
Abstract
Alternative methods and their use in planning and conducting toxicology experiments have become essential for modern toxicologists, thus reducing or replacing living animals. Although in vitro human co-culture models allow the establishment of biologically relevant cell–cell interactions that recapitulate the tissue microenvironment and better mimic its physiology, the number of publications is limited specifically addressing this scientific area and utilizing this test method which could provide an additional valuable model in toxicological studies. In the present study, an in vitro model based on central nervous system (CNS) cell co-cultures was implemented using a transwell system combining human neuronal cells (SH-SY5Y cell line) and glial cells, namely astrocytes (D384 cell line), to investigate neuroprotection of D384 on SH-SY5Y and vice versa. The model was applied to test acute (24-48 hours) cytotoxicity of 3 different neurotoxicants: (1) methyl mercury (1-2.5 μM), (2) Fe3O4 nanoparticles (1-100 μg/mL), and (3) methylglyoxal (0.5-1 mM). Data were compared to mono-cultures evaluating the mitochondrial function and cell morphology. The results clearly showed that all compounds tested affected the mitochondrial activity and cell morphology in both mono-culture and co-culture conditions. However, astrocytes, when cultured together with neurons, diminish the neurotoxicant-induced cytotoxic effects that occurred in neurons cultured alone, and astrocytes become more resistant in the presence of neurons. This human CNS co-culture system seems a suitable cell model to feed high-throughput acute screening platforms and to evaluate both human neuronal and astrocytic toxicity and neuroprotective effects of new and emerging materials (eg, nanomaterials) and new products with improved sensitivity due to the functional neuron–astrocyte metabolic interactions.
Keywords
Introduction
The application of an Integrated Approach for Testing and Assessment to test substances, 1,2 to fulfil the standard requirements of REACH (Registration, Evaluation, Authorisation, and Restriction of Chemicals) and the need of nonanimal methods in order to reduce the in vivo test according to the 3Rs, 3,4 ask for the development of new strategies and predictive models, including simple in vitro systems with single cell types, in vitro co-cultures, and complex 3-dimensional in vitro models.
Coculture systems are in vitro models that mimic in the best way the in vivo anatomical conditions to achieve a significant level of mimicry of in vivo tissue, including at least 2 cell types in order to obtain the spatiotemporal context of the original tissue. In many different studies, co-culture systems have been proven to react in a more realistic way and to be more predictive of the in vivo response. 5
In vitro co-culture models of brain, in particular human co-culture models, allow the establishment of biologically relevant cell–cell interactions that recapitulate the tissue microenvironment and better mimic the physiological conditions of this organ, whereby neurons are surrounded by glial cells. For this reason, co-cultures have gained increased attention as tools for neuroprotection assessment.
Astrocytes, in either mono-culture or co-culture with neurons, are a powerful tool for studying the underlying mechanisms driving the neuroprotective effects 6 –9 and cell type–specific interactions in the central nervous system (CNS). Astrocytes are known to regulate the activities of other normal CNS components modulating brain function and response to injury. 10 –13
Our study sought to set up a co-culture system (cell types separated by membrane) with glial cells namely astrocyte (human astrocytoma-D384 cell line) and neuronal cells (human neuroblastoma-SH-SY5Y cell line) for evaluating whether this human neuron–astrocyte co-culture can represent a more predictable cell model for obtaining acute cytotoxicity information on human CNS after chemical and new product exposure.
For this purpose, transwell co-culture system of SH-SY5Y and D384 cells, which permits the free exchanges of soluble factors without allowing any physical contact between the 2 cell populations, was adopted to study the best neuroprotection of astrocyte cells on neurons and vice versa after exposure to 3 different neurotoxic agents, used as model compounds, such as (1) methyl mercury (MeHg) as an appropriate model for neurotoxicity because of its well-known primary toxicity to the human CNS 14,15 ; (2) iron oxide nanoparticles (ie, magnetite [Fe3O4NP]) since they have attracted extensive interest due to their superparamagnetic physicochemical properties in the biomedical (ie, brain-targeted drug or gene delivery, magnetic resonance imaging, contrast agents, etc) and industrial fields (ie, audio speakers, position sensing, catalysis and magnetic storage, and water purification), 16 –19 although their effects on CNS are still unclear; and (3) methylglyoxal (MGO) as ubiquitous product of cellular metabolism inevitably formed as a by-product of glycolysis, therefore present in brain cells, and which is one of the most potent glycating agents present in cells making its accumulation highly deleterious. 20 For instance, MGO readily reacts with lipids, nucleic acids, and lysine and arginine residues of proteins to form advanced glycation end products (AGEs) that are associated with several pathologies including diabetes, aging, and neurodegenerative diseases. 20 –22
In the present study, human astrocyte-D384 cells and the human neuronal-SH-SY5Y cells were included, for the first time, into the co-culture model. To better understand the cross-talk between glial and neuronal cells, as it happens in in vivo situation, D384 and SH-SY5Y cells were cultured in different experimental settings: alone and transwell co-culture system.
After exposure of the individual cell lines or co-cultures to MeHg, Fe3O4NPs, and MGO for 24 and 48 hours, mitochondrial function was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and cell morphology by contrast-phase microscopy.
The findings of the present study provide further support for other published literature 6,7,23 –27 demonstrating the protective/ameliorative effects to toxins/toxicants of coculturing neuronal–astrocyte cells, and specifically does so in a transwell/restricted contact model (as applied in this study), for which there are limited other publications, 6,25,28 as well doing so using human-derived cells 8,9,29 instead of rodent-derived cells.
Materials and Methods
Materials
Human astrocytoma (D384 clonal cell line established from Balmforth et al 30 ) and human neuroblastoma (SH-SY5Y cell line, ECACC; Sigma-Aldrich, Milan, Italy) cells were grown in 75 cm2 tissue culture flask and in 6-well plates (for the mono-cultures) or in transwell 6-well plates with insert porous polyester membrane (pore size: 0.4 µm; for the co-cultures; Corning) purchased from VWR (Milan, Italy). Fetal bovine serum (FBS), culture medium, and all cell culture reagents were purchased from Carlo Erba Reagents S.r.l. (Cornaredo, Italy). Chemicals, MGO solution (∼40% in H2O), and methylmercury hydroxide were obtained from Sigma-Aldrich. Polyvinylpyrrolidone-coated Fe3O4 nanoparticles (Fe3O4NPs) were obtained from nanoComposix (San Diego, California; lot no MGM1837B) and physicochemical characteristics of the Fe3O4NP suspension in aqueous 2 mM citrate were provided by the company. Briefly, morpho-dimensional analysis of Fe3O4NP stock suspension by transmission electron microscopy indicated roughly spherical almost nonagglomerated particles. Size distribution by dynamic light scattering measurements showed an average diameter of 19.07 (±5.5) nm. For more specific details on physicochemical characterization of Fe3O4NP in Dulbecco modified Eagle medium (DMEM), see the study by Coccini et al. 31
Monoculture System
Cryopreserved D384 and SH-SY5Y cells were thawed at 37°C up to 120 seconds and then cultured following the appropriate culture condition. Specifically, SH-SY5Y cells were cultured in Eagle minimum essential medium and Ham F12 (1:1) with 15% (vol/vol) heat-inactivated FBS, 2 mM
Cell Coculture Model in a Transwell Plate System
To test the hypothesis that glial cells will exert a protective effect on neuronal cells exposed to toxic agents, co-cultures of neuronal SH-SY5Y and astrocytic D384 cells were grown in a transwell system with a 0.4 μm pore size (4 × 106 pores/cm2). Neuronal SH-SY5Y cells (2.5 × 105 cells) were seeded in the lower compartment of a 6-well transwell system, whereas astrocytic D384 cells (2.5 × 105 cells) were cultured in the insert (Figure 1). Cells were maintained in DMEM supplemented with 10% (vol/vol) heat-inactivated FBS, 50 IU/mL penicillin and 50 μg/mL streptomycin, 2 mM
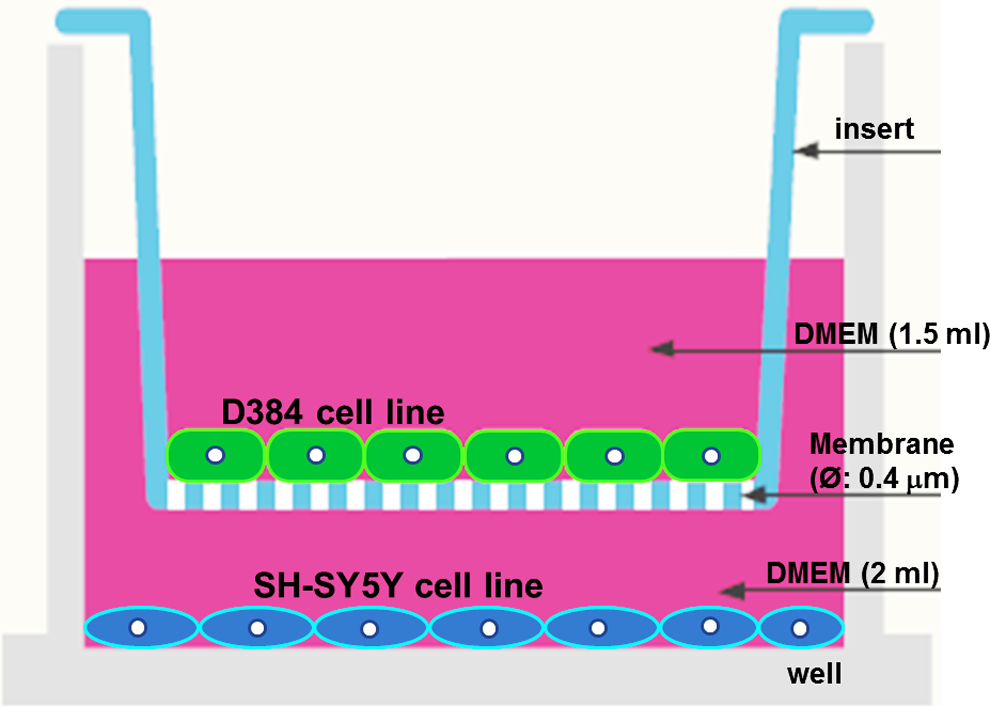
Six-well transwell system. Human neuroblastoma-SH-SY5Y cells were seeded in the lower compartment of a 6-well transwell system, whereas human astrocytoma-D384 cells were cultured in the insert.
