
Abstract
In support of a clinical trial in the pediatric population, available nonclinical and clinical data provide input on the study design and safety monitoring considerations. When the existing data are lacking to support the safety of the planned pediatric clinical trial, a juvenile animal toxicity study is likely required. Usually a single relevant species, preferably a rodent, is chosen as the species of choice, while a nonrodent species can be appropriate when scientifically justified. Juvenile toxicology studies, in general, are complicated both conceptually and logistically. Development in young animals is a continuous process with different organs maturing at different rates and time. Structural and functional maturational differences have been shown to affect drug safety. Key points to consider in conducting a juvenile toxicology study include a comparative development of the organ systems, differences in the pharmacokinetics/absorption, distribution, metabolism, excretion (PK/ADME) profiles of the drug between young animal and child, and logistical requirement in the juvenile study design. The purpose of this publication is to note pertinent points to consider when designing and conducting juvenile toxicology studies and to aid in future modifications and enhancements of these studies to enable a superior predictability of safety of medicines in the pediatric population.
Introduction
For most drug development programs, the indication targets adults initially and is later expanded to include a pediatric population. Owing to differences between the adult and the developing child, a pediatric clinical trial is most likely required to support the therapeutic use in children, unless a waiver is granted. Often, investigators must consider ethical and practical consideration of performing pediatric clinical trials. Available nonclinical and clinical data provide input on the study design and safety monitoring considerations of a pediatric trial. Completed nonclinical toxicology studies in mature animals supplemented with reproductive developmental toxicity studies, and combined with safety data from clinical trials in adults, provide the basis of predicting the safety of medicine in the pediatric population. When a pediatric clinical trial is required, the existing data may not be sufficient to support the safety of the planned trial. It is in these situations that a juvenile animal toxicity study is more than likely required prior to initiating the pediatric trial. Factors unique to juvenile toxicity studies present a challenge to the drug development team. In designing a juvenile toxicity study, many aspects must be considered including but not limited to the test species, target organs and age-relevant organ development, pharmacokinetics / absorption, distribution, metabolism, excretion (PK/ADME), pharmacology, and chemical structure characteristics. The timing of a juvenile study to support a pediatric trial (depending on whether a deferral is granted) differs among regulatory agencies. In both the European Union (EU) and United States, the pediatric plan begins during the program development stage. In the EU, a Pediatric Investigational Plan (PIP), approved by the Pediatric Committee, must be in place prior to submitting a marketing application. In the United States, a final pediatric commitment is often a part of the postmarketing requirement.
During the 2015 Annual Meeting of the American College of Toxicology (Summerlin, Nevada), a juvenile toxicology continuing education course was presented. This publication summarizes the 3 technical presentations from the continuing education course. The course included US and EU regulatory perspectives, but these are not presented herein as the regulatory perspectives can differ widely among health authorities and regulatory guidance documents are constantly evolving. The forthcoming International Council for Harmonization (ICH) S11 Guidance “Nonclinical Safety Testing in Support of Development of Pediatric Medicines” is expected to harmonize regulatory differences. 1 This topic was endorsed by the ICH Steering Committee in November 2014 followed by an ICH press release in December 2015. The guideline process is currently ongoing, and the anticipated milestones for the Expert Working Group developing this new guideline are anticipated in 2017 and 2018. Three technical presentations in the course were provided by Robert M. Parker, PhD, DABT, Gerhard F. Weinbauer, PhD, and Amera K. Remick, DVM, DACVP, DABT, and addressed considerations and challenges associated with conducting juvenile toxicity studies and included examples based on industry experience. Areas covered in this publication include discussions of rodent and nonrodent species used in juvenile toxicology studies and histopathologic considerations when performing studies in young developing animals. Much of what is currently known about the conduct and interpretation of juvenile toxicology studies comes from on-the-job experiences of toxicologists and pathologists. The purpose of this publication is to identify challenges and pertinent points to consider when designing and conducting juvenile toxicology studies and to aid in future modifications and enhancements of these studies which will enable a superior predictability of safety of medicines that will be used by the pediatric population.
The ICH M3 Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals 2 stated that the conduct of any juvenile animal toxicity studies should be considered only when previous animal data and human safety data, including effects from other drugs of the pharmacological class, are judged to be insufficient to support pediatric clinical trials. If a study is warranted, one relevant species preferably a rodent would be generally considered adequate by the regulatory agencies and scientific community. A study in a nonrodent species can be appropriate when scientifically justified.
Juvenile toxicology studies, in general, are complicated both conceptually and logistically for a number of reasons including the dynamic anatomical and physiological changes occurring during early postnatal development when compared to the relatively static adult anatomy and physiology; the potential for latency of effects on neurobehavioral and reproductive end points; the greater interdependence of end points (eg, the covariance of body weight); a myriad modes of action; and litter-based statistical analysis may be required. The reasons why juvenile toxicity studies are logistically complicated are related to the age range required to cover childhood development of the target organs: size and design of studies; litter composition; number and diversity of end points; difficulty in scoring end points; distinguishing direct versus latent effects; and small size of the animals for dose administration; and blood and tissue sample collection.
Three key knowledge points in conducting a juvenile toxicology study include the following: Understanding comparative development of the organ systems in the animal species versus that of the child. Understanding differences in the PK/ADME profiles of the drug in the test species versus that of the child and adult. Understanding of logistical requirement in study design (eg, cross-fostering requirements, multiple behavioral tests, brain perfusion of young pups, reproductive capacity assessment, etc).
Development in young animals is a continuous process with different organs maturing at different rates over time. Structural/functional maturational differences have been shown to affect drug safety profile. Postnatal toxicity is more likely in tissues undergoing postnatal development. Excretory organ (liver and kidneys) development has the greatest impact on drug disposition (pharmacokinetics). Therefore, one should anticipate age-related differences in drug disposition based on knowledge of ontogeny of the liver and kidney. The most dramatic changes occur during the first days to months of life. Unfortunately, the effect of ontogeny on tissue/organ sensitivity to drugs (pharmacodynamics) in animal models has been poorly studied. It is also important to keep in perspective that the disease state may alter a drug’s pharmacokinetic and pharmacodynamic profiles, therefore leading to its potential toxicity.
Juvenile Toxicology Study Design Considerations
Many different designs of juvenile toxicology studies have been performed over the years. Optimally, the study director needs the following background information to design the most appropriate juvenile toxicity study: indication and intended pediatric population, mechanism of action, sensitivity of species, justification of test species, toxicity and target organs, route of administration, and gap assessment of available reproductive and developmental toxicity and general toxicity study data including any previously generated juvenile toxicity data. Other information such as toxicokinetic data, metabolic profile, receptor–ligand binding assay results, and structural activity relationships information supplement the designing of an optimal study.
Principles for postnatal development, juvenile toxicity study design, and data interpretation
Often much diversity can exist between litters and is the largest contributor to physical and behavioral variability in juvenile studies that become more enhanced as animals age. Study design determines the conclusions that may be drawn from the data. Sample size may vary based on the study design complexity and logistical/technical issues. It is vital to know the test species well for the test molecule because patterns exhibited by the test species are exceedingly important.
