
Abstract
In a previously reported CD-1 mouse 2-year carcinogenicity study with the sodium glucose cotransporter-2 inhibitor empagliflozin, an increased incidence of renal tubular adenomas and carcinomas was identified only in the male high-dose group. Follow-up investigative studies have shown that the renal tumors in male high-dose mice were preceded by a number of renal degenerative/regenerative findings. Prior cross-species in vitro metabolism studies using microsomes identified an oxidative metabolite (M466/2) predominantly formed in the male mouse kidney and which spontaneously degrades to a metabolite (M380/1) and reactive 4-OH crotonaldehyde (CTA). In order to further evaluate potential modes of action for empagliflozin-associated male mouse renal tumors, we report here a series of in vitro investigative toxicology studies conducted to evaluate the cytotoxic and genotoxic potential of empagliflozin and M466/2. To assess the cytotoxic potential of empagliflozin and M466/2, a primary mouse renal tubular epithelial (mRTE) cell model was used. In mRTE cells, M466/2-derived in vitro 4-OH CTA exposure was cytotoxic, while empagliflozin was not cytotoxic or mitogenic. Empagliflozin and M466/2 were not genotoxic, supporting an indirect mode of action for empagliflozin-associated male mouse renal tumorigenesis. In conclusion, these in vitro data show that M466/2-derived 4-OH CTA exposure is associated with cytotoxicity in renal tubule cells and may be involved in promoting compound-related in vivo renal metabolic stress and chronic low-level renal injury, in turn supporting a nongenotoxic mode of tumor pathogenesis specific to the male mouse.
Introduction
Empagliflozin is a sodium glucose cotransporter-2 (SGLT2) inhibitor developed to improve glycemic control in type 2 diabetic patients by inhibiting SGLT2-mediated glucose uptake from the urine filtrate in the proximal convoluted tubules into the circulation. 1 In a CD-1 mouse 2-year carcinogenicity study with empagliflozin (dose levels of 100, 300, and 1,000 mg/kg/d), an increased incidence of renal tubular adenomas and carcinomas was identified only in the male high-dose group. 2 The renal tumors generally occurred in association with chronic and persistent tubular degeneration, necrosis, and regeneration and were considered secondary to these changes. A series of investigative toxicology and metabolism studies were conducted to (1) better characterize the pathogenesis of empagliflozin-associated renal tubular tumor formation in the male mouse, (2) identify the mode of action key events involved, and (3) evaluate the potential relevance of those key events to humans. 3 Five modes of action key events were hypothesized to underlie the male mouse-specific pathogenesis of empagliflozin-related renal tumors. The first 2 are related to mouse physiology and pharmacological action. The third mode of action key event is nonpharmacology-related renal stress, and the last 2 key events are proposed to explain progression to the observed lesions. Support for a nonpharmacology-related and nongenotoxic third key event was provided by the identification of a previously unobserved metabolite, M466/2 in in vitro metabolism studies incubating [14C]-empagliflozin with microsomal preparations derived from liver and kidney of male and female mouse, rat, and human. 4 These studies showed that M466/2 (considered a precursor to stable downstream oxidized metabolites identified in mice) is predominantly formed in male mouse kidney compared to female mouse kidney and not in human tissue microsomes. Synthesized M466/2 was found to be unstable and spontaneously degrades under physiological conditions, via a retro-Michael reaction, to M380/1 and 4-OH crotonaldehyde (CTA) (Table 1), resulting in >90% degradation over 24 hours. Furthermore, in vitro glutathione (GSH) trapping experiments performed with M466/2 resulted in the formation of a 4-OH CTA adduct with glutathione, supporting the possibility of 4-OH CTA reacting with cellular constituents. The reactivity of α, β-unsaturated aldehydes such as 4-OH CTA is known to cause genotoxicity, cytotoxicity, oxidative stress, and carcinogenicity. 5 However, characterizations of the cytotoxic and genotoxic effects of such a possible reaction were not previously performed 4 and are pivotal to understanding the role of nonpharmacology-related renal stress in the pathogenesis of empagliflozin-related renal tumors and the potential relevance to humans. Although empagliflozin was previously demonstrated to be nongenotoxic and noncytotoxic in nonrenal cells, 2 M466/2 may be directly genotoxic, invalidating the proposed nongenotoxic mode of action and necessitating the demonstration of a lack of M466/2 formation in humans. If M466/2 is found not to be genotoxic and not to be cytotoxic, the proposed mode of action will fail to fully explain the renal carcinogenicity and further investigation of alternative mechanisms would be required. Here, we present an evaluation of the genotoxic and cytotoxic potential of empagliflozin and M466/2 and their potential role in the formation of renal tubular tumors in male mice, as well as explore potential off-target activities for empagliflozin suggestive of alternative modes of action.
Cytotoxicity of Empagliflozin-Related Chemicals and Aldehydes.
