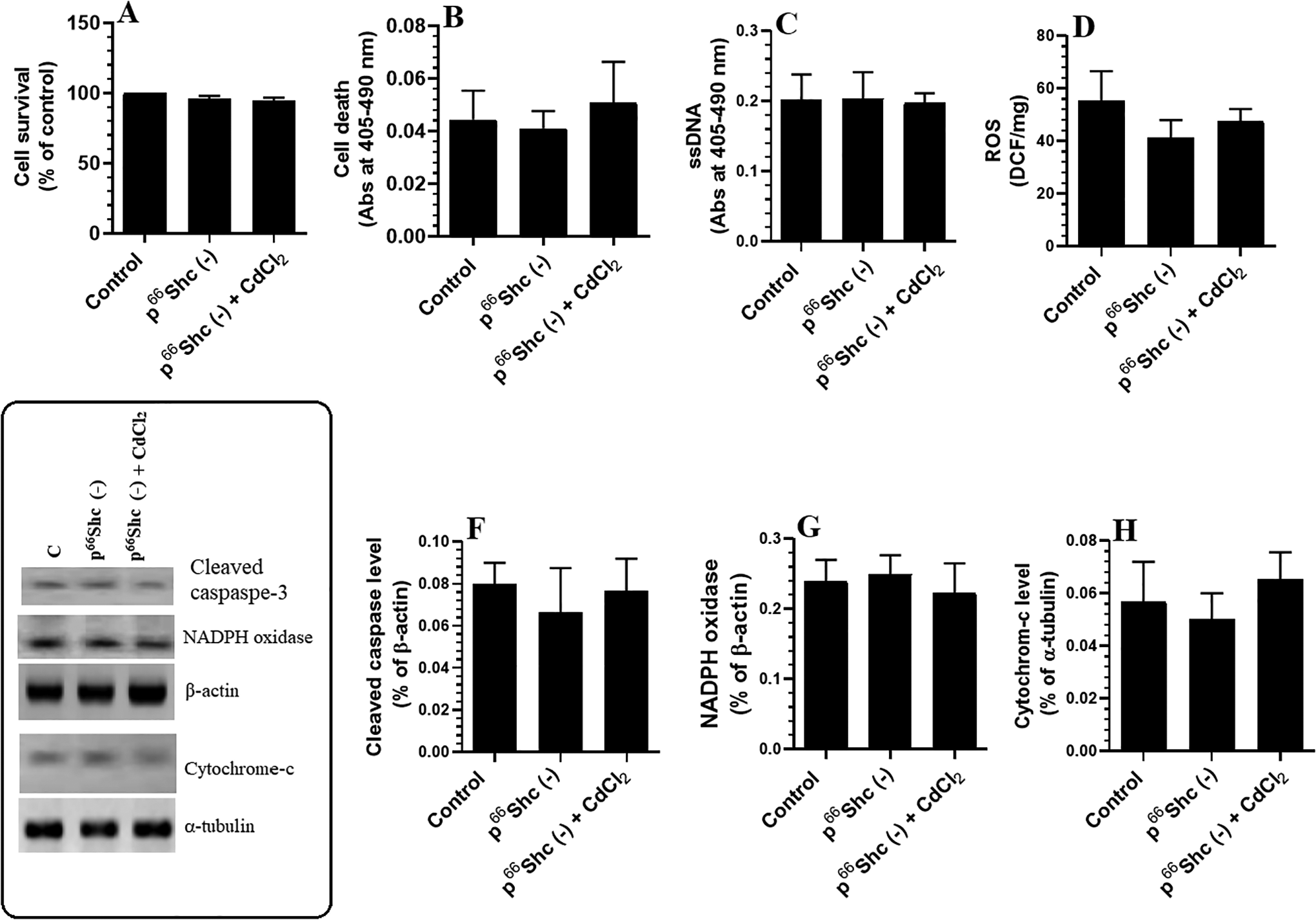
Levels of cell survival (A), cell death (B), single-stranded DNA (ssDNA, C), and reactive oxygen species (ROS, D), as well as protein levels of cleaved caspase-3 (F), nicotinamide adenine dinucleotide phosphate oxidase (G), and cytochrome C (H) in control or p66Shc knocked out cultured hippocampal cells. Cells were cultured in Dulbecco’s modified Eagle’s medium for 24 hours and treated with 25 μM cadmium chloride (CdCl2). Besides, some cells were also pretreated with 10 µM SP600125, a specific c-Jun N-terminal kinase inhibitor, and then were then incubated with the highest dose of CdCl2 (25 µM) for 24 hours. Control cells were treated only with dimethyl sulfoxide (0.1%) and received no CdCl2 (0.0 µM). Data were considered significantly different at P < 0.05. Data are presented as mean ± SD of n = 12 rats/group. aSignificantly different when compared with the 0.0 µM CdCl2-treated cells. bSignificantly different when compared with the 5 µM CdCl2-treated cells. cSignificantly different when compared with 10 µM CdCl2-treated cells. dSignificantly different when compared with 25 µM CdCl2-treated cells.