Biotechnology-derived pharmaceuticals
Biotechnology-derived pharmaceuticals are a diverse group of therapeutics derived from biological sources or complex biotechnological processes and commonly include monoclonal antibodies, peptides/proteins, vaccines, allergen extracts, antitoxins and blood-derived products. There are specific challenges involved in testing such as diverse structural and biological properties, species specificity, and the potential for immunogenicity. The ICH S6 Guidance “Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals” 3 provide a flexible, case-by-case, science-based approach; it does not address juvenile toxicity assessments but has a discussion of species specificity for safety testing of biological molecules. Pharmacological relevance and cross-reactivity of the biologic molecule should be established in the test species. The biological molecule should induce similar in vitro or in vivo pharmacological effects in test species. And the target of the molecule must be expressed in animal test species, and this implies that the target distribution, sequence homology, and affinity to drug candidate should be similar and the downstream effects should be comparable between the test species and human. In practice, this often means that the toxicologic testing of biologics is limited to the nonhuman primate (NHP) model.
Species selection
The selection of the test species for the juvenile study must be justified. There are several items to consider in the selection of the juvenile test species. The following are points to consider in selecting the most relevant species. Much of the available information comes from the human where differences are seen among various pediatric ages to adults.
The choice of the test species for the juvenile toxicology studies depends on the pharmacology, PK/ADME profile, test species used in mature animal toxicology studies, and target organs from the mature animal toxicology studies. Test species used by the pharmaceutical companies were assessed based on the pediatric investigational plans and industry-wide surveys. 4,5 The majority of juvenile studies use rodents (>70%), with the rat being the most prevalent model, followed by the dog (9%-12%), NHP (2%-4%), and minipig (1%-2%), with other species (rabbit and sheep) being the least used (Figures 1A and 1B).
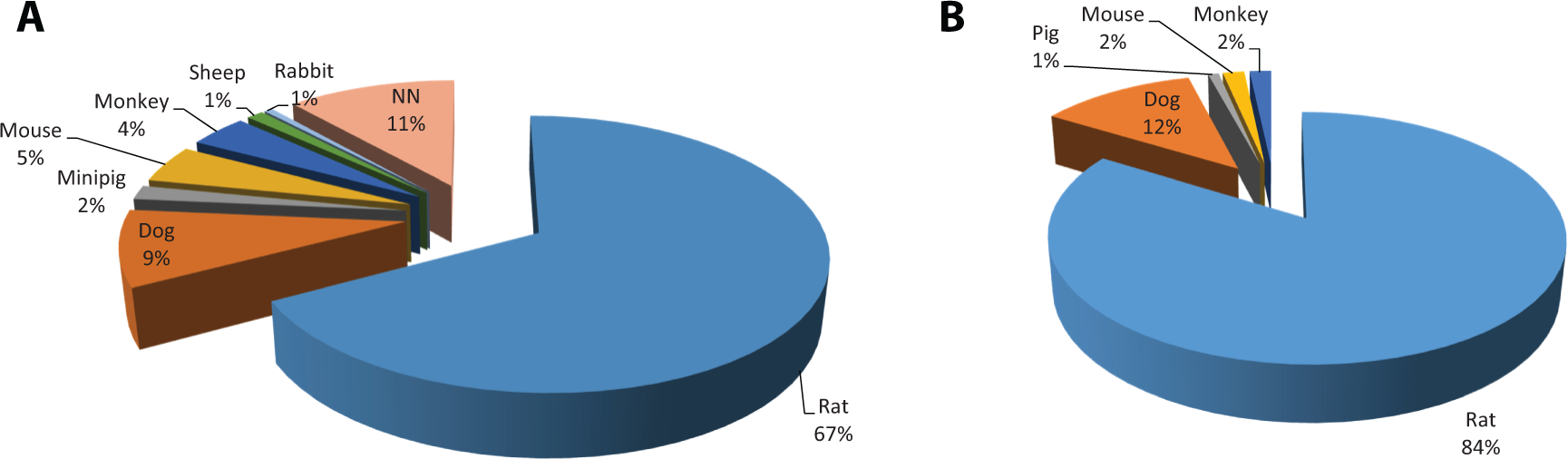
A, Species most commonly used in juvenile toxicology studies from pediatric investigational plans (PIP). 4 Of 164 PIPs submitted from 2007 to 2013, the rat (117) was the most common test species followed by the dog (16), mouse (8), monkey (7), minipig (3), sheep (2), and rabbit (1). Of these, 10 cases evaluated 2 species while others investigated a single species; 20 PIPs did not specify the test species (noted as NN). B, Species most commonly used in juvenile toxicology studies from industry survey. 5 Of 241 studies from 24 companies, the rat (202) was the most common, followed by the dog (29), mouse (4), monkey (4), and minipig (2).
Age and growth comparison of species
Although the organ development in growing animals can differ, a general age range at development phase and growth can be used as a comparison to the human (Table 1). Rodent species mature over days to weeks, while nonrodent species matures over months to years.
Age Comparison of Development Phase Among Species. 11
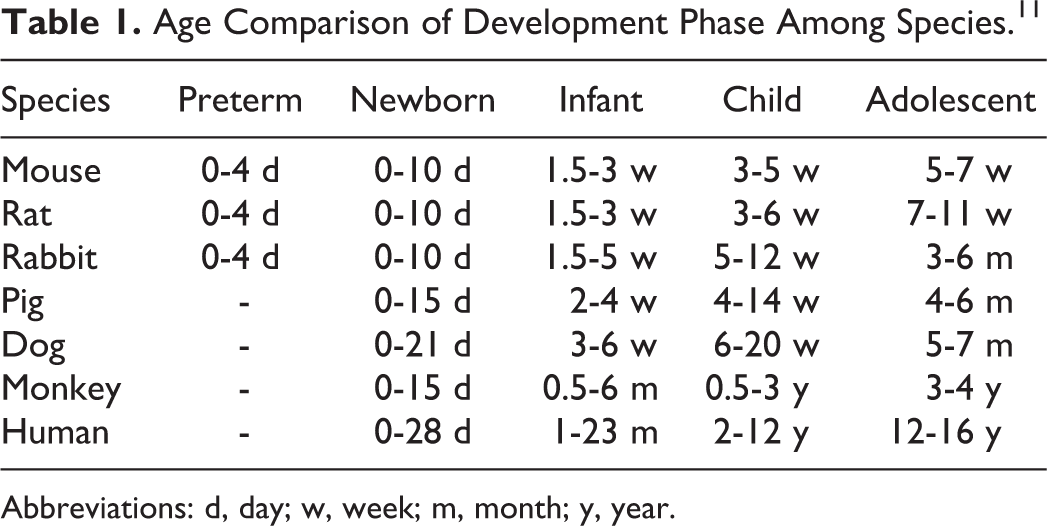
Abbreviations: d, day; w, week; m, month; y, year.
Organ development can vary in growing animals and should be considered when a target organ has been identified in toxicology studies with mature animals. The comparative ages based on central nervous system (CNS) and reproductive development in the rat, minipig, dog, NHP, and human are shown in Figure 2.