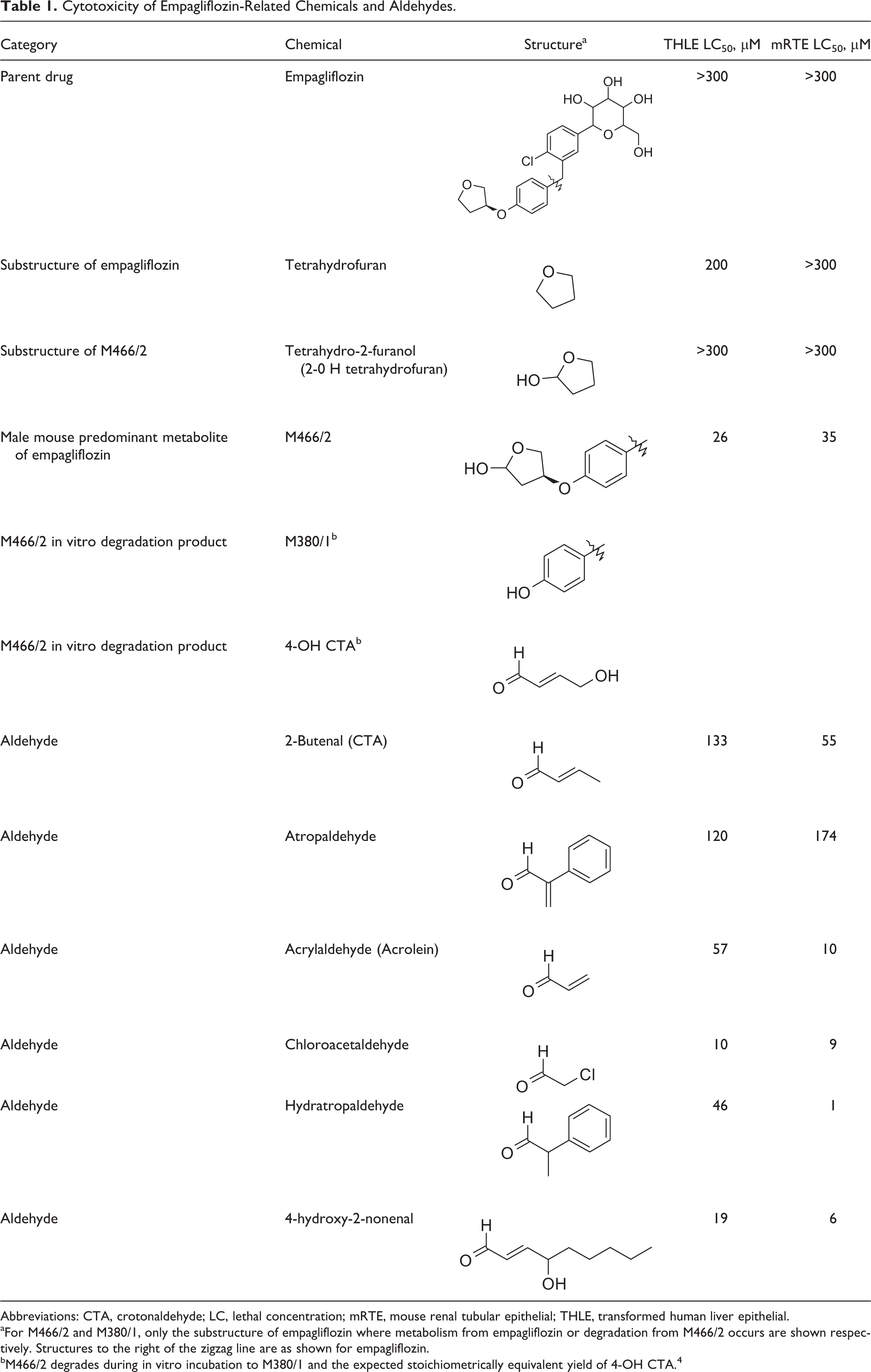
Abbreviations: CTA, crotonaldehyde; LC, lethal concentration; mRTE, mouse renal tubular epithelial; THLE, transformed human liver epithelial.
aFor M466/2 and M380/1, only the substructure of empagliflozin where metabolism from empagliflozin or degradation from M466/2 occurs are shown respectively. Structures to the right of the zigzag line are as shown for empagliflozin.
bM466/2 degrades during in vitro incubation to M380/1 and the expected stoichiometrically equivalent yield of 4-OH CTA. 4
Material and Methods
Chemicals
All chemicals were purchased from Sigma Aldrich Co, except 2-hydroxy-tetrahydrofuran from Santa Cruz Biotechnology Inc (Dallas, TX), 4-hydroxy-2-nonenal from Cayman Chemical Co (Ann Arbor, MI), 2-phenyl propenal from Novel Chemical Solutions (Crete, NE), CTA from Crescent Chemicals Co (Islandia, NY), and empagliflozin and M466/2 from Boehringer Ingelheim Pharmaceuticals Incorporated (Ridgefield, CT). M466/2 was synthesized and characterized by nuclear magnetic resonance to confirm the correct structure. 4
Computational Structure–Toxicity Relationship Analysis
Empagliflozin, M466/2, and other putative aldehyde intermediates were submitted to computational structure–activity relationship (SAR) analysis to initially evaluate potential structural alerts for toxicity, with the main focus on potential genotoxicity. Analyses were conducted using the following independent computer models: DEREK (Lhasa, Leeds, UK, www.Lhasalimited.org) and CASE Ultra (MultiCASE, Beechwood, OH, www.Multicase.com). In addition, public domain databases were searched for structurally similar compounds (Tanimoto method) with toxicity data (Leadscope; www.Leadscope.com).