The procedure for coculturing D384 and SH-SY5Y cells is shown in Figure 2. At first, astrocyte (D384 cells) and neuronal cells (SH-SY5Y cells) were prepared and maintained separately in mono-cultures (as described in “Monoculture System” paragraph); subsequently, D384 and SH-SY5Y cells were combined in co-cultures at the ratio of 1:1 (2.5 × 105 cells) in complete DMEM. Neuronal SH-SY5Y cells were seeded in the lower compartment of a 6-well transwell system in 2 mL of complete DMEM (Figure 2, step 1). The insert was equilibrated for 6 hours at 37°C, prior to seed the astrocytes, to improve cell attachment by adding 1 mL of complete DMEM medium to the insert (Figure 2, step 1). After this period, the medium was carefully removed from insert and D384 cells were then seeded over the porous membrane of the insert in fresh medium (1.5 mL DMEM) and transferred to the neuronal plates and returned to the incubator for 27 hours (Figure 2, step 2). After this period, both cells co-cultured in transwell system were exposed to toxicants (Figure 2, step 3).
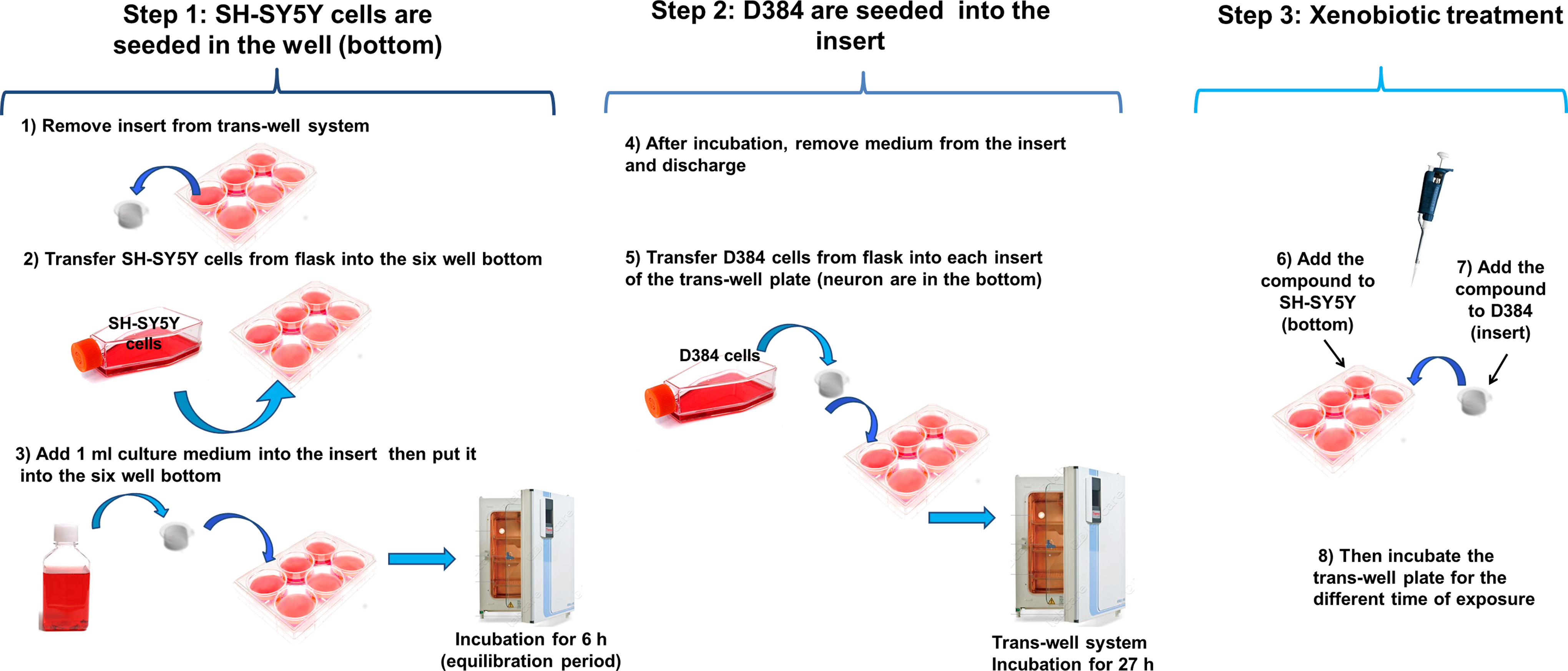
Human coculture model of neuronal and astrocyte cells. Step 1: Plate SH-SY5Y cells in the lower compartment of a 6-well transwell system. Step 2: Plate D384 cells over the porous membrane of the insert. Step 3: Exposure of D384 and SH-SY5Y cocultured in transwell system to toxicants.
Experimental well setup details were 16.8 mL of total volume/well, 9.5 cm2 of growth area/well, 2 mL of working volume/well, and 200 µL treatment volume/well. Experimental insert setup details were 24 mm of diameter, 4.67 cm2 of membrane growth area, 1.5 mL of volume added to inside of insert, and 150 µL treatment volume/well.
After treatments using this co-culture system, both astrocyte and neuronal cells were physically separated and each component was examined for cell morphology and mitochondrial function evaluations.
Chemical Treatments
The neurotoxicants were added to both sides of the transwell system (Figure 2, step 3). Fresh solutions of all compounds were prepared immediately before use.
Methyl mercury
This neurotoxic compound was used as a reference compound since several results have already been published applying mono-culture approach, 14,32 –35 and few with co-culture approach. 6,7
Methyl mercury concentrations of 1 and 2.5 μM were applied based on its cytotoxicity to cerebellar granular cells after 1-hour exposure, causing a 50% reduction in cell viability, respectively, as evaluated by MTT test. 32 In SH-SY5Y neurons, the lethal concentration 50 (LC50) of MeHg (evaluated by MTT) after 24-hour exposure was reported to be about 7 μM, 33 and in a more recent investigation, when SH-SY5Y cells were treated with MeHg for 24 hours, approximately 40% of cells died following 1 μM MeHg treatment and almost all cells died after 10 μM MeHg. 35
Methyl mercury stock solution (2.5 mM) was prepared by dissolving the powder in dimethyl sulfoxide (DMSO). From the stock solution, appropriate work dilutions were made in DMEM growth medium containing 10% FBS. The cells were exposed to different increasing MeHg concentrations (from 1-2.5 μM) for 24 and 48 hours.
Methylglyoxal
From an MGO stock solution (5.55 M), appropriate working dilutions were made in complete DMEM medium. Then cells were exposed to different increasing MGO concentrations (from 0.5 to 1 mM) for 24 and 48 hours.
Fe3O4NPs
Stock solution (20.6 mg/mL) was vortexed for 120 seconds at full speed before dilution into cell culture medium (DMEM). Cells were exposed to concentrations ranging from 1 to 100 μg/mL for 24 and 48 hours. Fresh suspensions of Fe3O4NPs were prepared and vortexed immediately before each use.