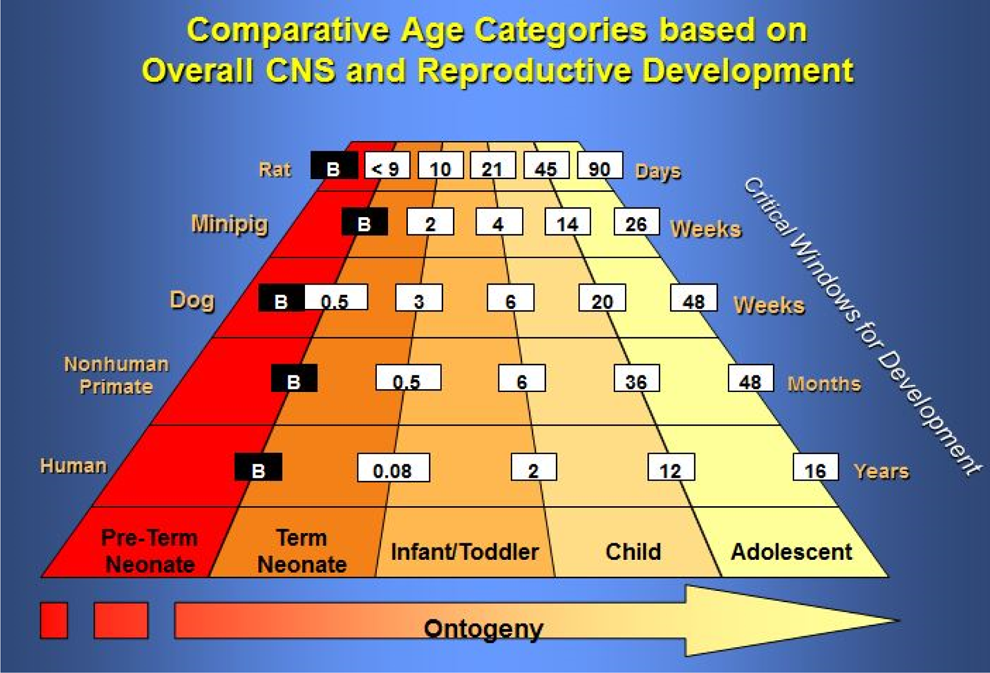
Comparative age categories based on overall central nervous system (CNS) and reproductive development in the rat, minipig, dog, nonhuman primate, and human. 7
Sexual maturation and growth can vary among species in relation to the life expectancy (Figure 3). The sexual maturation can range between species. Except for nonhuman primate and human, all other species reach sexual maturity within about 10% of the life expectancy. The growth plate closure can also vary between species. Although not presented in Figure 3, the sexual maturity of the minipig occurs at approximately 3% of the life span, and epiphyseal closure occurs very late in the minipig relative to sexual maturity (∼25%).
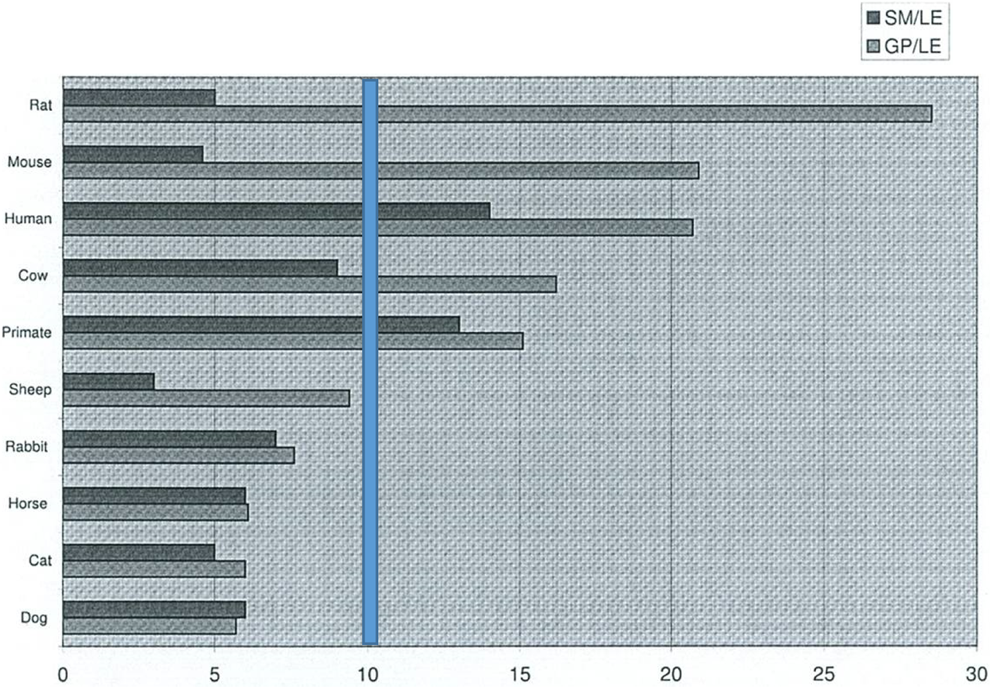
Age at sexual maturity and growth plate closure compared to life expectancy. 8 Figure shows the percentage of life span in relation to the growth and maturation process in various species. In the minipig, sexual maturity is at approximately 3% of life span, while growth plate closure occurs at approximately 25%. Primate data are from cynomolgus monkeys. Except for nonhuman primate and human, all other species reach sexual maturity within about 10% of the life expectancy (vertical bar). SM indicates sexual maturity; LE, life expectancy; GP, growth plate closure.
Toxicokinetics
In developmental and juvenile studies, systemic exposures are more relevant for assessing toxicity than the external dose. During the juvenile study, there may be several toxicokinetic intervals (with different dose levels) to correspond with different ages and maturity of ADME components. Toxicokinetic parameters are very useful for age and cross-species comparisons and characterizing plasma levels of parent drug, and relevant metabolites may clarify whether or not selective functional/developmental changes do occur or whether changes actually occur at same or even at greater internal exposures than in adults.
Absorption
Absorption is based on the physicochemical properties of the drug, for example, lipid versus water solubility. Compared to the adult, the child has a greater volume of distribution (Vd). Additionally, the child has a small size with a large surface area to body weight ratio. Although the body composition (extracellular water, intracellular water, fat, and protein) varies with age and diet, there are certain trends that are apparent (Figure 4). From newborn to adult, the following changes occur:
the percentage of extracellular water volume decreases from 38% to 17%;
intercellular water volume remains at approximately 40%;
fat content starts at 10% initially, increases to 25% during the first year of life, and then decreases to 18% by adulthood; and
protein amount increases from 10% to 20%.
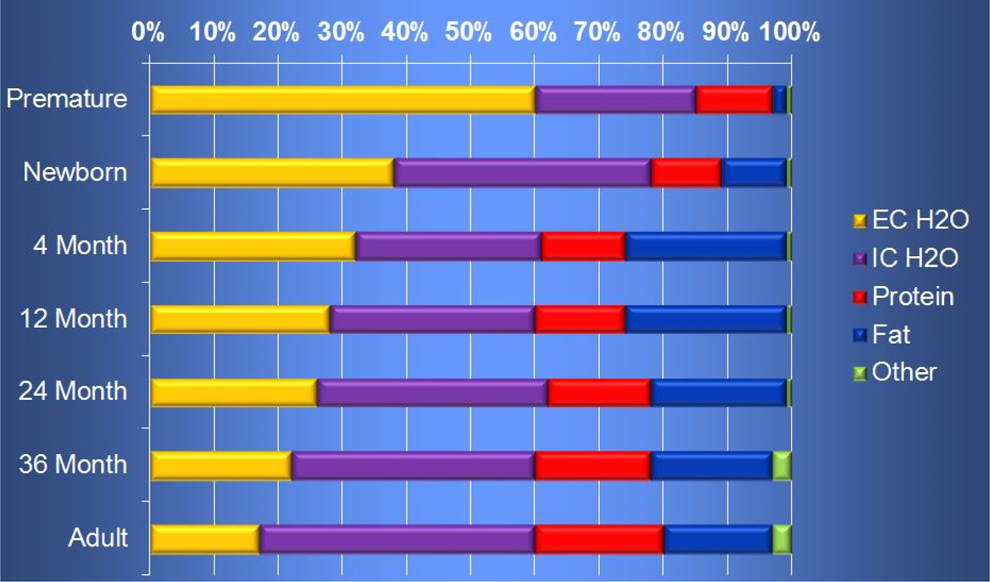
Ontogeny of body composition. 9 EC indicates extracellular; IC, intracellular.
