
Abstract
Mesalazine is widely used in the management of inflammatory bowel disease. Previous studies reported that mesalazine-induced cardiotoxicity is a rare, potentially fatal complication. Mitochondria play an important role in myocardial tissue homeostasis. Deterioration in mitochondrial function will eventually lead to cardiomyocyte death and consequently cardiovascular dysfunction. The aim of the current study was to investigate the effects of mesalazine on rat heart mitochondria. Rat heart mitochondria were isolated by mechanical lysis and differential centrifugation. Parameters of mitochondrial toxicity including succinate dehydrogenase (SDH) activity, reactive oxygen species (ROS) formation, mitochondrial membrane potential (MMP) collapse, mitochondrial swelling, and cytochrome c release were evaluated. Results revealed that mesalazine induced a concentration- and time-dependent rise in mitochondrial ROS formation, inhibition of SDH, MMP collapse, mitochondrial swelling, and cytochrome c release in rat heart mitochondria. These results indicate that the cardiotoxic effects of mesalazine are most likely associated with mitochondrial dysfunction and ROS formation, which finally ends in cytochrome c release signaling and induction of apoptosis.
Keywords
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder which is characterized by repetitive cycles of active and quiescent disease. Inflammatory bowel disease includes Crohn disease (CD) and ulcerative colitis (UC). 1 Crohn disease can affect any part of the gastrointestinal tract, whereas UC is limited to the colon. The features of CD are very heterogeneous and include abdominal pain, weight loss, and chronic diarrhea, whereas UC usually presents with tenesmus, rectal bleeding, along with urgency and bloody diarrhea. Both forms of IBD can involve other organs including the heart. 2 5-Aminosalicylic acid or mesalazine is widely used in the management of IBD. 3 Active disease, as well as maintenance of remission, by mesalazine reduces the risk of colorectal cancer by up to 75%. 4 Mesalazine is usually well tolerated by most patients due to its favorable safety profile. 5 Rarely, cardiac involvement including pericarditis and myocarditis (0.3%), fibrosing alveolitis, lupus erythematosus-like syndrome, blood disorders, nephrotoxicity, and pancreatitis have been reported after treatment with mesalazine. 6
The cardiovascular toxicity of drugs attracts remarkable attention from basic scientists to clinicians. 7 Today, cardiovascular toxicities and diseases are the most considerable determinants of morbidity and mortality in many countries. 8 Drugs causing heart rhythm disturbances can eventually result in impaired hemodynamic function of the heart 7 ; therefore, it is important to study any negative impact of drugs or toxins on cardiovascular system.
Mesalazine-induced cardiotoxicity is a rare but potentially fatal complication. Mesalazine-induced cardiotoxicity and mesalazine-induced death were reported in 1989 and 1990, respectively. 9,10 Cardiac events associated with mesalazine include myocardial infarction, conduction defects, ventricular dysfunction, and cardiomyopathy. 11 Cardiogenic shock owing to mesalazine-related cardiomyopathy has also been reported. 12 Over 100 cases of pericarditis and myocarditis due to mesalazine have been mentioned in the literature. 13 Mesalazine also induces hypersensitivity and myocardial inflammation in the treated individuals. Hypersensitivity is probably a result of accelerated metabolism of arachidonic acid to leukotrienes through cyclooxygenase-1 (COX1) enzyme inhibition. 14 The formation of antibodies against mesalazine due to humoral-mediated response is another mechanism of hypersensitivity. 15 The exact mechanism by which mesalazine induces pericarditis and myocarditis is not known. Mitochondria have an essential role in myocardial tissue homeostasis; thus, deterioration in mitochondrial function could result in cardiomyocyte and endothelial cell death and consequently cardiovascular dysfunction. 16 In this study, we investigated the effect of mesalazine on rat heart mitochondria as a possible mechanism of mesalazine cardiotoxicity.
Materials and Methods
Animals
Male Wistar rats (8-9 weeks old) weighting 250 to 300 g were purchased from the Pasteur Institute of Iran (Tehran, Iran). Animals were housed under standard conditions (temperature 22°C-21°C, humidity 50%-10%, 12-hour light–dark cycle, and free access to food and water). This study was approved by the Research Ethics Committee at the Shahid Beheshti University of Medical Sciences and performed strictly in accordance with institutional and international guide for animal care.
Chemicals
Trypan blue, 2′,7′-dichlorofuorescin diacetate (DCFH-DA), rhodamine123, bovine serum albumin, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES), 2,2′,2″,2‴-(Ethane-1,2-diyldinitrilo)tetraacetic acid (EDTA), sucrose, D-mannitol, Coomassie brilliant blue, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), 2-amino-2-hydroxymethyl-propane-1,3-diol (TRIS), 3-morpholinopropane-1-sulfonic acid (MOPS), ethylene glycol-bis(β-aminoethyl ether)-N, N, N′, N′-tetraacetic acid (EGTA), sodium succinate, monopotassium phosphate, magnesium chloride, potassium chloride, and rotenone were purchased from Sigma-Aldrich (St Louis, Missouri), and Cytochrome c Release Assay Kit was purchased from Abcam (Cambridge, United Kingdom). Mesalazine with a purity of about 98% and CAS number 89-57-6 was purchased from Dr. Abidi Pharmaceuticals Co (Iran). It was freshly prepared before use and dissolved in normal saline 0.9% wt/vol.