Discussion
The findings of this study indicate that p66Shc-mediated oxidative stress and apoptosis play a causative role in CdCl2-induced hippocampal damage and memory dysfunction. To the best of our knowledge, we are the first to show that the CdCl2 exposure is associated with sustained upregulation and activation of p66Shc in the rat hippocampus and in cultured hippocampal cells. In this study, p66Shc induced oxidative stress and apoptosis by increasing ROS generation, reducing the levels of the endogenous antioxidants GSH and MnSOD, upregulating NADPH oxidase, and stimulating cytochrome-c release. Notably, pretreating cultured hippocampal cells with SP600125, a specific JNK inhibitor, or silencing p66Shc prevented CdCl2-induced cell death, DNA damage, oxidative stress, and NADPH oxidase activation. Taken together, our data show that the CdCl2-induced activation of p66Shc is JNK-dependent.
In this study, although we used a CdCl2 dose that has previously been shown to primarily affect the brain, 31 it appears that other organs were also affected. Notably, 4 of the CdCl2-treated animals died, the body weight of the CdCl2-treated rats progressively reduced, and the levels of liver and kidney damage markers significantly increased. These data suggest that CdCl2 also induces systemic toxicity and support the findings of many other previous studies. 1,2,8,9,28,32 -43 Cadmium chloride has previously been reported to adversely affect the appetite and body weight of rats. These effects have been suggested to be a result of the systemic toxicity of Cd- or CdCl2-induced alterations in neurotransmitter levels and signaling in the hypothalamus. 35
The in vivo and in vitro neurotoxicity of CdCl2 is well established. Cadmium ions can cross the BBB and induce oxidative stress and inflammation in the brain. 1 -7 Notably, peripheral inflammatory cytokines and ROS can also cross the BBB and induce central inflammatory and oxidative stress responses leading to neurotoxicity in a variety of neurodegenerative disorders such as those triggered by obesity and ischemia. 36 -38 In addition, increased levels of peripheral markers of oxidative stress and inflammation are associated with neurodegeneration, dementia, and AD. 39 Therefore, although the oxidative stress and inflammatory response observed in the hippocampi of CdCl2-treated rats may be mediated by a direct effect of CdCl2 on the brain, as suggested by many authors, it is also possible that these effects may be secondary to the systemic toxicity/peripheral effects of CdCl2. In addition, it is likely that peripheral ROS and inflammatory cytokines facilitate the entry of CdCl2 into the central nervous system by altering the permeability of the BBB. However, several other studies have shown that CdCl2 can directly affect the brain and neural cells of rats independent of other tissue damage and peripheral factors. 1,6,7,28,40,41 Unfortunately, we could not confirm this in our study as it was beyond the scope of this article. Therefore, future research should be conducted to determine whether CdCl2-induced neurotoxicity is mediated via a direct or indirect route.
Oxidative stress and neuroinflammation promote neurodegeneration and memory deficits in a variety of neurodegenerative disorders by triggering apoptosis and impairing cholinergic function, long-term potentiation, and neural plasticity. 38,42 -45 These mechanisms are known to underlie memory dysfunction, neural damage, and apoptosis in the cerebral cortices, hippocampi, and other brain areas of CdCl2-treated rodents and promote cell death and apoptosis in CdCl2-treated cultured neurons. 2,3,5 -11,46 In this study, CdCl2-treated rats exhibited spatial, reference, and retention memory impairments. Indeed, many other studies have reported adverse effect of CdCl2 on memory function in rodents. 4,6,7,40 In addition, examination of the hippocampi of CdCl2-treated rats revealed a loss of pyramidal cells from the CA1 region and the existence of cells with shrunken and dark nuclei, which indicates cell apoptosis and explains the subsequent decline in rats’ memory function. Indeed, previous studies have shown that the increase in shrunk and dark pyramidal cells in the C1A area of rodent’s hippocampi is an indication of cell apoptosis due to ischemia and substantial abnormalities in the capillary walls of the BBB. 47 -49 Concomitantly, ROS, oxidative stress, inflammation, and mitochondria-mediated apoptosis were noted in the hippocampi of CdCl2-treated rats. Accordingly, our data confirmed that CdCl2-induced hippocampal damage and memory deficits are mediated, at least, by depleting endogenous antioxidants including MnSOD and GSH, activation of NF-κB, and increasing the release of cytochrome-c from the mitochondria, which support many other studies. 4,6,7,50 -53
Although we observed oxidative stress and inflammation in the hippocampi of CdCl2-treated rats, we were unable to determine whether oxidative stress triggers inflammation or vice versa. Previously, it was shown that ROS activate pro-inflammatory transcription factors and promote the synthesis and release of various inflammatory markers, thus inducing neuroinflammation and neurodegeneration. 54 -56 In addition, Cd ions induced inflammation and NF-κB activation in cultured midbrain neurons by promoting the generation of ROS. Furthermore, Cd ions stimulated neuroinflammation by increasing the levels of ROS. 52 However, scavenging ROS with pharmacological drugs or natural antioxidants protected the hippocampi and neural cells from CdCl2-induced neuroinflammation and apoptosis. Hence, these data suggest that oxidative stress is a key player in CdCl2-induced neurotoxicity in the rat hippocampus. However, the mechanism by which Cd ions induce ROS generation in the brains of rodents and humans is not completely understood, so elucidating this mechanism was a major aim of this study.