The child has increased gastric pH resulting in increased basic and decreased acidic drug absorption, and the child has reduced gastric emptying with variable gastrointestinal motility with the infant/neonate being reduced and the child greater. The bioavailability of the molecule is often unpredictable: Topical effect is increased in neonates, intramuscular absorption is faster, and inhalation absorption is increased (Table 2).
Trends in Pharmacokinetic Function in Human by Age-Groups Compared to Adults. 12

Abbreviations: NA, not applicable; U, uncertain; BW3/4, body weight at ¾ exponential power;
↑, increased; ↓, decreased; ↔, equivalent.
Metabolism
In the immature liver, the first pass effect is decreased. During phase I maturation (0-3 years of age), enzymatic activity varies with cytochrome P450 enzyme’s ontogeny and the substrate (Table 2). Phase II maturation occurs over the first 12 years.
Distribution
During the pre-/postnatal development, there are also some unique exposure routes (through placental transport and through lactation). Milk can concentrate lipophilic drugs up to 6-fold over maternal plasma levels. Transporter ontogeny affects the distribution of drugs (Figure 5). In addition, tissue/receptor/protein binding differences with age affect the concentration of free drugs available in circulation. The blood–brain barrier (BBB) tends to be immature in infants, and immature BBB allows more drugs to enter the CNS to induce potential neurobehavioral effects. 10,13,14 While often attributed to the lack of integrity of the tight junctions (Zona Occludens) of the immature brain, the most likely cause is the immaturity of the transporter systems at the BBB (endogenous BBB transporters including receptor-mediated transporters, carrier-mediated transporters and active efflux transporters).
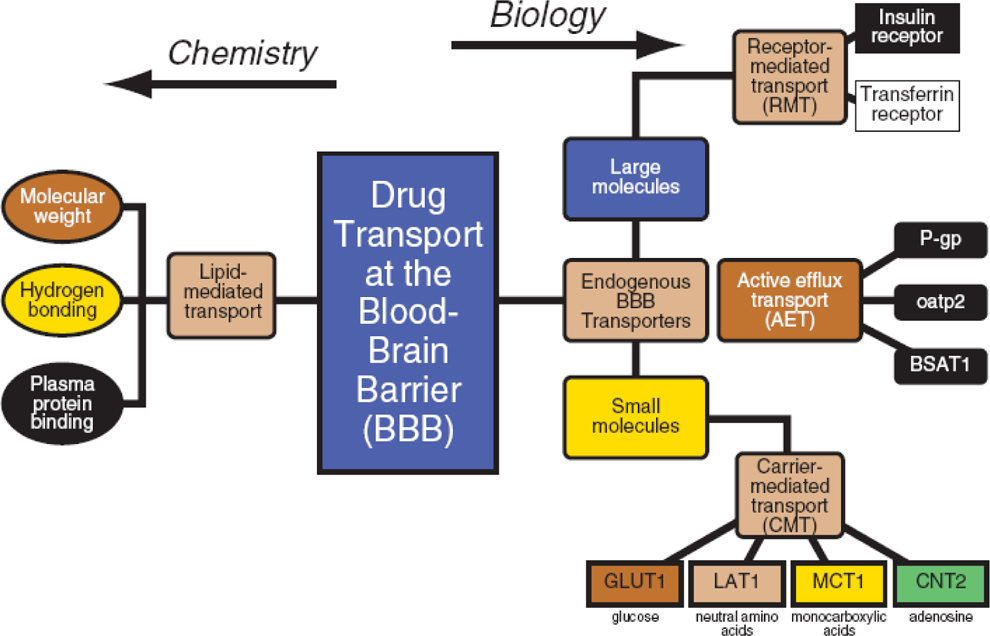
Distribution.
10
More drugs enter the immature blood–brain barrier (neonatal). Ontogeny of transporters involved is most likely to cause drug uptake and not likely related to zona occludens (tight junctions). P-gp indicates P-glycoprotein; oatp2, organic anion-transporting peptide 2; BSAT1, brain-specific anion transporter 1; GLUT1, glucose transporter 1; LAT1,
Excretion
Excretion generally depends on available transporters. The kidney develops full function by 2 to 3 years in humans. The immature human kidney has reduced glomerular filtration rate, reduced tubular secretion/reabsorption, and reduced kidney perfusion. The infant/neonate has slower clearance with a longer half-life, while the child has a rapid clearance with a shorter half-life. 6
Dose administration
When performing juvenile toxicology studies, the size of the young animals inherently leads to several technical challenges. Dose administration in young animals requires a sound technical procedure. The time of earliest possible dosing in young animals can vary depending on the species and the route of administration (Table 3).
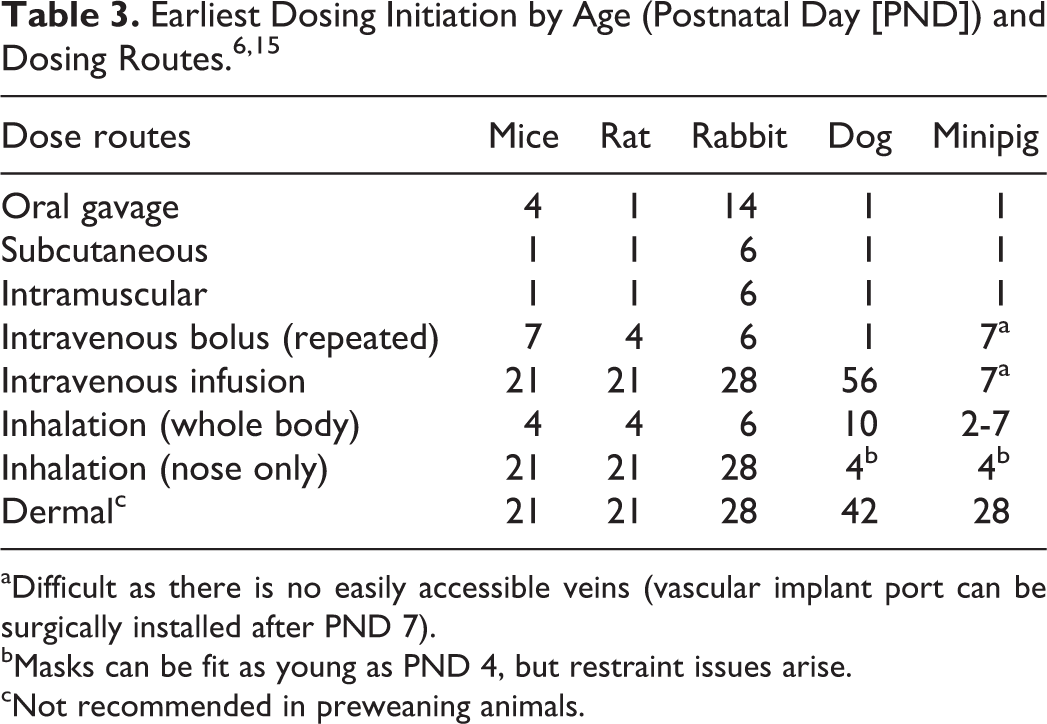
aDifficult as there is no easily accessible veins (vascular implant port can be surgically installed after PND 7).
bMasks can be fit as young as PND 4, but restraint issues arise.
cNot recommended in preweaning animals.