Preparation of Mitochondria
Rats were decapitated and the heart tissue was surgically harvested. The whole heart was cleared of blood, chopped, and homogenized with a glass homogenizer in a 10-fold volume of the medium containing 225 mM D-mannitol, 75 mM sucrose, and 0.2 mM EDTA at pH 7.4 in ice-cold bath. The homogenate was centrifuged at 1,000g for 10 minutes, and the pellet was removed. The mitochondria contained in the supernatant were sedimented at 10,000g for 10 minutes at 4°C. 17 The protein content in mitochondria was determined using the Bradford assay. 18 Protein concentration in the mitochondrial suspension samples was then adjusted to 1 mg/mL in the normalization process.
Measurement of Succinate Dehydrogenase Activity
The activity of succinate dehydrogenase (mitochondrial complex II) was assayed by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction in mitochondria isolated from rat heart. 19 Briefly, after incubation of mitochondrial suspensions with mesalazine (0, 25, 50, and 100 µM) at 30°C for 60 minutes and addition of MTT (0.4% wt/vol) to the medium, samples were incubated at 37°C for 30 minutes. Then, the purple formazan crystals were dissolved in DMSO and the absorbance was measured at 570 nm with an enzyme-linked immunosorbent assay (ELISA) reader (Tecan, Rainbow Thermo, Austria).
Quantification of Mitochondrial Reactive Oxygen Species Formation
After incubation of mitochondria isolated from rat heart with mesalazine (0, 25, 50, and 100 µM) in respiration buffer (0.32 mM sucrose, 10 mM TRIS, 20 mM MOPS, 50 μM EGTA, 0.5 mM MgCl2, 0.1 mM KH2PO4, 5 mM sodium succinate, and 10 µM DCFH-DA), mitochondrial H2O2 production was assayed by Schimadzou RF-5000U fluorescence spectrophotometer (Kyoto, Japan) in the period of 60 minutes (5, 30 and 60 minutes). Excitation and emission wavelengths were 485 and 530 nm, respectively. 20
Determination of Mitochondrial Membrane Potential Collapse
Rhodamine 123 as cationic fluorescent dye was used for the determination of mitochondrial membrane potential (MMP) collapse. 21,22 Briefly, mitochondria isolated from rat heart were suspended in MMP buffer including 220 mM sucrose, 68 mM D-mannitol, 10 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 50 μM EGTA, 5 mM sodium succinate, 10 mM HEPES, 2 μM rotenone, and 10 µM rhodamine123, then incubated with mesalazine (0, 25, 50, and 100 µM) at 30°C for 5 minutes. The fluorescence was measured using Schimadzou RF-5000U fluorescence spectrophotometer at the excitation and emission wavelength of 490 and 535 nm, respectively, in the period of 60 minutes (5, 30, and 60 minutes).
Determination of Mitochondrial Swelling
The change in mitochondrial volume, due to the colloidal osmotic effects of solute flux into and out of the mitochondrial matrix, was measured by monitoring the absorbance at 540 nm (A540). 23 Briefly, after incubation of mitochondria isolated from rat heart with mesalazine (0, 25, 50, and 100 µM) at 30°C for 5 minutes in swelling buffer (70 mM sucrose, 230 mM mannitol, 3 mM HEPES, 2 mM Tris-phosphate, 5 mM succinate, and 1 µM of rotenone), absorbance was measured at 540 nm at 60 minutes (5, 30, and 60 minutes) with an ELISA reader (Tecan, Rainbow Thermo). A decrease in the absorbance indicates an increase in mitochondrial swelling.
Determination of Cytochrome c Release
Mitochondria isolated from rat heart were incubated in 1.5-mL Eppendorf tubes at 37°C for 60 minutes. Inhibitor of mitochondrial permeability transition (MPT) pore, cyclosporine A, at the final concentration of 5 µmol/l and also antioxidant butylated hydroxytoluene (BHT) at the final concentration of 5 µM were added 15 minutes before the addition of mesalazine (50 µM). Following the incubation, tubes were centrifuged at 10,000g. The supernatant contained the cytochrome c released from the mitochondria into cytosolic fraction, and the pellet consisted of the mitochondrial fraction. The concentration of cytochrome c in the supernatant was determined by Cytochrome c Release Assay Kit according to supplier’s instruction with an ELISA reader (Tecan, Rainbow Thermo). 24
Statistical Analysis
All data were presented as mean ± standard deviation in 3 separate experiments. Data were analyzed by GraphPad Prism 5 (GraphPad Software, La Jolla, California) using 1- and 2-way analysis of variance followed by post hoc Tukey and Bonferroni tests, respectively. P value of less than 0.01 was considered statistically significant.