Mitochondrial Function Evaluation by MTT Test
The MTT assay was performed to evaluate the cytotoxicity induced by each neurotoxic agent. The choice of this cytotoxic test was based on the fact that it has long been regarded as the gold standard of cytotoxicity assays as it is highly sensitive and has been scaled for use as a high-throughput screening assay. This assay measures cell viability in terms of reductive activity as enzymatic conversion of the tetrazolium compound into water insoluble formazan crystals by dehydrogenases occurring in the mitochondria of living cells. 36
At the end of the each incubation period, the co-culture systems were processed as follows: The inserts containing the astrocytes were removed from co-culture system and transferred into a new 6-well plate with 1 mL/well of complete culture medium, then 0.5 mg/mL MTT was added to both the insert (astrocytes) and the bottom well containing neurons (Figure 3, step 1). Next, the 2 plates: the one plate plus insert (with astrocytes) and the other one with the bottom well only (with neurons) were incubated for 3 hours at 37°C to evaluate the astrocyte and neuron cell viability (Figure 3, step 2). After incubation, the culture medium was removed by aspiration from insert (top) and well (bottom), and the resulting formazan crystals were solubilized by DMSO solution (1 mL/well), and both cell types were gently shaken for 2 minute to mix the blue reaction product uniformly with the solvent (Figure 3, step 3). Subsequently, 125 μL of the colored DMSO was transferred from each insert (top) and each well (bottom) to the new 96-well plate for the quantification of cell viability by measuring absorbance at 550 nm (measurement) and 655 nm (reference) using a microplate reader (Bio-Rad, Segrate, Milan, Italy). Data were expressed as a percentage of control (Figure 3, step 4). In parallel, MTT evaluation was performed on mono-cultures (D384 and SH-SY5Y) treated with the same neurotoxic agents at the same concentrations applied to the co-culture system.
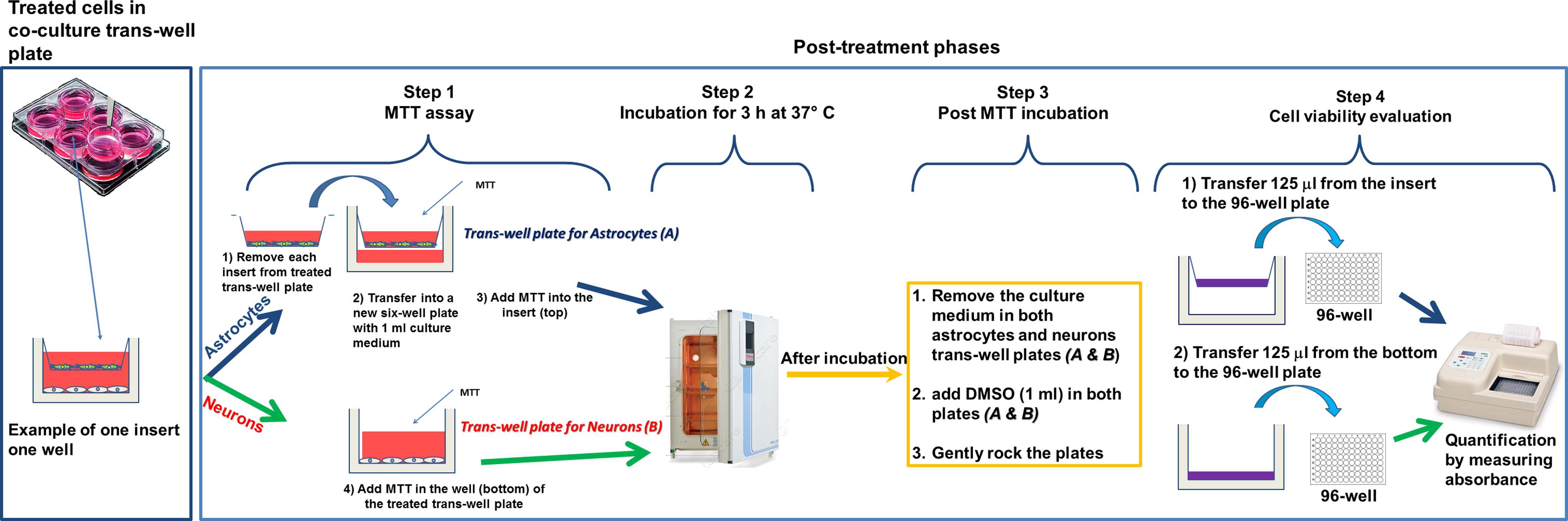
Post-treatments phases: Mitochondrial function evaluation by MTT assay. Step 1: MTT addition; step 2: incubation of SH-SY5Y (in well) and D384 (in insert) for 3 hours at 37 °C; step 3: DMSO solubilization of formazan crystals; step 4: cell viability evaluation. DMSO indicates dimethyl sulfoxide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Cell Morphology Evaluation by Phase-Contrast Microscopy
Both cell types (D384 and SH-SY5Y) in mono-culture or co-culture systems were observed in phase-contrast microscopy after MeHg (1-2.5 μM), MGO (0.5-1 mM), and Fe3O4NPs (0.05-100 μg/mL) exposure for 24 and 48 hours in order to obtain a better understanding of the effects on cell adherence, cell morphology, and culture growth. The cells were examined under a Zeiss Axiovert 25 microscope using phase-contrast objectives (×32 magnification) and combined with a digital camera (Canon powershot G8, Arese, Italy). Digital photographs were taken from each well and stored on the PC.
Statistical Analysis
Results are expressed as mean (±standard deviation; SD) of 3 independent experiments each carried out in 2 replicates. Lethal concentration 50 values (with 95% confidence limits) were calculated using the GraphPad Prism software. Statistical analysis was performed by 2-way analysis of variance followed by Dunnett test to compare the differences between mono-culture and co-cultures of each cell type (eg, D384 mono-culture versus D384 in co-culture) exposed to neurotoxic compounds at each time point. A value of P < 0.05 was considered statistically significant.
Results
Mitochondrial Function and Phase-Contrast Microscopic Evaluation
The DMEM, typically used for growing D384 cells, was also applied for the co-culture system. A preliminary experiment was performed to evaluate the SH-SY5Y cell proliferation in this medium instead of the Eagle minimum essential medium and Ham F12 (1:1) typically used for neuron cultures. Since no cell proliferation differences were observed between the cells grown on these 2 different media (data not shown), DMEM was employed for the co-culture system.
Methyl mercury data
In Figure 4, it can be observed that neurons in co-culture tolerated greater than 1.75 μM MeHg concentration compared to mono-cultures. In the present study, the mitochondrial activity in mono-cultured SH-SY5Y was decreased in a concentration-dependent manner: 30% to 75% at concentrations ranging from 1.75 to 2.5 μM after 24 hours (Figure 4, A1). The reduction in viability was not further exacerbated after 48 hours (15%-75% reduction), although the effects started at 1.5 μM (Figure 4, B1).
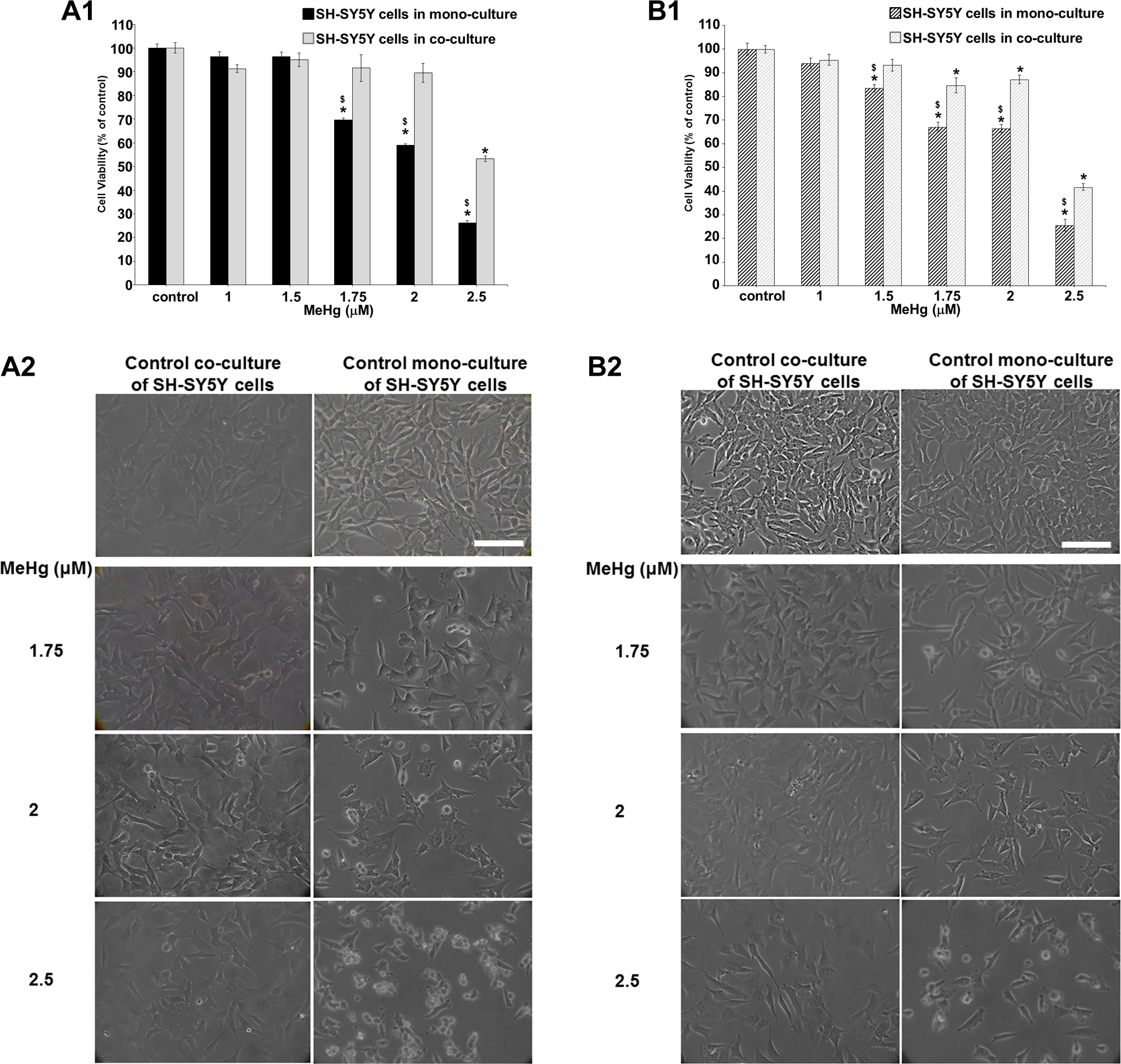
Effects of MeHg exposure (1-2.5 μM) after 24 and 48 hours in SH-SY5Y alone versus SH-SY5Y cocultured with D384. (A1-B1) Evaluation by MTT assay after 24 (A1) and 48 hours (B1). The mitochondrial dysfunction was significantly mitigated in neurons cocultured in the presence of astrocytes at both time points considered. Data are the mean (±SD) of 2 or 3 separate experiments each carried out in 2 replicates. *Different from control P < 0.05 and $different between monocultured and cocultured systems P < 0.05. Statistical analysis by ANOVA followed by Dunnett test. (A2-B2) Representative images of randomly selected microscopic fields by phase-contrast microscopy of SH-SY5Y cells alone or in SH-SY5Y cells cocultured with astrocytes exposed to increasing MeHg concentration (1.75-2.5 μM) after 24 (A2) and 48 hours (B2). Scale bar: 100 μm. ANOVA indicates analysis of variance; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation.