Bacterial Reverse Mutation Assay (Ames Assay)
The mutagenic potential of empagliflozin (reported previously 2 ) and M466/2 was assessed by measuring the reverse mutations at selected loci of Salmonella typhimurium tester strains TA98, TA100, TA1535, TA1537 and of Escherichia coli WP2 uvrA (pKM101). The assays were conducted via the plate incorporation methodology originally described by Ames 6 and Maron 7 and following the Organization for Economic Co-operation and Development (OECD) guideline TG471. 8 Up to 9 dose levels were tested in concentrations reaching 5,000 μg/plate, with and without metabolic activation (Araclor-induced rat liver S9 extract). For M466/2 bioanalysis, agarose plates treated with M466/2, without bacteria, were incubated in parallel under the same conditions as used in the Ames assay.
In Vitro Micronucleus Assay
The aneugenic and clastogenic potential of M466/2 was assessed in an in vitro micronucleus assay in Chinese hamster ovary (CHO) cells. The cells were treated for 4 hours with and without metabolic activation (Araclor-induced rat liver S9 extract) and for 24 hours without metabolic activation. In the initial assay, 19 doses were tested up to 500 μg/mL, and then, for the micronucleus assessment, 3 dose levels were evaluated for each condition (OECD TG487), 9 with the highest dose based on approximately 50% cytotoxicity. In the 4-hour treatment with metabolic activation, the highest dose tested was 118.7 μg/mL (51% cytotoxicity). In the 4-hour treatment without metabolic activation, the highest dose tested was 15.8 μg/mL (50% cytotoxicity). In the 24-hour treatment without metabolic activation, the highest dose tested was 8.9 μg/mL (62% cytotoxicity). In addition, the 4- and 24-hour treatment without metabolic activation was repeated to confirm initial results. The CHO cell culture supernatants from the repeat in vitro micronucleus assay were used for M466/2 bioanalysis.
Mouse Renal Tubular Epithelial Cell Preparation
Primary mouse renal tubular epithelial (mRTE) cells were isolated and cultured using an adaptation of methods described by Breggia and Himmelfarb. 10 Briefly, kidney cortex from CD-1 male mice (6-7 weeks old) was collected in phosphate-buffered saline (4°C), minced and digested with collagenase 4 (Worthington Biochemical Corp, Lakewood, NJ) and soybean trypsin inhibitor (Life Technologies, Carlsbad, CA) at 37°C. Renal tubules were isolated from the mixture by multiple rounds of centrifugation and filtration followed by digestion with trypsin-EDTA (Life Technologies) at 37°C to obtain a single-cell suspension. The cell suspension was diluted in growth media (Lonza Inc, Allendale, NJ) with added recombinant mouse epidermal growth factor (rmEGF) (Life Technologies) and plated on laminin-coated cell culture plates (BD Biosciences, San Jose, CA). The isolation method above and the experimental use of mRTE cells at passage 1 and between culture days 7 and 8 were guided by positive confirmation of the mRTE phenotype and SGLT2 relevant physiology using the following characterization procedures (data not shown): (1) The SGLT2 messenger RNA (mRNA) expression, as reported 11,12 : mRNA was isolated from mRTE cells using TRIzol (Life Technologies). The SGLT2 mRNA expression levels were demonstrated by TaqMan gene expression analysis. (2) The SGLT2 activity by [14C]-α-methyl glucose uptake, as reported. 13 (3) Morphology: Epithelial morphology was verified by phase-contrast microscopy as reported. 10 (4) Immunofluorescent staining: Positive staining (>75%) for tubular epithelial-specific markers cytokeratin, N-cadherin, and CD-13 (aminopeptidase N) as reported. 14 –16 (5) Proliferative responses: Positive responsiveness to rmEGF and fetal bovine serum (FBS) stimulation as measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation. 17 –19
In Vitro Cytotoxicity Assay
Mouse renal tubular epithelial cells in FBS- and EGF-free medium (Lonza Inc) or a nonrenal cell line (transformed human liver epithelial [THLE]-2 cells; American type culture collection) in complete medium (Lonza Inc) were plated on 96-well plates and dosed with 0.1 to 300 μM empagliflozin, M466/2, or relevant controls including aldehydes and chemicals with similar substructures to empagliflozin. Empagliflozin and M466/2 test concentration ranges were chosen to cover empagliflozin values for human therapeutic plasma exposures, pharmacological SGLT2 inhibition (1 µM), and the approximate maximum plasma exposure (Cmax) in the 2-year mouse carcinogenicity study (100 µM). Rotenone (100 μM) or valinomycin (125 μM) was included as a cytotoxicity positive control. After 1 to 3 days of dosing, terminal cell culture supernatants were collected for bioanalysis. Cell viability was determined by measuring cellular adenosine triphosphate (ATP) levels (CellTiter-Glo Luminescent Cell Viability Assay; Promega Corp, Madison, WI). The ATP values were expressed as a percentage of the dimethyl sulfoxide (DMSO) vehicle control. Cell viability assays were considered valid when controls induced ≥40% decrease in ATP levels.