Results
Measurement of Succinate Dehydrogenase Activity
Mitochondrial complex II or succinate dehydrogenase enzyme activity was evaluated by the MTT test after 60-minute incubation of fresh and living mitochondria of the rat heart with different concentrations of mesalazine (0, 10, 25, 50, and 100 µM). As shown in Figure 1, after 60 minutes of exposure, a significant decrease in the mitochondrial metabolism of MTT to formazan (P < 0.01) was seen after addition of different concentrations of mesalazine. This assay showed that mesalazine at 25, 50, and 100 µM concentrations could cause a significant decrease in succinate dehydrogenase activity.
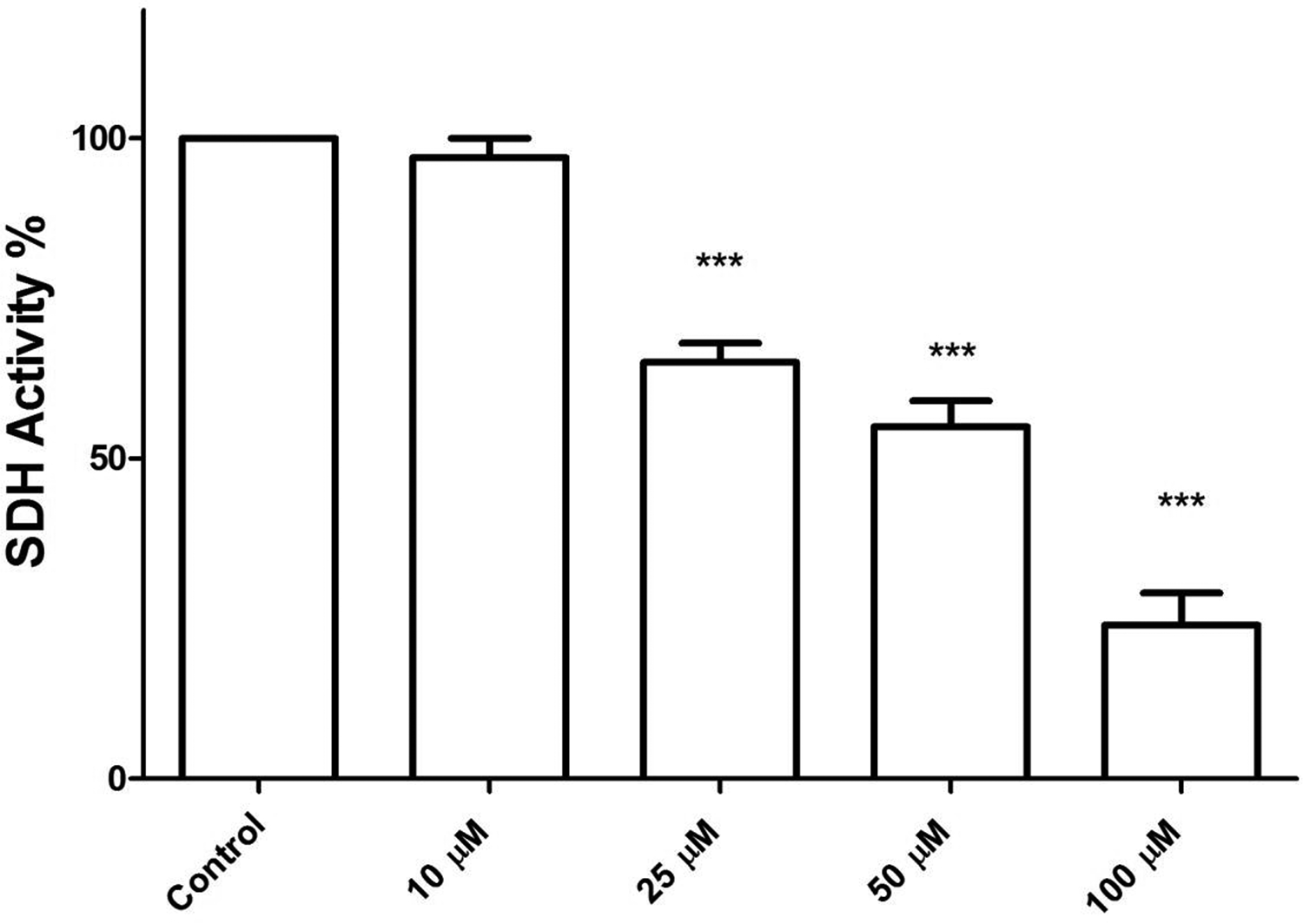
Effect of mesalazine on succinate dehydrogenase activity in rat heart isolated mitochondria. This figure demonstrates the effect of the mesalazine on succinate dehydrogenase activity isolated mitochondria. Mitochondrial succinate dehydrogenase activity was measured by 4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay within 1 hour after mesalazine exposure. Mitochondrial succinate dehydrogenase activity was significantly decreased (P < 0.01) by mesalazine in comparison to control. Values were expressed as mean ± standard deviation of 3 separate determinations. ***A significant difference (P < 0.01) from the control group.