However, the mitochondrial dysfunction was significantly mitigated in neurons co-cultured in the presence of astrocytes (at concentrations from 1.75 to 2.5 μM; Figure 4, A1); the effects on cell viability was still observed at 2.5 μM (50% of cell death in co-cultured neurons vs 75% in mono-cultured neurons). No effect was observed up to 1.75 μM. In order to quantify the difference, MTT data after 24 and 48 hours were used to calculate LC50. The 24-hour LC50 values (with 95% confidence intervals) were estimated to be significantly different: 2.10 μM (1.94-2.32) versus 2.55 μM (2.34-3.52) for neuronal mono-culture and neuronal co-culture system, respectively. Again, the LC50 values, based on mortality data after 48 hours to MeHg exposure, indicated that SH-SY5Y under mono-culture conditions was more susceptible compared to SH-SY5Y under co-culture conditions: 2.1 μM (1.80-2.3) versus 2.4 μM (2.2-2.90), respectively.
As evaluated by phase-contrast microscopy, the cell morphology of SH-SY5Y co-cultured with D384 cells was less affected by MeHg, from 1.75 to 2.5 μM, than that of SH-SY5Y mono-cultures after 24 and 48 hours (Figure 4, A2 and B2): The effects started from 1.75 μM MeHg, which caused a cell density decrease in SH-SY5Y cells co-cultured with D384, with cells grew more spread in the culture plate without morphological alteration. Notably, MeHg-treated SH-SY5Y cells in mono-culture conditions (1.75-2.5 μM MeHg concentration) appeared with marked morphological cell alterations: roundish cells, shortened neurites, clusters, and a small number of substrate-adherent cells (Figure 4, A2 and B2).
Control SH-SY5Y cells (both co-cultured and mono-cultured) showed flattened-like phenotype with some cytoplasmic projections (neurites, Figure 4, A2 and B2). No effects (ie, morphological alterations) were observed at 1 to 1.5 μM MeHg after 24 and 48 hours (not shown) in both mono-culture and co-cultured neurons.
Similar to neurons, D384 astrocytes in co-cultures tolerated more MeHg than in mono-cultures. In particular, from 1.75 to 2.5 μM MeHg, cell viability decreased from 20% to 70% for D384 mono-cultured versus 10% to 55% for D384 co-cultured with neuronal cells after 24 hours (LC50: 2.2 μM [2.16-2.32] vs 2.5 μM [2.33-2.63] for astrocytes mono-culture and astrocytes co-culture system, respectively; Figure 5, A1), as well as 20% to 80% versus 5% to 70% for mono-culture and co-culture systems, respectively, after 48 hours from 1.5 to 2.5 μM MeHg (LC50: 2.0 μM [1.9-2.2] vs 2.3 μM [2.1-2.4] for D384 in mono-culture and co-culture conditions, respectively; Figure 5, B1).
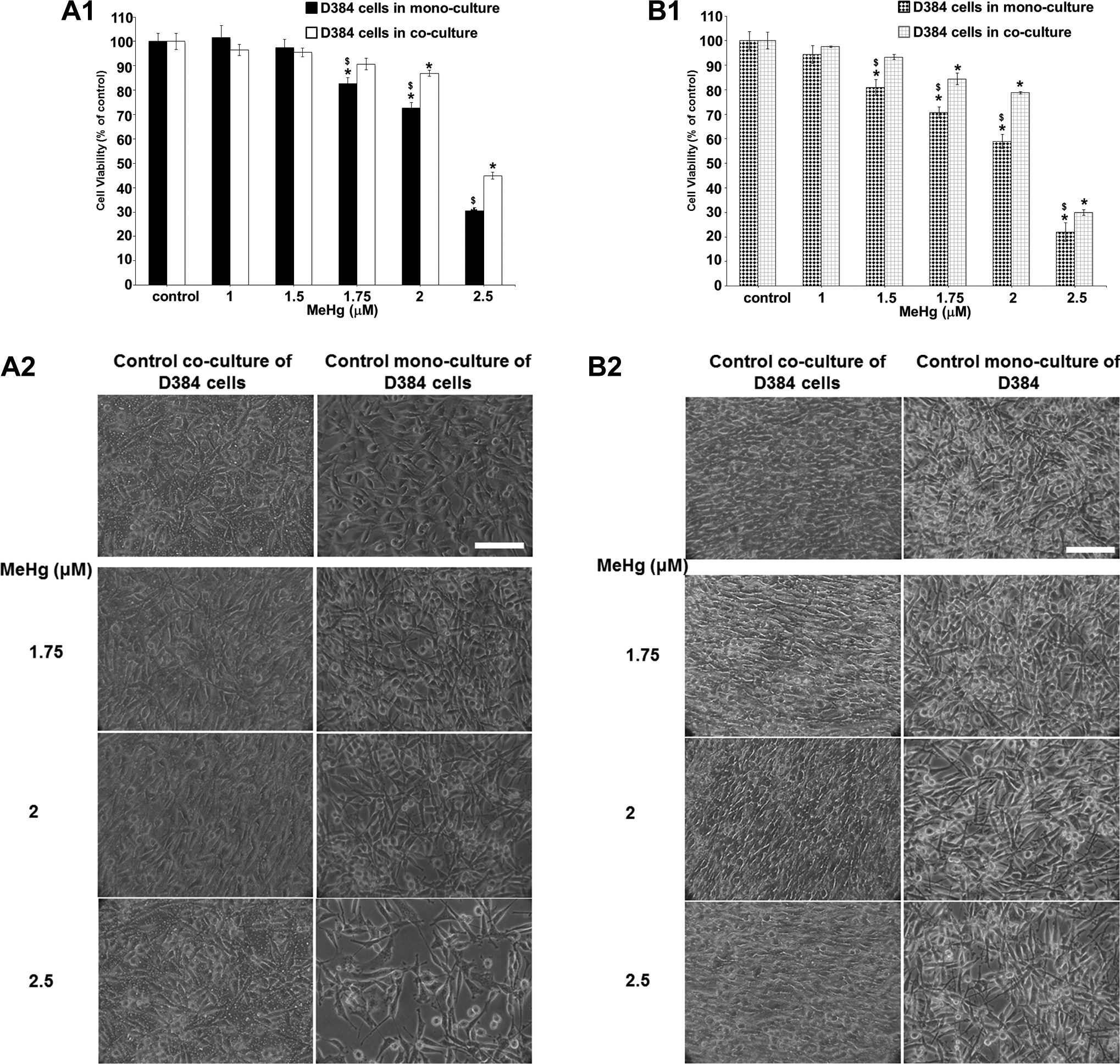
Effects of MeHg exposure (1-2.5 μM) after 24 and 48 hours in D384 cells monocultured versus D384 cocultured with neurons. (A1-B1) Evaluation by MTT assay after 24 (A1) and 48 hours (B1). MeHg treatments caused in monocultured astrocytes a time–concentration-dependent decrease of cell viability. The mitochondrial dysfunction was significantly mitigated in astrocytes cocultured in the presence of neuronal cells at both time points considered. Data are the mean (±SD) of 2 or 3 separate experiments each carried out in 2 replicates. *Different from control P < 0.05 and $different between monocultured and cocultured systems P < 0.05. Statistical analysis by ANOVA followed by Dunnett test. (A2-B2) Representative images of randomly selected microscopic fields by phase contrast microscopy of D384 cells alone or cocultured with neurons treated with MeHg (1.75-2.5 μM) after 24 (A2) and 48 hours (B2). Scale bar: 100 μm. ANOVA indicates analysis of variance; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation.