Litter composition
Litter composition has a direct impact on the study design. Simply stated, the “litter effect” considers pups within a litter to show less diversity of response than pups from different litters and therefore are not “independent subjects” while they remain in their original litter.
6
Several options are available to avoid the litter effect: Within-Litter Design—Each litter has all treatment groups. Split-Litter Design—Each litter has some of the treatment groups. Between-Litter Design—Each litter has the same treatment group. Fostering Design—Each litter is composed of pups from other litters without using any same sex siblings. All pups within new litter receive the same treatment. One Pup per Sex per Litter Design—Select one pup per sex per litter.
Each method has its advantages and disadvantages. Logistically, one must consider technical challenges, potential cross-contamination, Institutional Animal Care and Use Committee (IACUC) queries to address, and statistical evaluation of the data.
The “within-litter design” has the animals within the litter assigned to the control and each treatment group. While statistically sound, this method is logistically difficult; each pup requires dosing from a different dose level causing difficulties for the technicians in identifying the pups and administering the dosage correctly. This method is also fraught with cross-contamination issues.
The “split-litter design” is a variation of the within-litter design and has been used when the number of treatment groups exceeds the number of pups per litter. For example, if the study design requires 7 groups and the litter size is limited, then the litter may be insufficient to be represented in all groups.
The “between-litter design” has all pups in the litter treated with the same dosage. While minimizing technical efforts with less complicated process and no contamination issues, it is not statistically optimal due to the well-known “litter effect” (siblings tend to respond more similarly than nonsiblings).
The “fostering design” has each litter composed of pups from other litters without using any same-sex siblings, and all pups within the new fostered litter receive the same treatment. The cross-fostering procedure eliminates the “litter effect” and therefore is statistically acceptable. There would be no contamination issues and the daily technical effort would be simplified. Utilization of careful planning and selection of healthy pups allows for cross-fostering pups to be accepted by the dams. However, the cross-fostering procedure is a time-consuming and labor-intensive process.
The “one pup per sex per litter design” eliminates the “litter effect” and therefore is statistically acceptable. There would be no contamination issues, and the technical effort would be simplified; however, the drawback is the number of litters potentially required (eg, ∼ 48 litters for a typical study) and number of pups not selected and therefore discarded (eg, ∼ 288 pups for a standard study). The “one pup per sex per litter design” would be very difficult to justify to the IACUC.
Study design
There are basically four components of juvenile toxicity study designs: (1) general toxicity (including specific targeted organ juvenile toxicity), (2) reproductive toxicity, (3) CNS toxicity, and (4) immunotoxicity. The study designs based on the evaluation parameters are included subsequently.
Rodents as a Juvenile Safety Testing Model
Rodents are the most common species tested in juvenile toxicity studies due to a number of factors, including the extensive experience and historical data across laboratories and the ability to test the full span of postnatal development. However, there are limitations as well, which can present challenges to designing and executing successful studies. The use of rodents has both advantages and disadvantages:
Advantages are that rodents are most frequently used for juvenile testing, extensive experience/historical data exists across testing laboratories and can test full span of postnatal development, ability to procure appropriate numbers of animals even for early age assessments, and a wide range of neurobehavioral tests are available.
Disadvantages are that rodents may not be pharmacologically relevant, small size limits number of routes of administration, the earliest day of dosing initiation is often based on the dosing route, the potential immunogenic response, small size limits ability to collect multiple biologic specimens, and different age categories based on overall CNS and reproductive development among species.
Examples of various juvenile toxicology study designs in rodents are shown subsequently. These include for general toxicity (Figure 6), for toxicity with CNS and reproductive development evaluation (Figure 7), and toxicity with immunological evaluation (Figure 8).
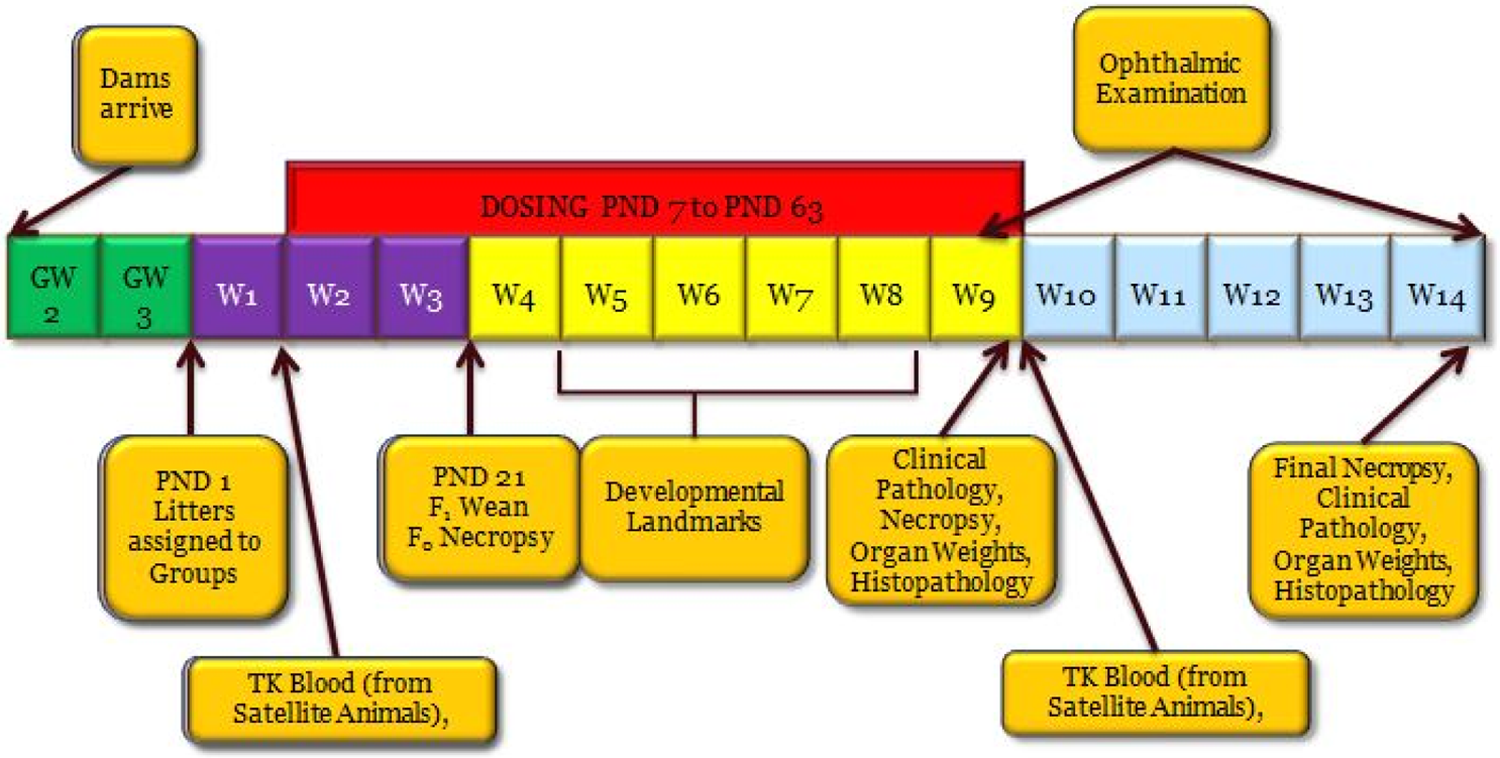
Juvenile toxicity study design—general toxicity. PND indicates postnatal day; GW, gestation week; W, week; F0, parent generation; F1, offspring (juvenile animals); TK, toxicokinetics.