Quantification of Mitochondrial Reactive Oxygen Species Formation
As shown in Figure 2, mesalazine (25, 50, and 100 µM) induced significant increase in the reactive oxygen species (ROS; H2O2) formation in rat heart mitochondria over 60 minutes of exposure (5, 30, and 60 minutes). Mesalazine-induced mitochondrial ROS formation was concentration and time dependent. Hydrogen peroxide (H2O2) was used as a positive control (100 µM).
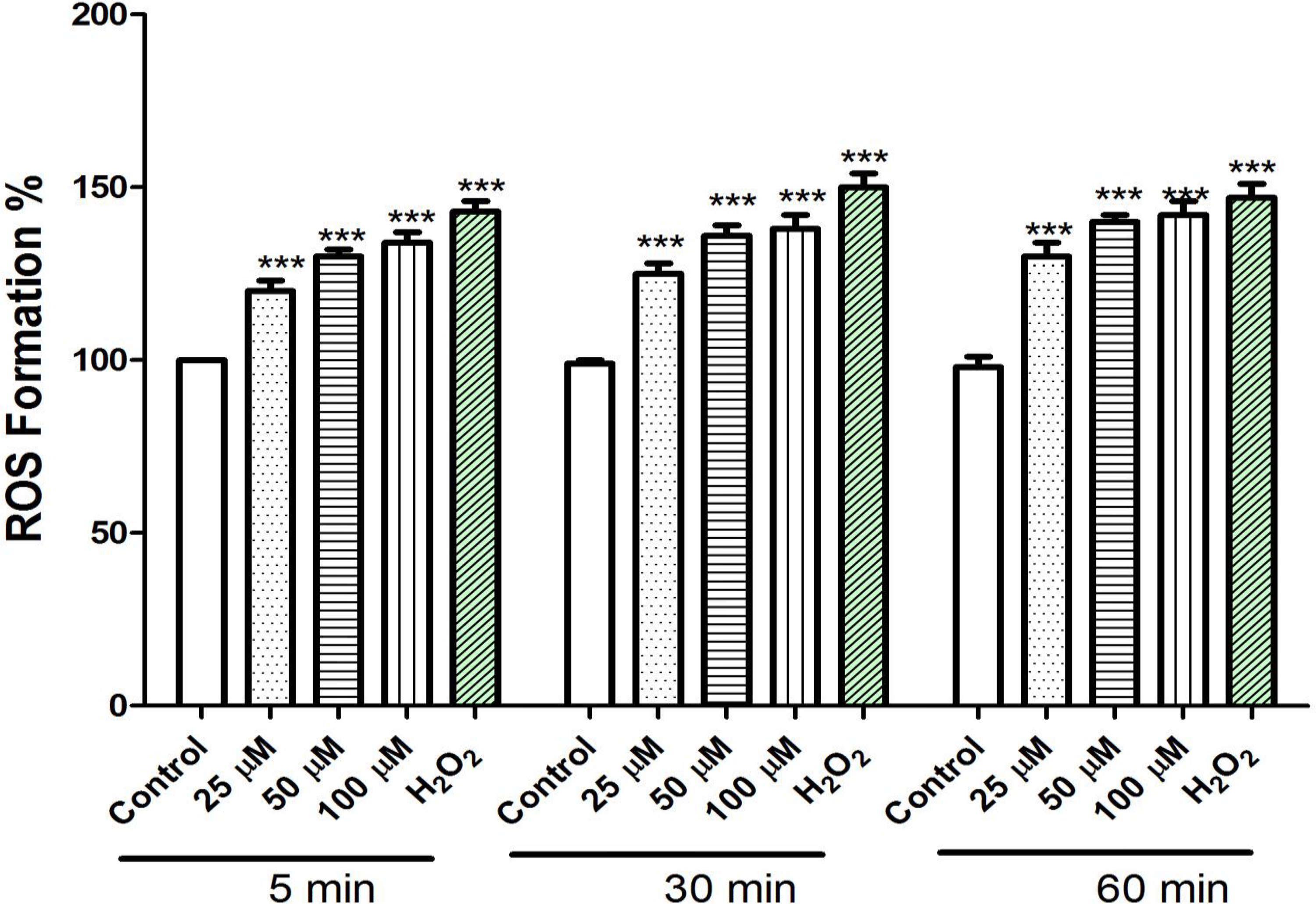
Effect of mesalazine on reactive oxygen species formation in isolated rat heart mitochondria. Freshly isolated mitochondria were incubated with the various concentrations of mesalazine (25, 50, and 100 µm) for 1 hour. Reactive oxygen species was measured by 2′,7′-dichlorofuorescin diacetate staining with spectrofluorescence method. The ROS formation percentage was significantly increased (P < 0.01) by mesalazine in comparison to control. Hydrogen peroxide (H2O2) was used as a positive control (100 µM). Values were expressed as mean ± standard deviation of 3 separate determinations. ***A significant difference (P < .01) from the control group.