The morphological analysis showed strong D384 morphological alterations starting from 1.75 μM MeHg in mono-culture conditions, such as the cells lost the typical star shape becoming rounded, while D384 in the co-culture system displayed reduction in cell density only (Figure 5, A2 and B2). Again, no morphological alterations were observed from 1 to 1.5 μM MeHg after 24 and 48 hours (not shown).
Altogether, the results largely confirmed that MeHg-induced cell viability decreases in mono-culture conditions, starting from 1.5 and 1.75 μM (after 48 and 24 hours), were significantly attenuated in the co-culture model.
Methylglyoxal data
Results in Figure 6, A1 and B1 showed mitochondrial function in SH-SY5Y cells co-cultured with D384 astrocytes after 24 and 48 hours of exposure to increasing concentrations of MGO (0.5-1 mM). In human neuronal cells in mono-culture conditions, MGO treatments caused a marked concentration-dependent decrease in cell viability: 30% to 90% at concentrations ranging from 0.5 to 1 mM after 24 hours (Figure 5, A1). The reduction in viability was not exacerbated after 48 hours (40% to 95% at ranging concentration from 0.5 to 1 mM; Figure 6, B1).
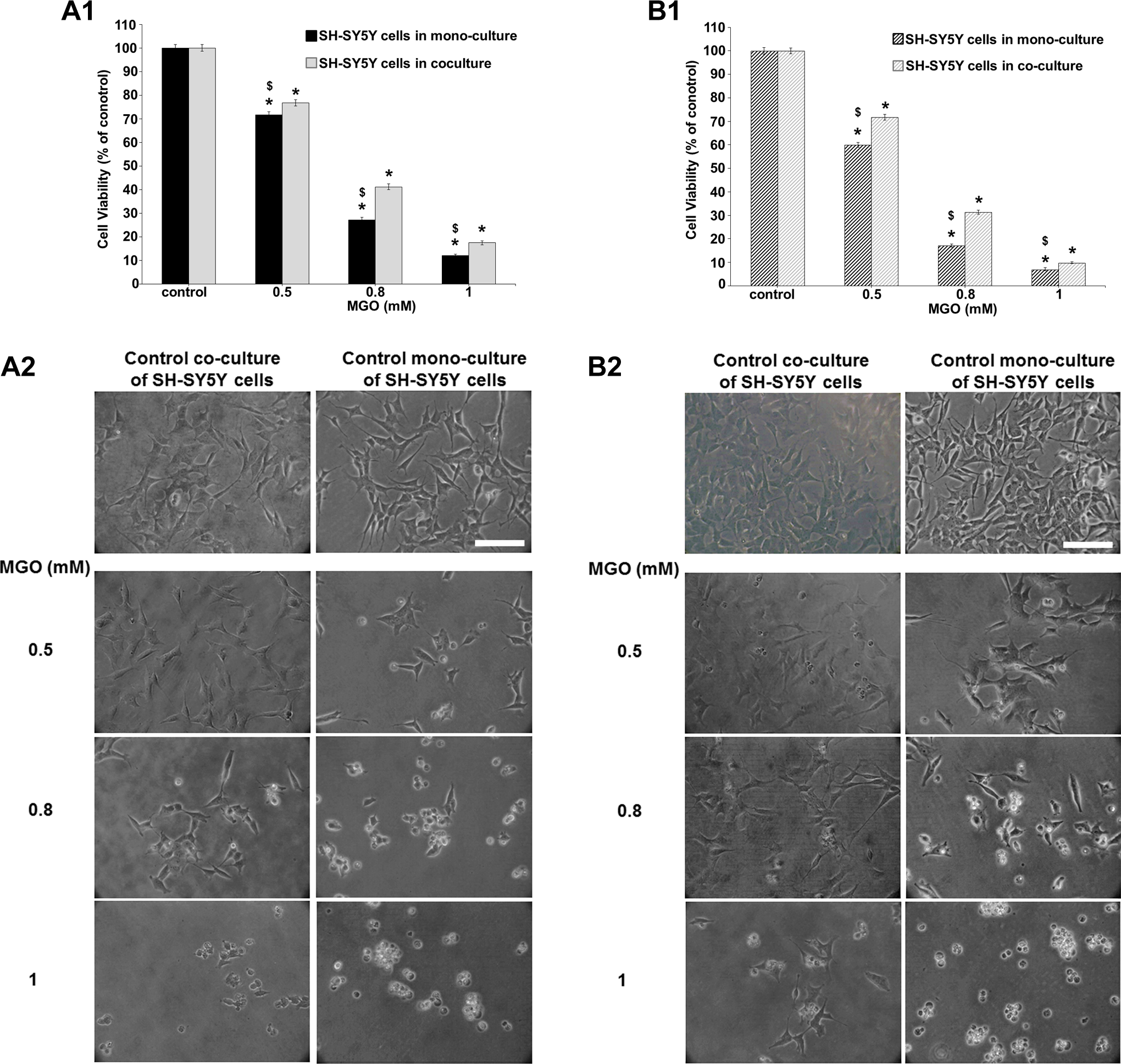
Effects of MGO exposure (0.5-1 mM) after 24 and 48 hours in SH-SY5Y cells under monoculture condition versus SH-SY5Y cocultured with astrocytes. (A1-B1) Evaluation by MTT assay after 24 and 48 hours. The protective effects of D384 cells on SH-SY5Y cells were observed after both 24 (A1) and 48 hours (B1). Data are the mean (±SD) of 3 separate experiments each carried out in 2 replicates. *Different from each control P < 0.05 and $different between monocultures and cocultures P < 0.05. Statistical analysis by ANOVA followed by Dunnett test. (A2-B2) Representative images of randomly selected microscopic fields by phase-contrast microscopy of SH-SY5Y cells alone or cocultured with D384 cells exposed to MGO after 24 (A2) and 48 hours (B2). Scale bar: 100 μm. ANOVA indicates analysis of variance; MGO, methylglyoxal; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation.
Notably, SH-SY5Y cell viability in the co-culture system with D384 cells was significantly less affected after MGO treatment when compared to SH-SY5Y cells in mono-culture conditions (cell death: 25%-80% vs 30%-90% for co-culture systems and mono-culture conditions, respectively, after 24 hours; Figure 6, A1). The protective effects of D384 cells on SH-SY5Y cells were also observed after 48-hour exposure to MGO (cell death: 30%-90% vs 40%-95% for co-culture and mono-culture conditions, respectively; Figure 6, B1). In addition, within each incubation time, LC50 values were significantly different (0.62 [0.53-0.64] mM vs 0.70 [0.66-0.79] mM for SH-SY5Y mono-cultured and SH-SY5Y co-cultured with astrocytes, after 24 hours, respectively; and 0.54 [0.48-0.63] mM vs 0.64 [0.59-0.72] mM after 48 hours, respectively).
The morphological analysis by phase-contrast microscopy showed that MGO treatments (0.5-1 mM) after 24 and 48 hours induced morphological alterations in both types of SH-SY5Y culture systems (ie, mono-culture and co-culture) in that cells became round and formed clusters, and neurite outgrowth was degenerated. These effects were associated with a decrease in cell density. However, the cell morphology alterations were more evident in mono-culture conditions compared to SH-SY5Y co-cultured with D384 cells (Figure 6, A2 and B2).
Once again, similar to neurons, D384 astrocytes in co-cultures tolerated more MGO than in mono-cultures. In particular, MGO treatment induced significant changes in cell viability of D384 cells in mono-culture conditions (compared to control; cell death: 50% to 85% at concentrations ranging from 0.5 to 1 mM after 24 hours; Figure 7, A1), whereas the D384 viability (40%-70%) co-cultured with SH-SY5Y cells was significantly less affected when compared to D384 mono-cultured at the same exposure condition (LC50 values of MGO in D384 mono-cultures and co-cultured with neurons were 0.49 [0.42-0.52] mM and 0.61 [0.55-0.76] mM, respectively; Figure 7, A1). A similar trend of MGO toxicity on cell viability was also observed after 48 hours (LC50 values were 0.45 mM [0.31-0.49] and 0.60 mM [0.48-0.72] for D384 mono-cultured and co-cultured with neurons, respectively; Figure 7, B1).