In Vitro Cell Proliferation Assay
Mouse renal tubular epithelial cells were plated on 96-well plates in FBS- and EGF-free growth medium (Lonza Inc) and dosed with 0.1 to 100 μM empagliflozin. The dose range of empagliflozin was chosen as described above (see section “In Vitro Cytotoxicity Assay”). The addition of FBS (Lonza Inc) or rmEGF (R&D Systems, Minneapolis, MN) were included as positive proliferative controls. Within 18 to 24 hours of dosing, the BrdU incorporation cell proliferation enzyme-linked immunosorbent assay (Roche Diagnostics, Indianapolis, IN) was performed. Proliferation values are expressed as a percentage of the DMSO vehicle control. Cellular proliferation assays were considered valid when positive controls triggered a ≥40% increase in proliferation. For each batch of cells used in a proliferation assay, a confirmatory count was performed with cells plated on 60-mm plates and treated with a subset of the doses used in the proliferation assay.
Bioanalysis of M466/2 and M380/1 Metabolites In Vitro
Mouse renal tubular epithelial and CHO cell culture supernatants and bacterial agar (Ames assay) samples were mixed with quench solution (acetonitrile with 1 μM of [13C6]-empagliflozin [internal standard] and 0.1% acetic acid), filtered, and analyzed by liquid chromatography/dual mass specrometry (LC/MS/MS) (API 4000 QTRAP mass spectrometer with Atlantis dC18 high-performance liquid chromatography column [Waters, Milford, MA], column temperature maintained at 30°C). Mobile phase A was 20 mM ammonium acetate in water (pH 6.5) and mobile phase B was acetonitrile. The percentage recovery of M466/2 and percentage of M380/1 formed were then calculated.
Off-Target Profiling
Empagliflozin was tested in (1) a focused panel of 50 kinase assays spanning the human kinome (Invitrogen Life Technologies’ (Carlsbad, CA) SelectScreen Profiling) at 3 μM and (2) a broad panel of 107 proteomic assays (Cerep, St Charles, MO) including assays of receptor binding, ion channel binding, enzyme and transporter activity, in vitro metabolism, and cellular and nuclear receptor function at 10 μM. Assays were conducted according to standard protocols. In both assay systems, off-target hits were defined as those showing >30% inhibition at the single test concentrations tested.
Results
Computational Structure–Toxicity Relationship Analysis
The results of the genotoxicity-focused computer model analyses (DEREK and MultiCASE) for toxicity structural alerts in empagliflozin and M466/2 revealed the presence of an aldehyde or aldehyde precursor genotoxicity structural alert in M466/2 and a putative aldehyde intermediate (Supplemental Table 1). The toxicological relevance of the presence of the aldehyde precursor substructures in complex compounds such as M466/2 is unclear due to lack of SAR for such compounds and the literature precedent for simpler α, β-unsaturated aldehydes. 5 Analysis of metabolites using the Leadscope database was either negative or the metabolites were not in the structure domain for standard genotoxicity tests. Similarly, no alerting substructures were identified in the metabolites using the mutagenicity modules in MultiCASE and CASE Ultra.
Genotoxicity Assessment
The genotoxicity assessment of empagliflozin (previously reported 2 ) and M466/2 revealed that neither of the compounds were genotoxic. Empagliflozin was negative in the Ames assay, mouse lymphoma assay, and the in vivo micronucleus assay in rats. 2 M466/2 was not mutagenic in the Ames assay and showed a weak thresholded increase in micronuclei at cytotoxic doses (∼50% viability) only at the 24-hour time point (Figures 1 and 2) in the in vitro micronucleus assay. This increase was considered not to be biologically meaningful, since it was observed only after 24-hour treatment at cytotoxic levels. Furthermore, the in vitro micronucleus assay demonstrated that the level of cytotoxicity due to M466/2 depended on the testing conditions, with cytotoxicity increasing with longer exposure time and without metabolic activation. As M466/2 was demonstrated to degrade to M380/1 and the expected stoichiometrically equivalent yield of 4-OH CTA during the time course and conditions of this assay (bioanalysis results [see section “Bioanalysis of M466/2 and M380/1 Metabolites In Vitro”]), 4-OH CTA is the likely ultimate cytotoxic entity consistent with data for CTA. 5,20 In contrast to studies with M466/2, empagliflozin did not show a comparable cytotoxicity profile, albeit in a different in vitro mammalian cell genotoxicity test system. 2
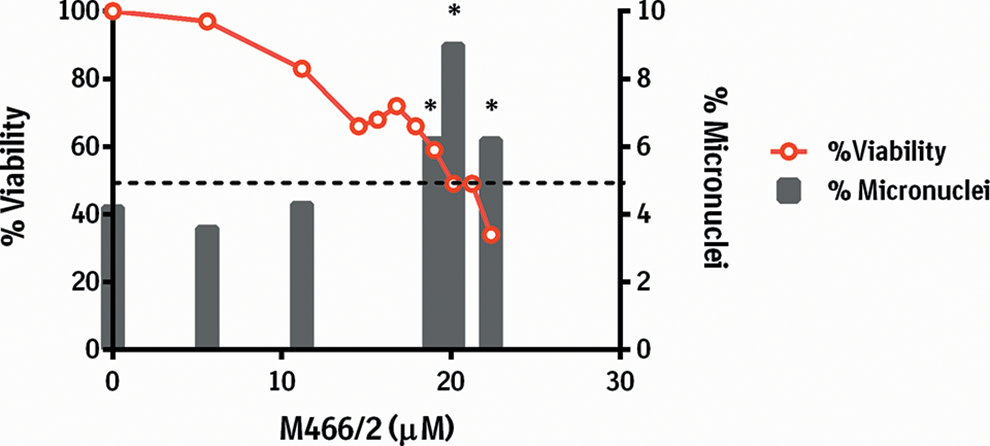
Percentage micronuclei and cell viability after 24-hour incubation with M466/2 in the absence of metabolic activation (S9) (Chinese hamster ovary (CHO) cell micronucleus assay). Representative result of 2 replicate experiments. *Statistical significance versus historical control range (Fisher test), P < 0.05.