Determination of MMP Collapse
Rhodamine123, as a cationic fluorescent dye, was used for the evaluation of MMP collapse. As shown in Figure 3, different concentrations of mesalazine (25, 50, and 100 µM) significantly increased rhodamine123 release from rat heart mitochondria over 60 minutes of exposure (5, 30, and 60 min). Also, mesalazine at 100 µM significantly increased rhodamine123 release from mitochondria (P < 0.0001). The rhodamine123 release is an indicator of MMP collapse. CaCl2 (100 µM), as a known inducer of MPT, was used as a positive control.
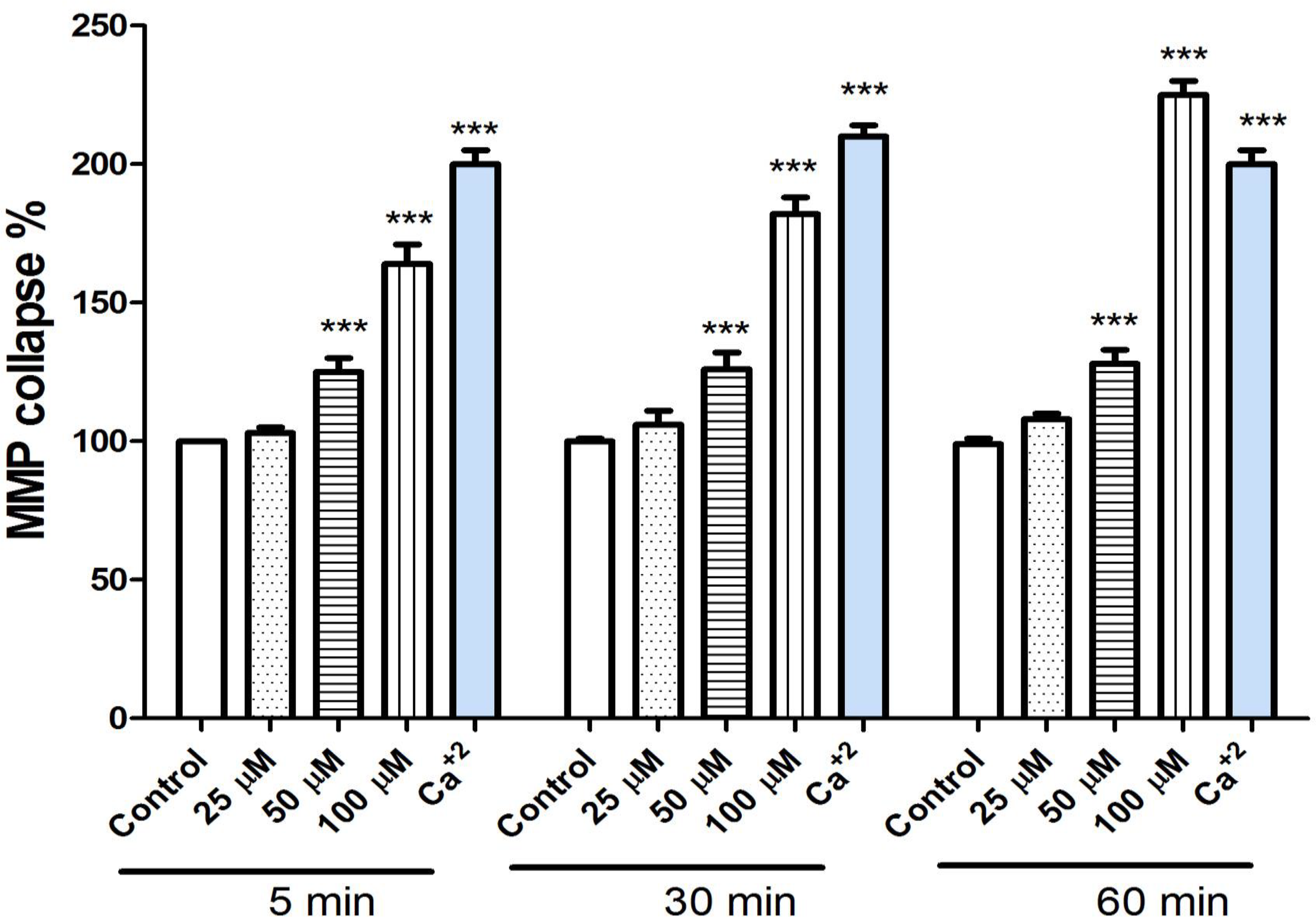
Effect of mesalazine on ΔΨm in isolated rat heart mitochondria. Freshly isolated mitochondria were incubated with various concentrations of mesalazine (25, 50, and 100 µM) for 1 hour. ΔΨm was measured spectrophotometrically by rhodamine 123 staining. Mesalazine induced a decrease in ΔΨm rat heart isolated mitochondria. Values were expressed as mean ± standard deviation of 3 separate determinations. ***A significant difference (P < 0.01) from the control group.