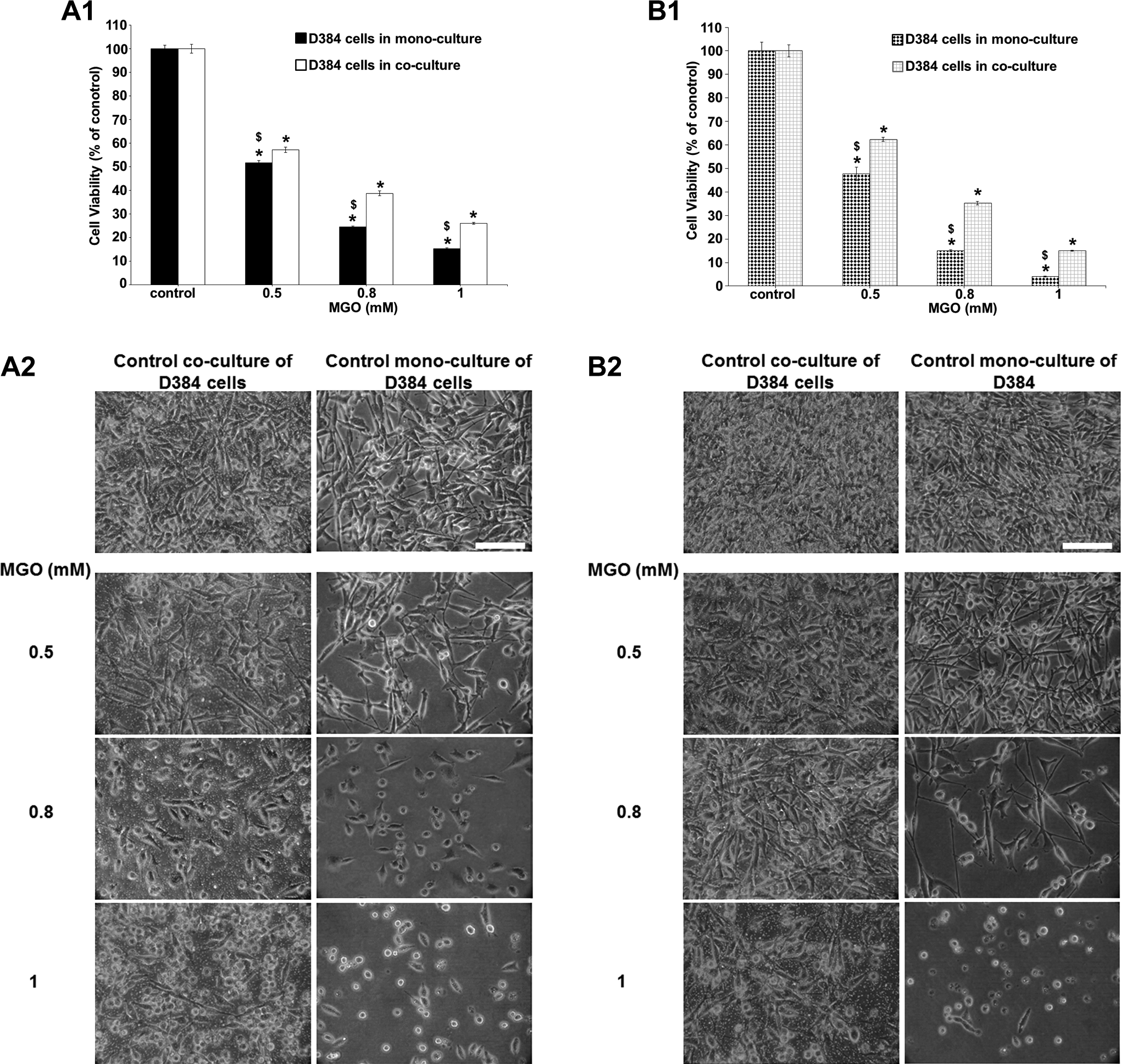
Effects of MGO exposure (0.5-1 mM) after 24 and 48 hours in D384 alone versus D384 cocultured with SH-SY5Y. (A1-B1) Evaluation by MTT assay after 24 and 48 hours. Astrocytes in monoculture conditions showed a strong concentration-dependent decrease in cell viability after 24 (A1) and 48 hours (B1) that was mitigated in coculture with SH-SY5Y. Data are the mean (±SD) of 3 separate experiments each carried out in 2 replicates. *Different from each control P < 0.05 and $different between monocultures and cocultures P < 0.05. Statistical analysis by ANOVA followed by Dunnett test. (A2-B2) Representative images of randomly selected microscopic fields by phase-contrast microscopy of D384 cells alone or in coculture with neurons exposed to MGO after 24 (A2) and 48 hours (B2). Scale bar: 100 μm. ANOVA indicates analysis of variance; MGO, methylglyoxal; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation.
In accordance with MTT results, stronger morphological alterations were observed in D384 mono-cultured compared to D384 co-cultured with neurons after 24-hour MGO treatment (0.5-1 mM): star-shaped cells became round and formed clusters and cell density decreased (Figure 6, A2). These effects were persistent after 48-hour exposure (Figure 7, B2).
Fe3O4NPs data
Fe3O4NPs (from 1 to 100 μg/mL) did not induce any significant effect on cell viability in neuronal mono-culture cells after 24-hour treatment (Figure 8, A1), whereas 35% to 45% cell death was observed after 48 hours from 10 to 100 μg/mL (Figure 8, B1). A maximal effect on neuronal cells was reached at 10 μg/mL (since not dose–response was observed, the LC50 value was not calculated). Indeed, previously, our study, employing Perls Prussian blue staining to directly visualize the cell-associated iron oxide particles, showed that Fe3O4NPs apparently accumulated in SH-SY5Y in much less manner than in D384: Very few neurons appeared stained, the scant blue spots started to be detectable at ≥10 μg/mL after 4 hours and faintly increased after 48 hours. Accordingly, less mitochondrial effect was observed in SH-SY5Y cells compared to astrocytes in mono-cultures (Figure 8, A1 vs Figure 9, A1 after 24 hours as well as Figure 8, B1 vs Figure 9, B1 after 48 hours).
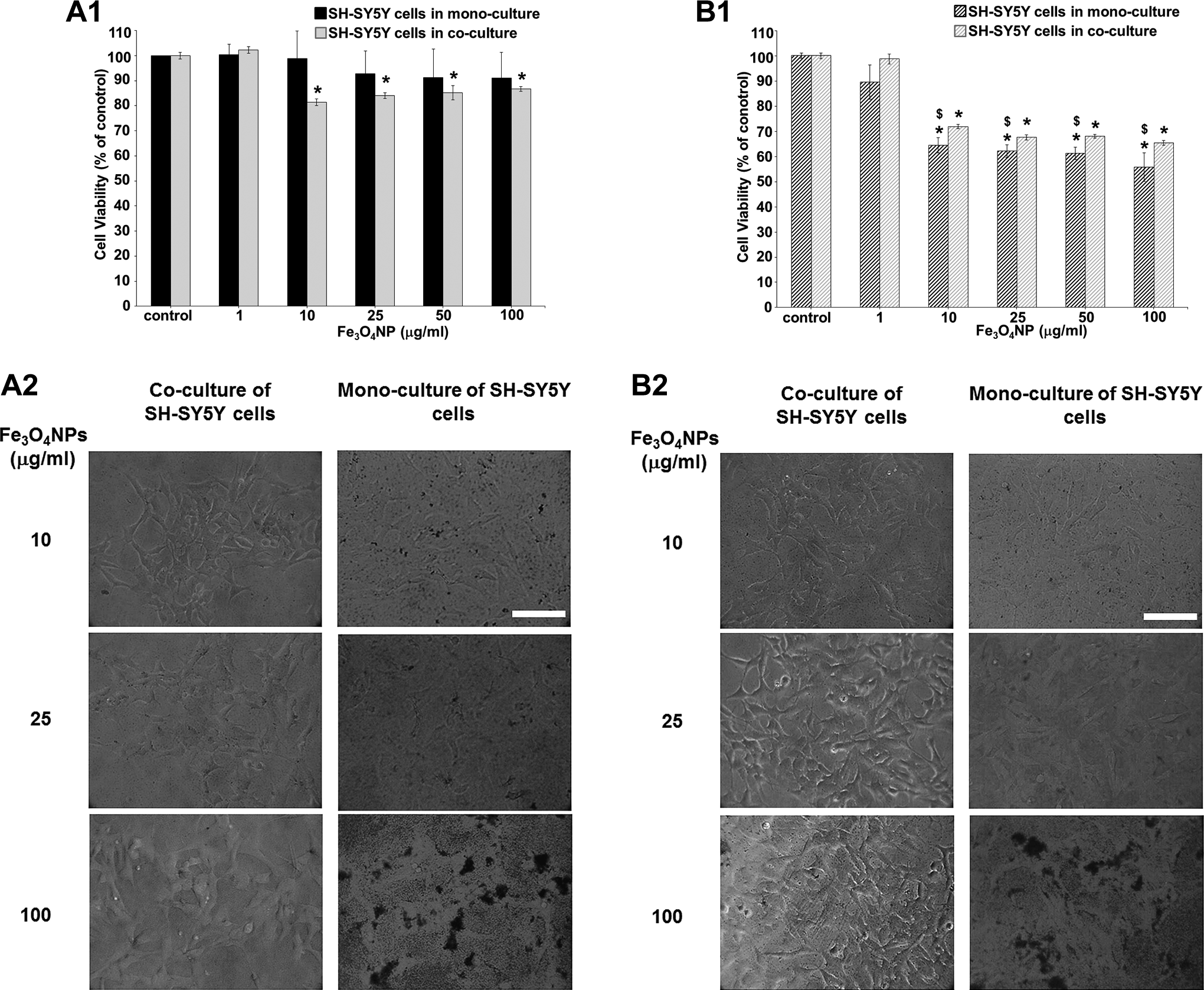
Effects of Fe3O4NP exposure (1-100 μg/mL) after 24 and 48 hours in SH-SY5Y monocultured versus SH-SY5Y cocultured with D384. (A1-B1) Evaluation by MTT assay after 24 (A1) and 48 hours (B1). An ameliorative effect was observed after 48 hours to Fe3O4NPs in coculturing neuronal–astrocyte cells compared to neuronal cells alone. Data are the mean (±SD) of 3 separate experiments each carried out in 2 replicates. *Different from each control P < 0.05 and $different between monocultures and cocultures P < 0.05. Statistical analysis by ANOVA followed by Dunnett test. (A2-B2) Representative images of randomly selected microscopic fields by phase-contrast microscopy of SH-SY5Y cells alone or in SH-SY5Y cells cocultured with astrocytes exposed to Fe3O4NPs after 24 (A2) and 48 hours (B2). Scale bar: 100 μm. ANOVA indicates analysis of variance; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation.