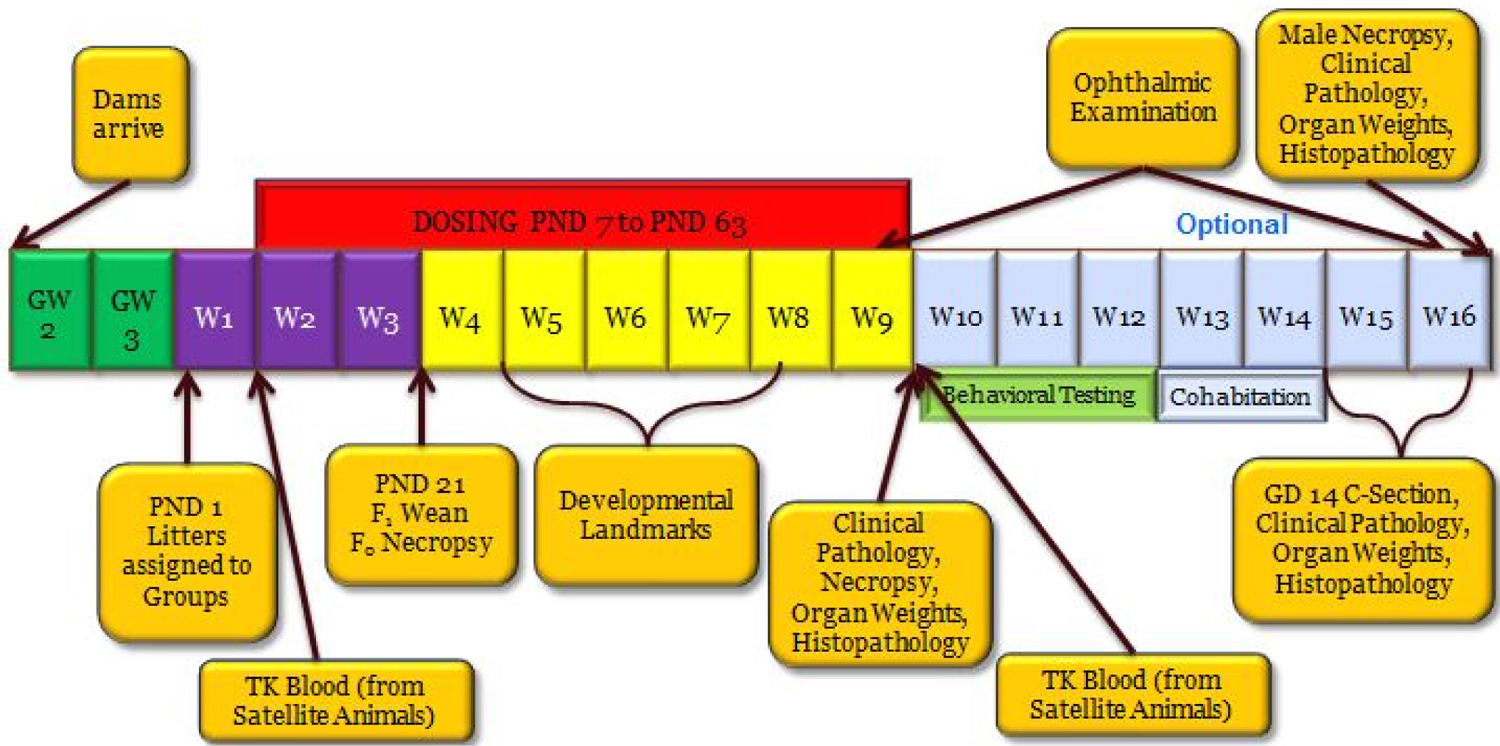
Juvenile toxicity study design—targeted CNS and reproductive evaluation. PND indicates postnatal day; GW, gestation week; W, week; GD, gestation day; F0, parent generation; F1, offspring (juvenile animals); TK, toxicokinetics; CNS, central nervous system.
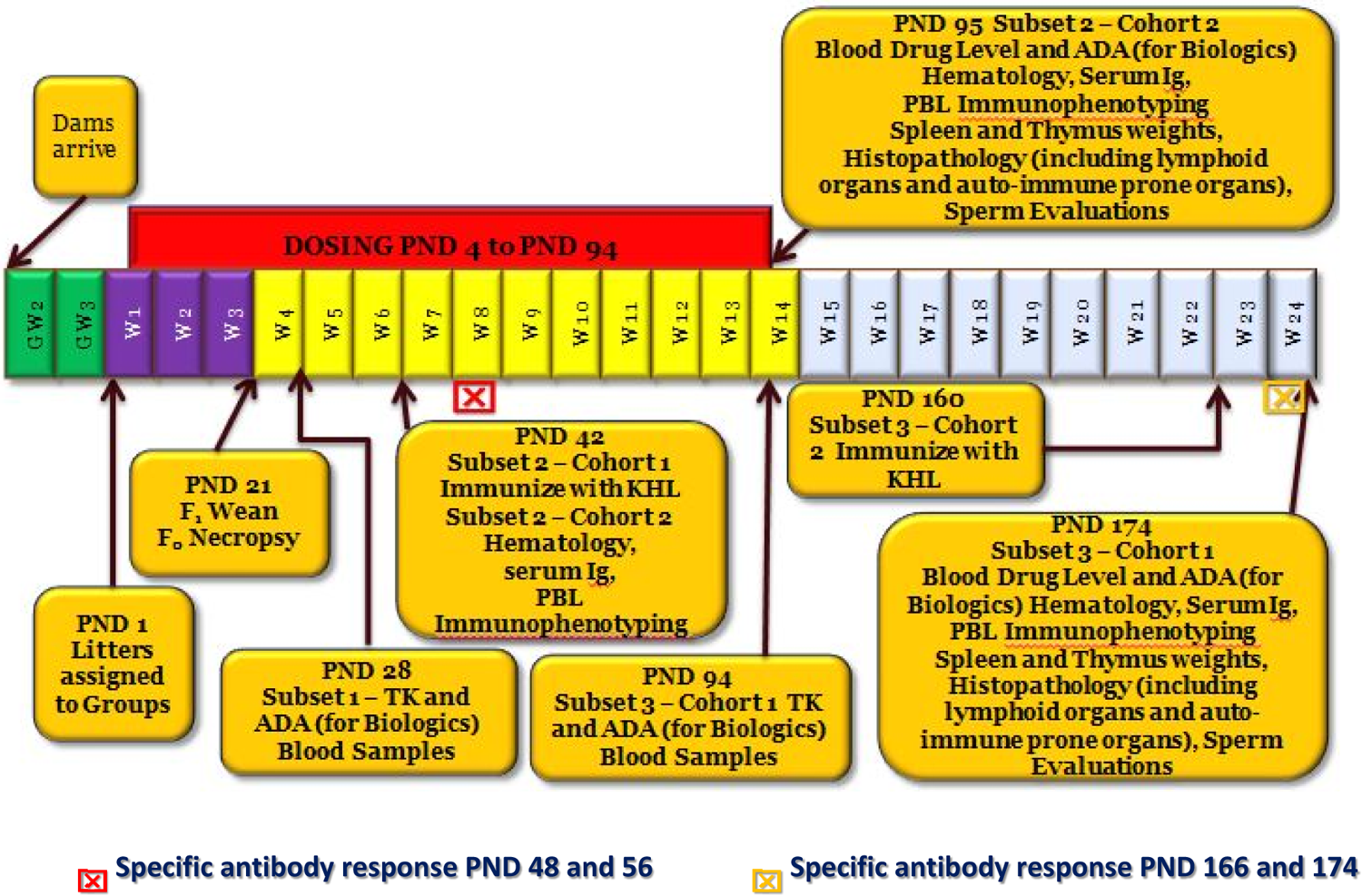
Juvenile toxicity study design—targeted immunological evaluation. PND indicates postnatal day; GW, gestation week; W, week; F0, parent generation; F1, offspring (juvenile animals); ADA, anti-drug antibody; Ig, immunoglobulin; KHL, keyhole limpet hemocyanine; PBL, peripheral blood lymphocyte; TK, toxicokinetics
Nonrodents as a Model for Juvenile Toxicology Studies
Three most commonly used nonrodent animal models are the dog, NHP (with cynomolgus monkey as predominant species), and minipig models. From an overall perspective, the choice of species is driven by the same considerations as for general safety assessment, in that the most relevant species should be used in order to provide meaningful identification of hazard and risk. 11,16 –18 Beyond that, species selection may also be driven by the maturation timing of specific targets organs, making sure that relevant postnatal development matches exposure periods. Similarly, the choice of study end points is influenced by considering feasibility, age-appropriate end points, and timing of testing relative to exposure.