Determination of Mitochondrial Swelling
Mitochondrial swelling as a criterion of mitochondrial membrane permeability was measured by monitoring the decreased absorbance of mitochondrial suspension at 540 nm (A540). As shown in Figure 4, addition of different concentrations of mesalazine (25, 50, and 100 µM) to the rat heart mitochondria over 60 minutes of exposure (5, 30, and 60 min) leads to mitochondrial swelling in a concentration- and time-dependent manner. CaCl2 (100 µM), a known inducer of MPT, was used as a positive control.
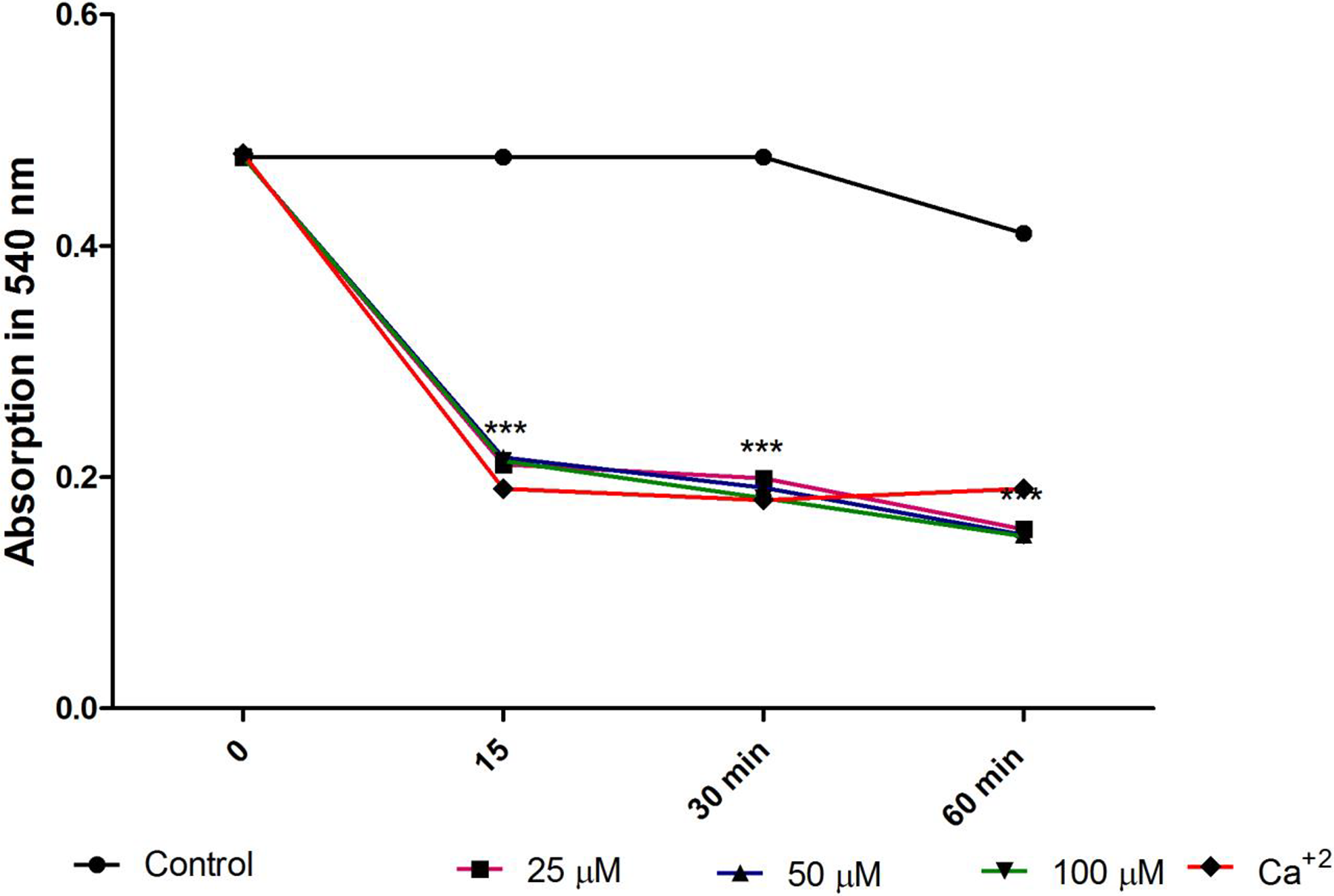
Effect of mesalazine on mitochondrial swelling in isolated rat heart mitochondria. Mesalazine at various concentrations (25, 50, and 100 µM) induced mitochondrial swelling in a concentration-dependent manner. Mitochondrial swelling was monitored by following 540 nm absorbance decrease. Values were expressed as mean ± standard deviation of 3 separate determinations. ***A significant difference (P < 0.01) from the control group.