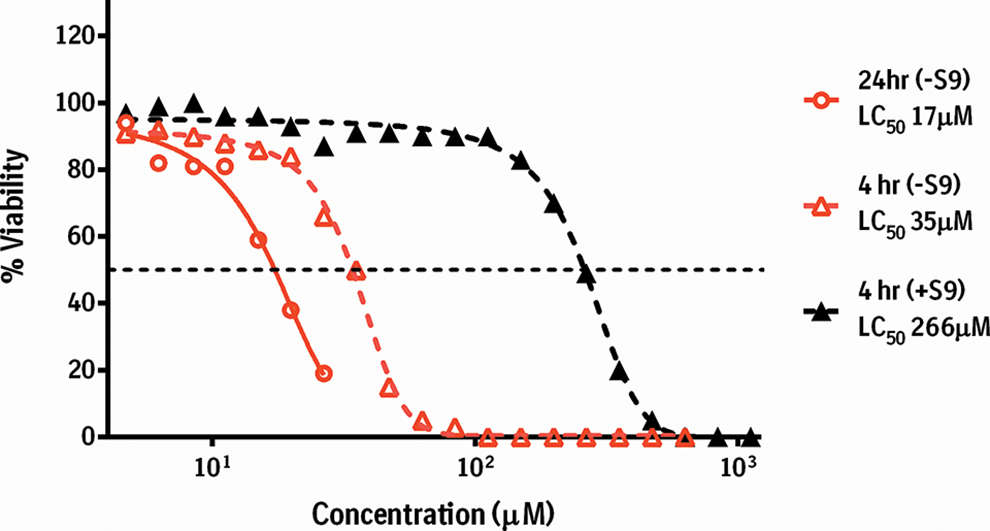
Percentage viability after incubation with M466/2 for 4 hours with and without metabolic activation (S9) and 24 hours without metabolic activation (Chinese hamster ovary (CHO) cell micronucleus assay). Dotted line is 4-parameter logistic fit curve. LC, lethal concentration.
In Vitro Cytotoxicity
The cytotoxicity profile of M466/2 in CHO cells in the micronucleus assay (see section “Genotoxicity Assessment”) and the investigative toxicology study finding of single-cell necrosis in renal proximal tubular cells in male mice dosed with empagliflozin (1,000 mg/kg/d; data not shown) prompted further comparative evaluation of the cytotoxic potential of empagliflozin and M466/2. For this evaluation, both a primary mRTE cell line, relevant to the renal cell target, and a nonrenal-derived cell line, THLE, were used. We also profiled other relevant compounds including simpler aldehydes and compounds containing substructures of empagliflozin or its metabolites that may be implicated in cytotoxicity. The results of repeated cytotoxicity assays in both THLE and mRTE cells showed no evidence of empagliflozin-related effects on cell viability (reflected by cellular ATP levels) over the concentration range and treatment durations evaluated (Figure 3 and Table 1 and Supplemental Figure 1). In contrast, incubation with M466/2 was cytotoxic in both THLE (LC50 26 μM) and mRTE cells (LC50 35 μM) with rather steep cytotoxicity dose–response profiles. The bioanalysis results (below [see section “Bioanalysis of M466/2 and M380/1 Metabolites In Vitro”]) demonstrated that M466/2 was unstable under the conditions of both of these assays and degraded quickly to M380/1 and the presumed stoichiometric equivalent yield of 4-OH CTA. Thus, the development of cytotoxicity corresponded to the formation of M380/1 and presumably 4-OH CTA. Overall, the cytotoxicity results for both cell types were similar for all chemicals tested (Table 1). The cytotoxic nature of the selected simpler aldehydes tested also aligns with literature reports 20 –24 and points to 4-OH CTA as the likely ultimate in vitro cytotoxicant. Although intracellular ATP values may decrease under noncytotoxic conditions, the ATP assay results described in this article are accompanied by an independent demonstration of cytotoxicity: trypan blue exclusion in the in vitro micronucleus assay (Figure 2). There is also a large body of work that demonstrates correlations between ATP levels and cytotoxicity, although the assay lacks the ability to differentiate between cell death types. 25
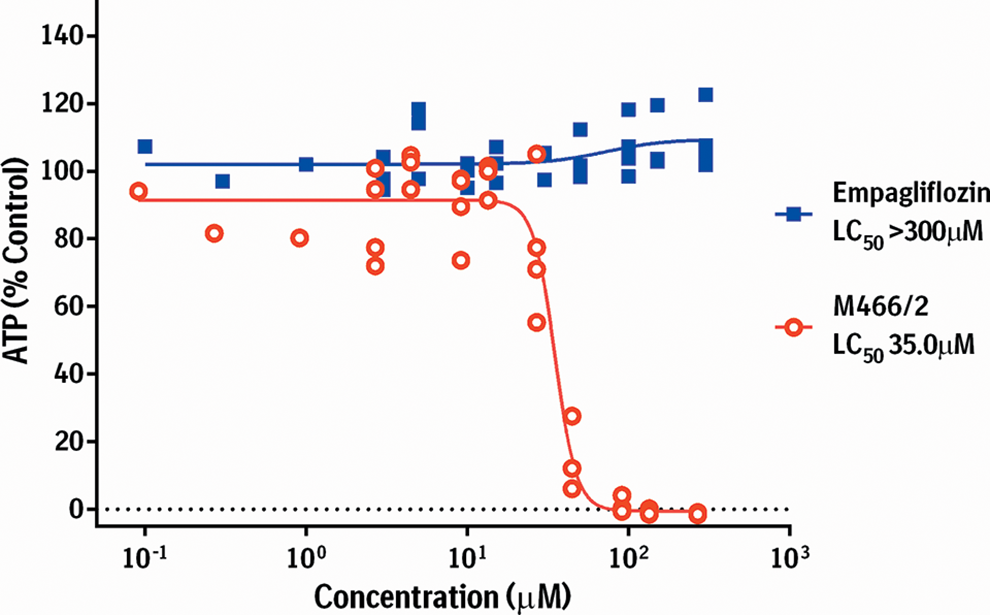
The ATP-depletion cytotoxicity assay for primary mRTE cells treated with empagliflozin or M466/2. Dotted line is 4-parameter logistic fit curve of n = 4 experiments. ATP, adenosine triphosphate; LC, lethal concentration; mRTE, mouse renal tubular epithelial.
Bioanalysis of M466/2 and M380/1 Metabolites In Vitro