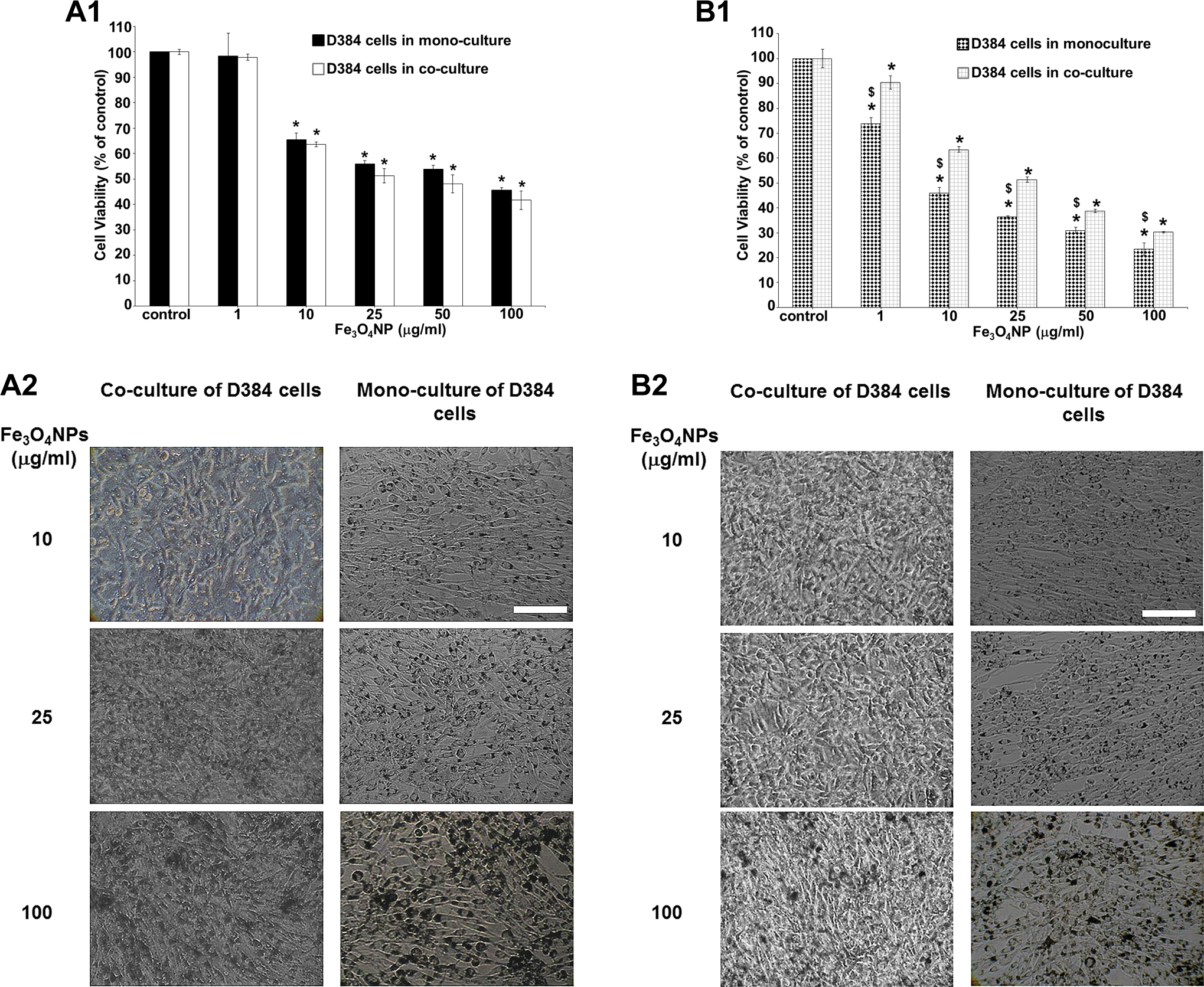
Effects of Fe3O4NP exposure (1-100 μg/mL) in D384 monocultured versus D384 cocultured with SH-SY5Y after 24 and 48 hours. (A1-B1) Evaluation by MTT assay after 24 (A1) and 48 hours (B1). A more pronounced toxicity effect was observed in D384 monocultured compared to D384 cocultured with neurons after 48 hours (B1). Data are the mean (±SD) of 3 separate experiments each carried out in 2 replicates. *Different from each control P < 0.05 and $different between monocultures and cocultures P < 0.05. Statistical analysis by ANOVA followed by Dunnett test. (A2-B2) Representative images of randomly selected microscopic fields by phase-contrast microscopy of D384 cells alone or in D384 cells cocultured with neurons exposed to Fe3O4NPs after 24 (A2) and 48 hours (B2). Scale bar: 100 μm. ANOVA indicates analysis of variance; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SD, standard deviation.
Peculiarly, Fe3O4NPs caused impairment of mitochondrial function in SH-SY5Y cells co-cultured with astrocytes with a decrease in cell viability of about 10% to 20% after 24 hours (Figure, 8 A1) and about 30% after 48 hours (Figure 8, B1) at concentrations ranging from 10 to 100 μg/mL, although the latter effect in co-cultured neurons was significantly less marked than those observed in mono-cultured neurons (Figures 8, A1 and B1). Once again, maximal effect on neuronal cells was reached at 10 μg/mL (no concentration–response was still observed and LC50 value was not calculated). The ameliorative effects to Fe3O4NP exposure of coculturing neuronal–astrocyte cells were observed at 48 hours.
The morphological analysis indicated no alterations in cellular shape and structure in both culture systems (mono-culture and co-culture) after 24- and 48-hour exposure to Fe3O4NPs (from 1 to 25 μg/mL; Figures 8, A2 and B2). Notably, Fe3O4NP brownish sediments of particles (detected by phase-contrast microscopy) in the culture medium were observed at concentrations of 10 and 25 μg/mL for SH-SY5Y mono-cultures and co-cultures, respectively. At higher concentrations, these sediments covered cell surface masking the light microscopy cell visualization of SH-SY5Y mono-cultured and co-cultured as shown in Figure 8, A2 and B2 for 100 μg/mL. This phenomenon was more pronounced in SH-SY5Y mono-cultures than that in neurons co-cultured with D384 cells (Figure 8, A2 and B2).
With respect to D384 astrocytes, Fe3O4NP induced concentration- and time-dependent changes of the mitochondrial function in D384 cultured in both mono-culture and co-culture systems. In particular, similar cytotoxicity was observed between the mono-culture and the co-cultures after 24-hour exposure to Fe3O4NP concentrations ranging from 10 to 100 µg/mL (cell death: about 35%-60%; Figure 9, A1), as demonstrated by similar LC50 values (50 [23-100] µg/mL in mono-culture and 49 [17-110] µg/mL in astrocytes co-cultured with neurons).
However, the cytotoxicity after 48-hour exposure to Fe3O4NPs was effectively more pronounced in D384 mono-cultured compared to D384 co-cultured with neurons (Figure 9, B1) as indicated by significant differences in LC50 values between mono-culture and co-culture conditions (ie, 8 [6.6-9.7] µg/mL vs 25.8 [22.3-29.8] µg/mL for astrocyte mono-cultured and astrocyte co-cultured with neurons, respectively).
The image analysis showed a concentration- and time-dependent Fe3O4NP accumulation in D384 mono-cultured and co-cultured cells: Intracellular brown bodies were visible at ≥10 μg/mL after 24 hours (Figure 9, A2) with marked increase at higher concentrations (25-100 μg/mL) and extended time of exposure (48 hours; Figure 9, B2). In addition, cell morphology alterations (ie, roundish-shaped instead of star-shaped cells) were detected in D384 mono-culture at higher concentrations (100 μg/mL) after 48 hours (Figure 9, B2), whereas in D384 co-cultured with neuronal cells, a cell density decrease only was observed (Figure 9, B2).