Determination of Cytochrome c Release
Our previous results showed that mesalazine significantly caused collapse of the MMP and mitochondrial swelling. These events could result in MPT and release of cytochrome c from mitochondria. As shown in Figure 5, mesalazine (50 µM) induced significant (P < 0.01) release of cytochrome c in the rat heart mitochondria. Pretreatment of the rat heart mitochondria with BHT, as an antioxidant, and cyclosporine (Cs. A), as an MPT inhibitor, inhibited mesalazine- (50 µM) induced cytochrome c release (P < 0.01), indicating the key effect of oxidative stress and MPT pore opening in the mesalazine-induced mitochondrial toxicities.
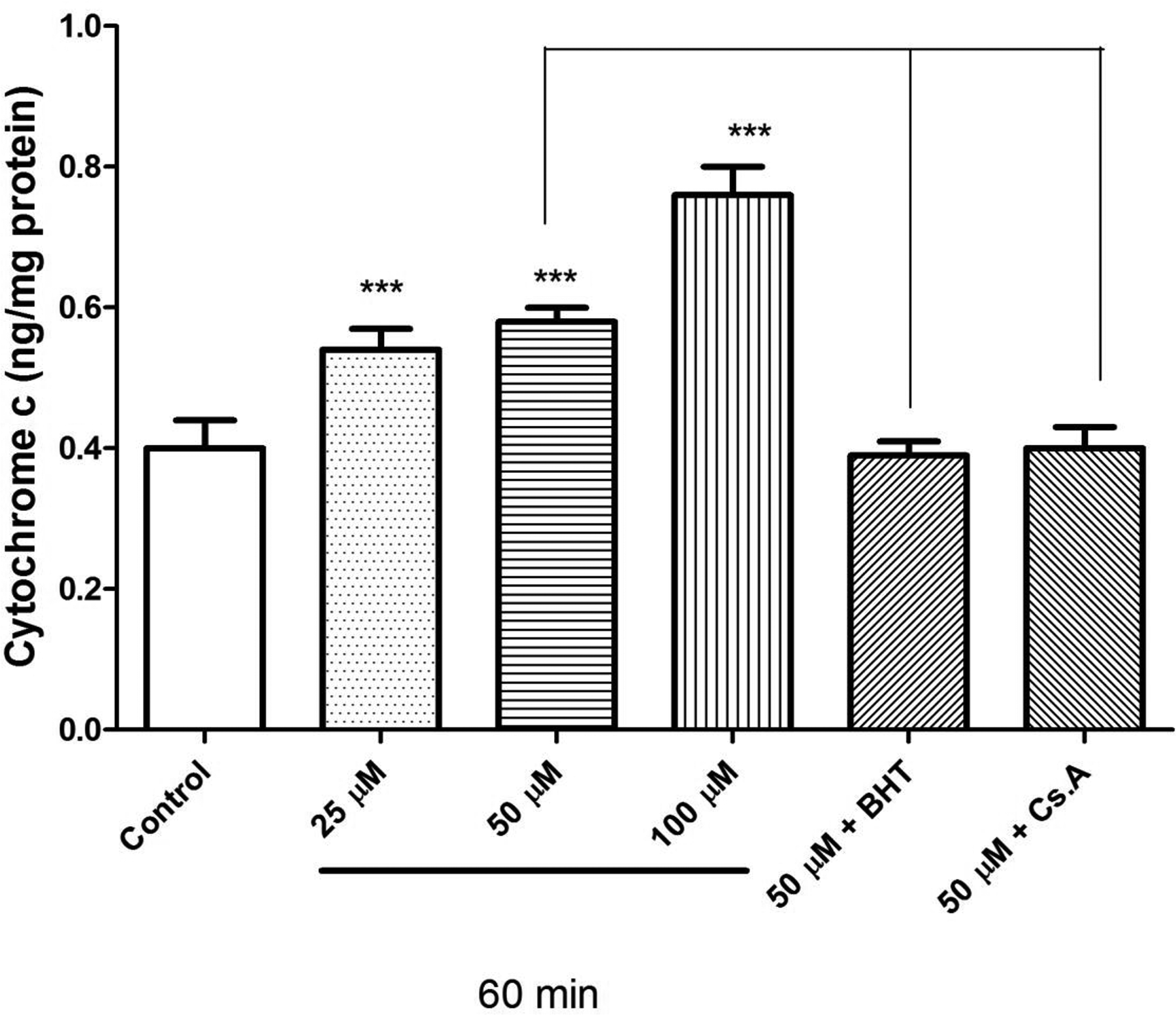
Effect of mesalazine on the cytochrome c release in isolated rat heart mitochondria. As shown in this figure, treatment with mesalazine significantly induced cytochrome c release. The amount of expelled cytochrome c from the mitochondrial fraction into the suspension buffer was determined using human cytochrome c enzyme-linked immunosorbent assay kit. Values were expressed as mean ± standard deviation of 3 separate determinations. ***A significant difference (P < .01) from the control group.