For biopharmaceuticals, there is an increased demand for studies in juvenile NHPs. In sharp contrast to most other laboratory animal species, primates achieve sexual maturity much later in life and consist of less than 10% of their life span (Figure 3). 8 Therefore, covering the entire postnatal development through sexual maturity and beyond in juvenile toxicity testing is not feasible in NHPs.
Alternatively, various phases and timing of postnatal development are well described for nonrodent models, 7,11,18,19 and the appropriate organ development phases that correspond to age of the human target population can be assessed.
Similar to rodents, a variety of study design options can be considered for the dog and minipig and may be decided on a case-by-case basis. 20 –22 Examples of study designs include within-litter design, split-litter design, between-litter design (whole litter), fostering design, and also pre- and postweaning approaches. Each of these designs can be complex and require a large number of animals. A typical design of a juvenile toxicology study in dogs for general toxicity (Table 4) integrates a 3-month treatment regimen along with intermediate groups of animals at key developmental ages for evaluation of safety parameters. When assessing a specific target organ, additional parameters will add to the complexity. With a molecule that targets CNS (Figure 9), neurobehavioral evaluation and neuropathology will be included. Because dogs have the potential to vomit, within-litter designs may result in cross contamination. In a recent case study using within-litter design and a successful preweaning approach, a separate set of control animals was used to account for this, and minor but no serious cross contamination was observed. 20
Example of Juvenile Toxicology Study Design in Dogs. 20
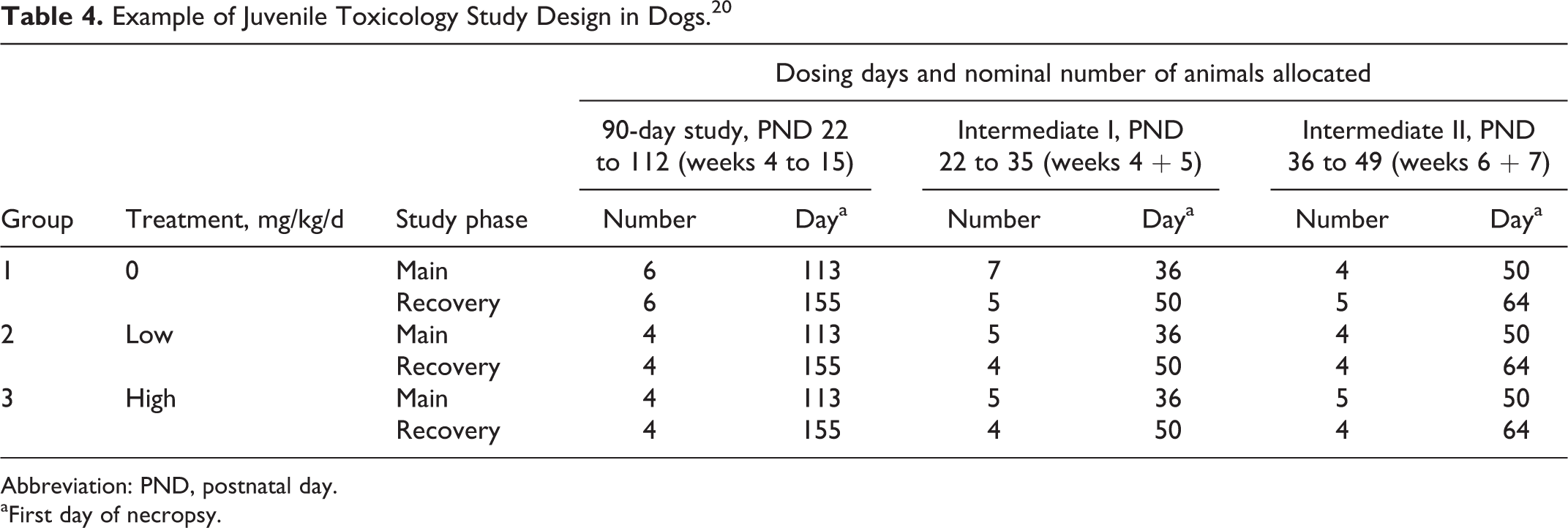
Abbreviation: PND, postnatal day.
aFirst day of necropsy.