Discussion
Cell-based in vitro models have become indispensable tools in toxicology as in vivo experiments are not only ethically debated but are also expensive and time-consuming and are unsuitable for screening large numbers of chemicals 37 and, for the complexities of intercellular relationships that mediate responses to toxic chemicals, is necessary to gain a better understanding of the progression of neurotoxic states in the CNS.
Thus, there is an urgent need to better understand whether in vivo system (where neurons are surrounded by glial cells) could provide neuroprotection from indirect effect of toxicant exposure because interactions between astrocytes and neurons contribute to the maintenance of homeostasis in CNS. 38 –41
Astrocytes play an integral role in the maintenance of CNS homeostasis and are associated with both neuroprotection and neurodegradation when they are activated in response to toxic substances or disease states; on the other hand, neurons may affect the proliferation and maturation of glial elements 42,43 and dynamically regulate the glial signaling pathway through the release of substances such as glutamate. 44
The present study was designed to assess the effects of 3 different neurotoxic test compounds namely MeHg, MGO, and Fe3O4NPs in mono-cultures of human neurons (SH-SY5Y cells) and astrocytes (D384 cells) and to determine whether these effects could be modulated or mitigated by the coculturing conditions.
The results clearly evidenced that all tested compounds affected the mitochondrial activity and cell morphology in both mono-culture and co-culture conditions. However, astrocytes, when cultured together with neurons, diminish/attenuate the cytotoxic effects of all tested agents that occur in neurons cultured under solitary conditions, and astrocytes were more resistant in the presence of neurons.
In particular, MeHg results are in agreement with the few literature data on MeHg cytotoxicity evaluation in the co-culture system. For example, Yin et al 7 demonstrated that astrocytes could mitigate the effects of MeHg (10 μM after 6 hours) on glutamate concentrations and other amino acids that occur in neurons cultured in solitary conditions, and moreover, the astrocytes became more resistant in the presence of neurons. In another study using transwell system, Morken et al 6 demonstrated that when neurons and astrocytes were incubated in the co-culture model, neurons showed less changes compared to respective cell types in mono-cultures for mitochondrial activity, cell membrane integrity, and cellular content of glutathione and amino acids after MeHg exposure (25 μM for 24 hours).
With respect to MGO, there is strong evidence showing that the glyoxalase pathway, which represents the most important pathway for the detoxification of MGO, differs significantly between astrocytes and neurons in a way that renders neurons more vulnerable to MGO and AGE accumulation. 20 These remarks pinpoint the concept that a metabolic specialization is taking place in brain cells, specifically the astrocytes being more glycolytic than neurons, which may help protecting neurons from MGO toxicity. In this context, the co-culture approach seems a more reliable test method than mono-culture to better evaluate MGO in vitro toxicity.
Regarding Fe3O4NPs, it is known that the uptake of Fe3O4NPs is different in different cells. 45 In particular, cultured astrocytes are able to accumulate iron from various exogenous iron sources including Fe3O4NPs in a time- and concentration-dependent manner (for an overview, see the study by Hohnholt and Dringen 46 ) with saturable uptake kinetics. 28 Too much iron can compromise cell viability, as well as the transport and storage of iron. 46 Fe3O4NPs might have produced iron liberation that exceeded the iron homeostasis capacity. In fact, our results indicate that neurons are less susceptible, compared to astrocytes, to Fe3O4NPs in accordance with much less iron accumulation in SH-SY5Y compared to D384. 31 A possible reason that suggests a greater iron (from heme or IONP [Magnetic Iron Oxide Nanoparticles]) accumulation in astrocytes compared to neurons is that astrocytes are considered key regulators of the iron metabolism of the brain. Indeed, they are able to accumulate and store large amounts of iron and are largely responsible for distributing iron in the brain. 47
Differences between LC50 values from mono-culture compared to co-culture systems, particularly after 48 hours, for the 3 specific toxins/toxicants, support the valuable use of this approach. Cocultures of neurons and astrocytes may indeed improve in vitro neurotoxicity testing as an additional valuable model in pharmacological and toxicological studies.
These findings are in agreement with literature data, although still limited in this area, on neuroprotection effects by astrocytes on neurons, that evaluated different end points and compounds (eg, 6-hydroxydopamine, tributyltin, trimethyltin, 2,5-hexanedione, MeHg, MGO, m-dinitrobenzene, and small iron oxide particles) using different co-culture models. 6,7,23 –27 In particular, these CNS co-culture models apply primary rodent astrocytes and neurons or mixture of primary/immortalized rodent astrocytes and human neurons mainly by using 2 methods of co-culture:
“Full contact method” in which neurons and astrocytes are in complete physical contact (neurons are over astrocytes). 7,23,24,26,27,48 Recently, Efremova et al 48 demonstrated, using this model, that murine astrocytic cell line (immortalized murine astrocytes—IMA2.1 cells) protected human neurons (LUHMES [Lund human mesencephalic] cells) from external toxic stimuli. This protective effect was also reproduced using human primary astrocytes co-cultured with LUHMES cells. 48
“Restricted contact method” namely “transwell system” (as that presently applied) in which 2 types of cells are in contact with each other through a semi-permeable membrane providing a more representative human in vivo-like tissue model. 6,25,49 In this model type, astrocytes and neurons face each other without touching exhibiting features of the blood–brain barrier such as low permeability to small hydrophilic molecules, restoring the cell to cell interaction signaling present in vivo, and thus reflecting the physiological environment.
This type of in vitro model is still in their initial stage. So far, there exist so few studies that have applied cell co-culture model in a transwell plate system and, in particular, those using human astrocytes and neurons, 8,9,29 the latter is more reliable compared to those of rodent origin since the interspecies extrapolation is not necessary. Astrocytes in humans are larger and more abundant 50,51 than in rodent brains, having 10 times more processes and different signaling, 52 and are organized in more complex domains than their rodent counterparts. Varicose projection and interlaminar astrocytes are present in human but not in rodent brains. 53 Furthermore, co-culture models with human immortalized cell lines offer other several advantages, such as cost-effectiveness, ease of manipulation, and an essentially unlimited supply of cells with similar genotypes and phenotypes. Their predominant strength is the ability to repeat studies with a well-characterized culture system that can be used in multiple laboratories and they are widely available through a number of large cell banks. 54
The data obtained in the present study by using human CNS co-culture model in which different cerebral cell types separated by semi-permeable membrane support the evidence of the neuroprotective effects induced by astrocytes to neurons (and vice versa) following short-term exposure (24 and 48 hours) to neurotoxic compounds (MeHg, MGO, and Fe3O4NPs). The cultures did not have astrocytic overgrowth and demonstrate progressive neuronal survival over time.
This co-culture system represents a suif (simple and rapid) initial stage tool to study human CNS pathophysiology. The coexistence of human astrocytes and neurons in this human CNS co-culture model provides a system in which interactions between different cerebral cell populations can be used for high-throughput in vitro testing based on the use of complementary cell-based assays for the definition of the neurotoxicological profile for a wide range of investigations of relevance to neurotoxicological and neurodegenerative fields.
Conclusion
Cells are the simplest models that allow us to study relations between molecular events and cellular physiology. Cocultures may provide a useful model system, because they combine the ease of an in vitro co-culture with the ability to perform detailed molecular analyses which may contribute to design the strategy for prevention of neurotoxicity and they can better mimic the in vivo situation.
To date, in vitro experiments designed to study neurotoxicity risk assessment using human CNS co-culture in transwell system of neurons and astrocytes have been limited, and it is well-accepted that astrocytes are integral in providing spatial and metabolic support to neurons and play a critical role in modulating several neuronal functions, including neuronal migration, differentiation, and synaptic activity.
Human CNS co-culture model is a good choice to investigate neurotoxic effects of different toxicants on CNS, it provides a more representative human in vivo-like tissue, and it permits species-specific mechanistic information on the neurotoxicant toxicity avoiding many of the uncertainties and problems that are inherent in interspecies extrapolations.
In summary, this human CNS co-culture system seems an amenable cell model to feed high-throughput acute screening platforms and to evaluate both human neuronal and astrocytic toxicity and neuroprotective effects of new and emerging materials (eg, nanomaterials) with improved sensitivity due to the functional neuron–astrocyte metabolic interactions.
Footnotes
Author Contributions
Teresa Coccini conceived and designed the study and supervised the research, involved in the interpretation of the results and data analyses, edited the manuscript, and performed the final version to be submitted. Uliana De Simone performed all the in vitro experiments, acquired and elaborated the data, and drafted the manuscript. Laura Gribaldo and Francesca Caloni contributed to conception and design of the study and manuscript revision. All authors read and approved the final version of the manuscript for the submission.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Grants from Italian Ministries of Health, Research and Education, and “5 ×1000 project 2011” (Italian Ministry of Health).