In order to determine the relative exposures to M466/2 and its associated degradation pathway to 4-OH CTA in the in vitro test systems used above (see sections “Bacterial Reverse Mutation Assay (Ames Assay),” “In Vitro Micronucleus Assay” and “In Vitro Cytotoxicity Assay.]), bioanalyses of test media (in vitro micronucleus assay and mRTE cytotoxicity assays) and agar plates incubated without bacteria under the same conditions as the Ames assay were carried out using LC/MS/MS. In agar plates, M466/2 was extensively degraded to M380/1 in all doses tested with the M380/1 formed representing >79% of the combined M466/2 and M380/1 concentrations. Bioanalysis of incubation media from the in vitro micronucleus assay showed that M380/1 is the predominant component within 4 hours (>97% of the combined M466/2 and M380/1 concentrations) and M466/2 was completely degraded within 24 hours. Bioanalysis of incubation media from the mRTE cytotoxicity assays revealed that the majority of M466/2 was degraded to M380/1 (≥90.5% of the combined M380/1 and M466/2 levels) after being incubated for 24 hours. Together, these analyses showed that, upon in vitro incubation of M466/2 in test systems, >75% of the M466/2 was degraded to M380/1 and the expected stoichiometrically equivalent yield of 4-OH CTA, consistent with observations by Taub et al. 4
Cell Proliferation
To rule out a potential direct pro-proliferative role for empagliflozin in mRTE cells, these cells were utilized in a standard cell proliferation assay. The results of repeated in vitro BrdU-incorporation assays with mRTE cells showed no evidence of empagliflozin-related effects on cell proliferation over the concentration range and treatment durations evaluated (Supplemental Figure 2). These results indicate no direct mitogenic role for empagliflozin.
Off-Target Profiling
To assess potential off-target pharmacological activities, empagliflozin was tested in a broad proteomic panel of 107 binding and functional assays (Cerep) 26 and a focused panel of 50 kinase assays (Invitrogen) including specific kinases chosen to investigate effects on renal pathways associated with proliferation, apoptosis, injury, ion transport, and the development of carcinoma. The results revealed that empagliflozin had very low protein binding potential and can be considered to be nonpromiscuous against this sample set of the human proteome and kinome (Supplemental Figure 3A and 3B).
Discussion
A series of in vitro investigative toxicology studies were conducted to further evaluate a mode of action for empagliflozin-associated male mouse renal tumors. The in vitro data presented demonstrate no genotoxic, cytotoxic, or mitogenic role for empagliflozin. However, the data indicate that an oxidative metabolite of empagliflozin predominantly formed in male mouse kidney, M466/2, is cytotoxic to multiple cell types, including renal tubule cells, but is not genotoxic.