Discussion
Adverse cardiac manifestation in patients diagnosed with IBD undergoing therapy with mesalazine is infrequent. When cardiotoxicity occurs, it can be difficult to recognize the primary cause, as both mesalazine and IBD can cause cardiotoxicity separately from each other. The main mechanism of mesalazine-induced cardiotoxicity is unknown, but there are several possible mechanisms in this regard. 25 One hypothesis is inhibition of COX enzyme by mesalazine and accelerated metabolism of arachidonic acid through lipoxygenases. Inhibition of COX leads to the overproduction of cytokines and initiating a hypersensitivity reaction. 26 Another suggested mechanism is antibodies formed against mesalazine that react with cardiac tissues. 27 Antibodies formed against mesalazine cross-react with cardiac tissue causing inflammation. 26 Direct toxic effect of mesalazine on the myopericardium is another hypothesized mechanism, which occurs through a cell-mediated hypersensitive reaction and an allergic reaction by immunoglobulin E. 15 For the first time, in the current study, we investigated the toxic effects of mesalazine on rat heart mitochondria. Our results confirmed that mesalazine causes mitochondrial toxicity through ROS formation (Figure 2) most likely leading to MMP collapse (Figure 3), mitochondrial swelling (Figure 4), release of cytochrome c (Figure 5), and finally mitochondrial dysfunction.
Mesalazine is a major metabolite of sulfasalazine, which is generally used for the treatment of IBD and rheumatoid arthritis. 28 Around 90% of a dose of sulfasalazine reaches the colon, where most of it is metabolized by bacteria into mesalazine and sulfapyridine. Sulfasalazine therapy is responsible for the majority of the side effects, whereas mesalazine is known to be the active moiety in the treatment of UC. 29 A previous study showed that sulfasalazine caused oxidative stress and renal injury. 30 Also, some studies have shown mesalazine administration is associated with renal injury. 31,32 Similar to the present findings, mesalazine treatment in vivo showed decreased kidney mitochondrial SDH activity, mitochondrial glutathione (GSH) depletion, mitochondrial depolarization, increase in mitochondrial swelling mitochondrial ROS, and lipid peroxidation in treated animals. 33 Isolated mitochondria, an in vitro system, were recently proposed to be a useful tool to predict drug-induced cardiotoxicity in humans, and previous results indicated that the performance of mitochondrial assays for the prediction of cardiotoxicity is 33%. 34 Mitochondrial toxicity is caused by several mechanisms, such as interference with the important mitochondrial enzymes and the inhibition of mitochondrial respiratory chain. The possible final step in mitochondrial dysfunction involves the loss of MMP and an increase in mitochondrial oxidative/nitrative stress and cytochrome c release, eventually culminating in cell death. 16,35 Our results indicate that mesalazine causes mitochondrial swelling, permeabilizing of the mitochondrial membrane, and cytochrome c release as the “point of no return” in the cascade of events leading to apoptosis 36 in isolated rat mitochondria.
Mitochondria are a major source of ROS production. 37 Previous published works demonstrated that chronic tumor necrosis factor (TNF) administration 38 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) blockade induce mitochondrial damage by altering mitochondrial respiration and decreasing ATP production. It has been demonstrated that NF-κB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. 39 Also, other work shows mesalazine inhibits activation of transcription factor NF-κB and TNF growth in inflamed mucosa of patients with UC and mouse colonocytes. 40,41 Our results in the current study on ROS formation and mitochondrial damage are consistent with mesalazine effect on inhibition of transcription factor NF-κB, where inhibition of NF-κB increase oxidative stress and mitochondrial damage. However, the underlying mechanisms by which NF-κB and TNF can contribute to oxidative stress are unclear and require further investigation. Finally, the results obtained from this study indicate that mesalazine increases ROS formation in isolated rat heart mitochondria and leads to mitochondrial dysfunction. Although the findings of this study are in isolated mitochondria, further studies in cellular, animal, and clinical trials are recommended. Results of this study suggest that the combination therapy of mesalazine with other antioxidant and mitochondrial protective agents could decrease cardiotoxicity effects of this drug.
Footnotes
Authors’ Note
The data provided in this article were extracted from the PharmD thesis of Dr Farnaz Bahreini. The thesis was conducted under supervision of Prof. Jalal Pourahmad at Department of Toxicology and Pharmacology, Faculty of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Author Contributions
A. Salimi contributed to interpretation of data and critically revised the manuscript for important intellectual content. F. Bahreini contributed to acquisition of data and drafted the manuscript. Z. Jamali contributed to analysis of data and drafted the manuscript. J. Pourahmad substantially contributed to conception or design and critically revised the manuscript for important intellectual content. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Shahid Beheshti University of Medical Sciences, Deputy of Research.