Previous studies have shown that the neurotoxic effect of Cd ions involves the activation of numerous pathways including the ERK, p38, JNK, and mTOR pathways. 5,53 However, it is possible that other unidentified mechanisms are also involved in the neurotoxic effect of Cd ions. Associated with the increased production of ROS and activation of intrinsic cell apoptosis in the hippocampi of CdCl2-treated rats, the novel finding of this study is that CdCl2 not only upregulated p66Shc and NADPH oxidase in the hippocampi of rats but also promoted the phosphorylation and activation of p66Shc. These effects were also observed in cultured cells alongside an increase in ROS levels and decrease in MnSOD levels. Moreover, in vivo and in vitro, CdCl2 significantly increased the activation of JNK, a well-known stimulator of p66Sch. These data suggest that the prooxidant and apoptotic effects of CdCl2 may be mediated via the activation of both JNK and p66Shc. Activation of CdCl2-induced p66Shc may account for the increase in ROS generation, mitochondrial damage, higher levels of NADPH oxidase, and increased release of cytochrome c observed in the hippocampi of CdCl2-treated rats. Therefore, these findings add to those previously reported in the literature and show that the neurotoxic effect of CdCl2 in the rat hippocampus involves activation of the JNK/p66Sch/NADPH oxidase axis.
To confirm these findings, we silenced both p66Shc-1 and p66Sch-2 in cultured fetal hippocampal cells. In addition, we incubated the cells with SP600125, a specific JNK inhibitor, prior to CdCl2 treatment. Interestingly, p66Shc silencing and JNK inhibition prevented CdCl2-induced cell death, DNA damage, ROS generation, cytochrome c release, NADPH upregulation, and cleaved caspase-3. In a previous study, p66Shc deletion prevented the systemic generation of ROS and increased the lifespan of rats by 30%. In addition, mouse embryonic fibroblasts derived from p66Shc−/− mice were protected from H2O2 and UV light-induced apoptosis. 17 Unfortunately, we did not measure the activity of PKCβ or p53 in the hippocampi or cultured cells and thus could not determine whether they are involved in the CdCl2-induced activation of p66Shc. However, given that JNK inhibition completely prevented all CdCl2-induced adverse events, it appears that Cd-induced hippocampal damage involves the activation of JNK .
Despite these findings, this study has some limitations. Given the large population of cells in the central nervous system, further studies are needed to investigate the expression of p66Shc in other cells such as glial cells, which express large quantities of this protein 13 and play a significant role in oxidative stress and inflammation. In addition, the major source for activation of JNK after CdCl2 exposure needs to be identified. Hence, the involvement of other peripheral factors such as peripheral oxidative stress and inflammatory cytokines in this process needs to be evaluated.
In conclusion, we have shown for the first time that CdCl2-induced memory deficits and hippocampal damage involve activation of the JNK/p66Sch/NADPH oxidase axis. Hence, targeting JNK, p66Shc, and NADPH oxidase may be a novel therapeutic strategy for patients with idiopathic dementia who have a history of Cd exposure.
Footnotes
Author Contributions
A. El-kott substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, drafted the manuscript, and critically reviewed the manuscript for important intellectual content. A. Alshehri contributed to acquisition, analysis, or interpretation of data and drafted the manuscript. H. Khalifa contributed to acquisition, analysis, or interpretation of data and critically reviewed the manuscript for important intellectual content. A. Abd-Lateif substantially contributed to conception or design, drafted the manuscript, and critically reviewed the manuscript for important intellectual content. M. Alshehri contributed to acquisition, analysis, or interpretation of data and drafted the manuscript. M. AbdEl-Maksoud contributed to acquisition, analysis, or interpretation of data and drafted the manuscript. R. Eid substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, drafted the manuscript, and critically reviewed the manuscript for important intellectual content. M. Bin-Meferij substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, drafted the manuscript, and critically reviewed the manuscript for important intellectual content. All authors gave final approval and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
All existing international, national, and institutional guidelines for animal care and use were followed.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University, Abha, Kingdom of Saudi Arabia for funding this work through the research group program (Grant No. R.G.P.1/46/40). This work was also funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-track Research Funding Program.