Empagliflozin and M466/2 were not genotoxic when tested in the Ames assay and in vitro mammalian cell genotoxicity assays (mouse lymphoma and micronucleus). 2 In the CHO cell micronucleus assay, M466/2 demonstrated a weak and biologically irrelevant thresholded increase in micronucleus formation closely associated with cytotoxicity. The cytotoxicity observed on dosing with M466/2 in this assay was decreased with S9 metabolic activation, without induction of micronuclei, and increased with longer exposure (in the absence of S9 metabolic activation; Figure 2). Due to the rapid degradation of M466/2 under the conditions of the assay, it is anticipated that toxicity in the presence of S9 would be the same as in the absence of S9. The difference in toxicity observed with S9 may be due to metabolism of M466/2 to another metabolite which does not generate 4-OH CTA or due to trapping of 4-OH CTA by S9.
Following a demonstration of cytotoxic effects of M466/2 in CHO cells (in vitro micronucleus assay), a more comprehensive evaluation of cytotoxicity was carried out for empagliflozin and M466/2 in a nonrenal (THLE) cell line and a mRTE cell system. M466/2 was cytotoxic in both THLE and mRTE cells (Figure 3 and Table 1 and Supplemental Figure 1). The cytotoxicity results for M466/2 concur with the computational SAR analysis identification of an aldehyde or aldehyde precursor toxicity structural alert in M466/2 (Supplemental Table 1). The toxicological relevance of the presence of the alerting aldehyde precursor substructure in M466/2 is unclear, since structure–toxicity relationships that include more complex aldehyde compounds like M466/2 are lacking. Furthermore, M466/2 was demonstrated to degrade to M380/1 and 4-OH CTA under the physiological conditions used in the in vitro assays with the formation of 4-OH CTA corresponding to the cytotoxicity observed due to M466/2 dosing. Direct confirmation of the formation of 4-OH CTA in biological systems was precluded in this study by its high reactivity. However, it is known that binding of α, β unsaturated aldehydes such as 4-OH CTA to cellular constituents via Michael addition causes the cytotoxic effects of aldehydes 5,20–24,27 and that, based on their electrophilicity, aldehydes can be categorized into subgroups with differing toxicity. 28 The reactive potential of M466/2-derived 4-OH CTA with cellular constituents was previously confirmed by the formation of 4-OH CTA adducts with glutathione (GSH) in cell-free in vitro GSH trapping experiments using M466/2. 4 In contrast, M380/1 was not found to be reactive in the trapping studies and was not considered to be a direct contributor to these cytotoxic effects and was not synthesized for individual evaluation of cytotoxicity. Due to the inability to directly measure 4-OH CTA in the in vitro test systems used in this study, M380/1 was employed as a bioanalytical indicator of the exposure of the expected stoichiometric equivalent 4-OH CTA. Bioanalysis from these studies demonstrated that after 4 hours more than 75% of the total M380/1 was present in all assays, implying a stoichiometric release of reactive 4-OH CTA and aligning with previous demonstrations of the degradation of M466/2 after incubation under physiological conditions. 4 Therefore, the appearance of M380/1 corresponds to cytotoxicity related to M466/2-derived in vitro 4-OH CTA exposure. This cytotoxicity is in concordance with cytotoxicity induced by other simple aldehydes related to CTA in THLE and mRTE cells (Table 1) and other cell types. 5,20 Direct determination of the cytotoxicity of 4-OH CTA was precluded by unavailability of this highly reactive aldehyde.
In order to closely approximate mouse renal tubular cell and SGLT2 physiology, mRTE cell isolation and culture were optimized so that under test conditions the cell populations were predominantly tubular epithelial cells with intact SGLT2 function and proliferative capability. In addition, mRTE cells demonstrated susceptibility to aldehyde toxicity similar to THLE cells (Table 1) and other cells as reported. 20 –24 However, diminished or lost cellular metabolic potential, while retaining their primary physiological phenotype, is an expected and commonly observed feature of removing cells from their in vivo environment to be used as models for in vitro studies. 29 Therefore, one limitation of this ex vivo cellular model is the loss of metabolic capacity to generate M466/2, supported by bioanalysis of cell culture supernatants from mRTE cell experiments that demonstrates no empagliflozin metabolism after 24 hours at 37°C (data not shown). While this artifact does not allow for an ideal ex vivo test system, it also does not limit our ability to test directly the influence of the specific metabolites in this model. We proceeded to test empagliflozin and its male-specific renal metabolite M466/2 independently with the added understanding that cellular drug metabolism would not play a significant role in interpreting the outcomes.
The mRTE cell model was used to investigate a potential direct mitogenic effect of empagliflozin on RTE cells as the mechanism underlying the proliferative phenotypes of increased mitoses, tubular hyperplasia, and increased Ki67 staining observed in high-dose male mice. 2 Empagliflozin showed no evidence of influencing mRTE cell proliferation, as determined by BrdU incorporation and cell counts, across the concentration range and treatment durations evaluated (Supplemental Figure 2). These results indicate no direct mitogenic role for empagliflozin.
Other potential off-target binding and functional effects of empagliflozin that would support alternative mechanisms of action leading to renal cell proliferation were investigated by testing empagliflozin in a broad panel of in vitro pharmacology screening assays. The results from this testing revealed that empagliflozin had very low protein binding potential and can be considered to be nonpromiscuous against this sample set of the human kinome and proteome. Thus, there were no off-target activities for empagliflozin suggestive of alternative modes of action for the proliferative findings observed in vivo. Given that M446/2 was shown to be cytotoxic, it was not tested for pro-proliferative or off-target binding or functional effects.
The findings presented here concur with previously published observations of the lack of cytotoxicity and genotoxicity of empagliflozin 2 and those from the in vivo investigative toxicology studies in mice (unpublished data). The in vitro cytotoxic responses provide additional evidence that the in vivo observations of early subtle degenerative changes in the renal proximal tubule of male mice treated with 1,000 mg/kg of empagliflozin for 13 weeks are due to low-level chronic renal tubular injury (unpublished data). Similar findings have been previously demonstrated for nongenotoxic nephrotoxicants, including those that induce tumors through a sustained cytotoxicity/cell regeneration mode of action. 30 The absence of genotoxicity for empagliflozin and M466/2, presented here, is supported by gene expression profiles from the in vivo studies (unpublished data) demonstrating responses to renal injury and oxidative stress followed by proliferative and cell cycle genes in the absence of clear drug-related renal DNA damage/repair signaling (unpublished data). The lack of a direct in vitro proliferative effect for empagliflozin is consistent with observations in the renal proximal tubule of male mice treated with empagliflozin (1,000 mg/kg for 13 weeks) which show an indirect regenerative proliferative response secondary to low-level renal injury. An underlying degenerative/regenerative mechanism for the observations in the mouse study is further supported by the absence of renal cell proliferation in studies with empagliflozin in other species 2 or in SGLT2 knockout mice or human deficiencies in SGLT2. 1,31 –33
The in vitro profiles of empagliflozin and M466/2 presented here are key mechanistic information supportive of the comprehensive mode of action and human relevance assessment of the finding of male CD-1 mouse renal adenocarcinoma associated with lifetime exposure to empagliflozin (unpublished data). The weight of evidence when considering gender and mouse strain specificity, pharmacology-related stress, and the increased cellular oxidative burden due to the male mouse-specific renal production of M466/2 is consistent with a nongenotoxic mode of action. The specificity of this male mouse renal oxidative metabolism pathway of empagliflozin also indicates that the findings in mice have no relevance to humans. 4 In addition, the male mouse-specific metabolic basis for formation of a cytotoxic moiety within the renal proximal tubule differentiates empagliflozin from the mechanisms believed responsible for carcinogenicity findings with another SGLT2 inhibitor. 34,35
In summary, empagliflozin is not cytotoxic, genotoxic, or mitogenic in our in vitro investigative toxicology studies or prior studies. 2 The male mouse predominant oxidative renal metabolite M466/2 is not genotoxic, but these in vitro data show that M466/2-derived 4-OH CTA exposure is cytotoxic to multiple cell types including renal tubule cells. This cytotoxicity may be involved in promoting additional compound related to in vivo renal metabolic stress and further chronic low-level renal injury that support a nongenotoxic mode of tumor pathogenesis. These findings associate the male mouse predominant empagliflozin renal metabolite M466/2 with the male mouse specificity in renal injury observed in carcinogenicity studies which precede the late appearance of renal tumors in high-dose male mice.
Footnotes
Authors’ Note
All animal use described herein was performed under animal protocols that were reviewed and approved by an institutional panel and complied with the guidelines and principles listed by the International Journal of Toxicology.
Acknowledgments
The authors would like to acknowledge the contributions of Dr Jaromir Mikl for statistical analysis, Dr John Ginn for preparation of figures, Andrea Whitcher-Johnstone for analysis of empagliflozin metabolism in mRTE cells, Leah Her for coordination of genotoxicity analysis, Lalitha Podila for analysis of SGLT2 activity in mRTE cells, and Regina Nardi for bioanalysis.
Authors’ Contributions
J. Smith contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. Z. Huang contributed to conception and design, contributed to acquisition, analysis, and interpretation, and critically revised the manuscript. P. Escobar contributed to conception and design, contributed to acquisition, analysis, and interpretation, and drafted the manuscript. P. Foppiano contributed to acquisition, analysis, and interpretation and critically revised the manuscript. H. Maw contributed to conception and design, contributed to acquisition, analysis, and interpretation, and critically revised the manuscript. W. Loging contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. H. Yu contributed to conception, contributed to analysis and interpretation, and critically revised the manuscript. J. Phillips contributed to conception, contributed to analysis and interpretation, and critically revised the manuscript. M. Taub contributed to conception, contributed to analysis and interpretation, and critically revised the manuscript. W. Ku contributed to conception, contributed to analysis and interpretation, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: When the data were generated, all authors were employed by Boehringer Ingelheim Pharmaceuticals Inc, which manufactures and sells empagliflozin.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Boehringer Ingelheim Pharmaceuticals Inc.
Supplementary Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.