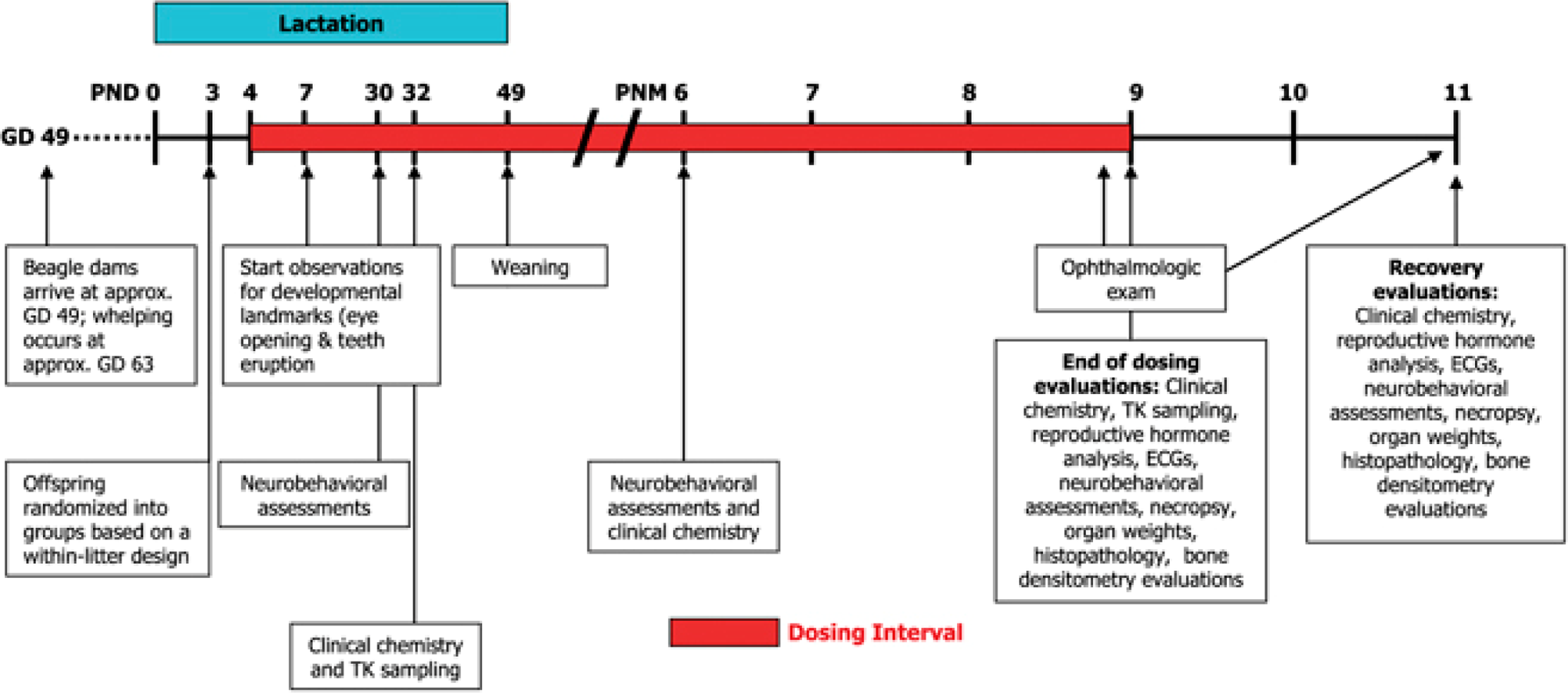
Example of juvenile toxicology study design in dogs for a CNS active molecule. 21 GD indicates gestation day; PND, postnatal day; PNM, postnatal month; TK, toxicokinetics; ECG, electrocardiography; CNS, central nervous system.
When considering the nonrodent species, minipigs may provide a more practical approach for a juvenile study than preweaned dogs in consideration of the following: currently pregnant sows are easier to obtain, litter size is more consistent, piglets can readily be cross fostered, and they wean earlier. 23 The growth of the cross-fostered piglets is comparable to that of the hand-reared piglets separated from the dam ∼24 hours after birth (after initial suckling—colostrum) and entirely artificially fed thereafter using a milk replaced formula (Figure 10). However, one should be aware that juvenile minipigs are larger and already weigh 5 to 10 kg at the start of the study and can get much larger as they age (Figure 11), with more challenges in handling of pigs. Minipigs achieve sexual maturity rather early within 3 to 5 months, but other work suggests that this might take 6 to 8 months 26 which would be similar timing to the dog. An example of juvenile toxicology study design in minipigs is shown in Figure 12. Certain dosing routes such as oral in minipigs can be a challenge while dermal method is easier, and parenteral dosing may require alternative delivery mode such as the use of vascular access port (VAP) for intravenous delivery.
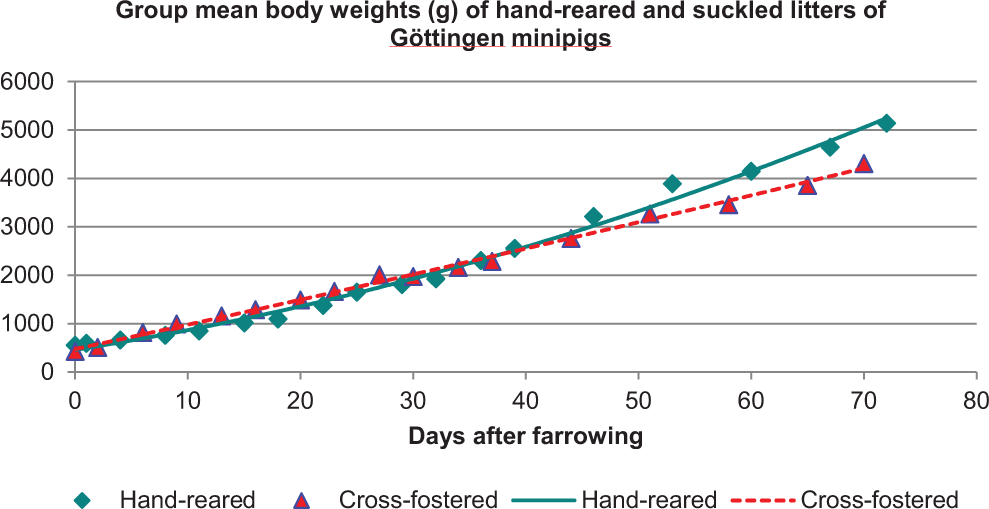
Body weight difference between bottle-raised versus suckling piglets. 24 Hand-reared piglets separated from the dam ∼24 hours after birth (after initial suckling—colostrum), and entirely artificially fed thereafter using a milk-replaced formula.
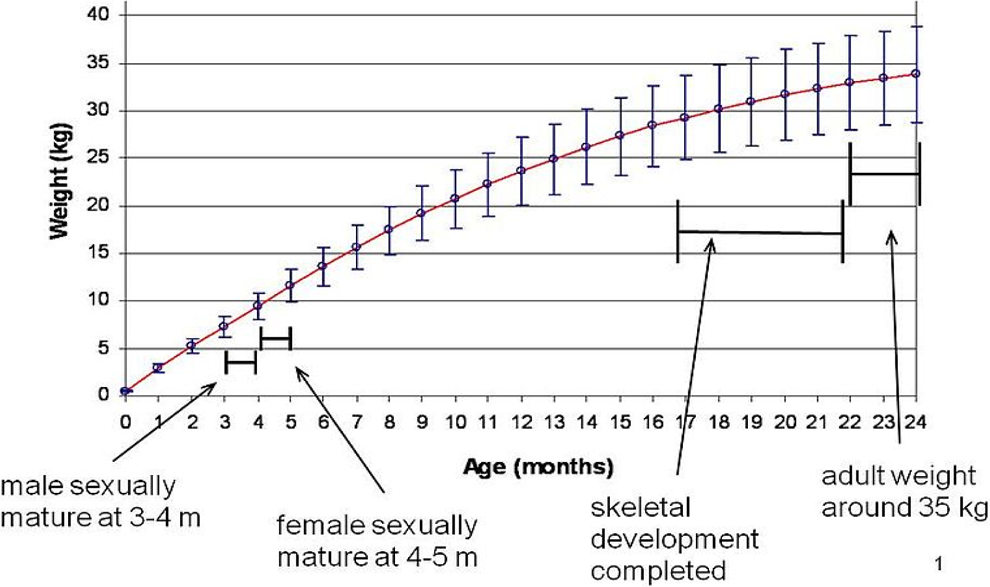
Growth curve of Göttingen minipig. 25 Growth curve and key development landmarks for Göttingen minipig.
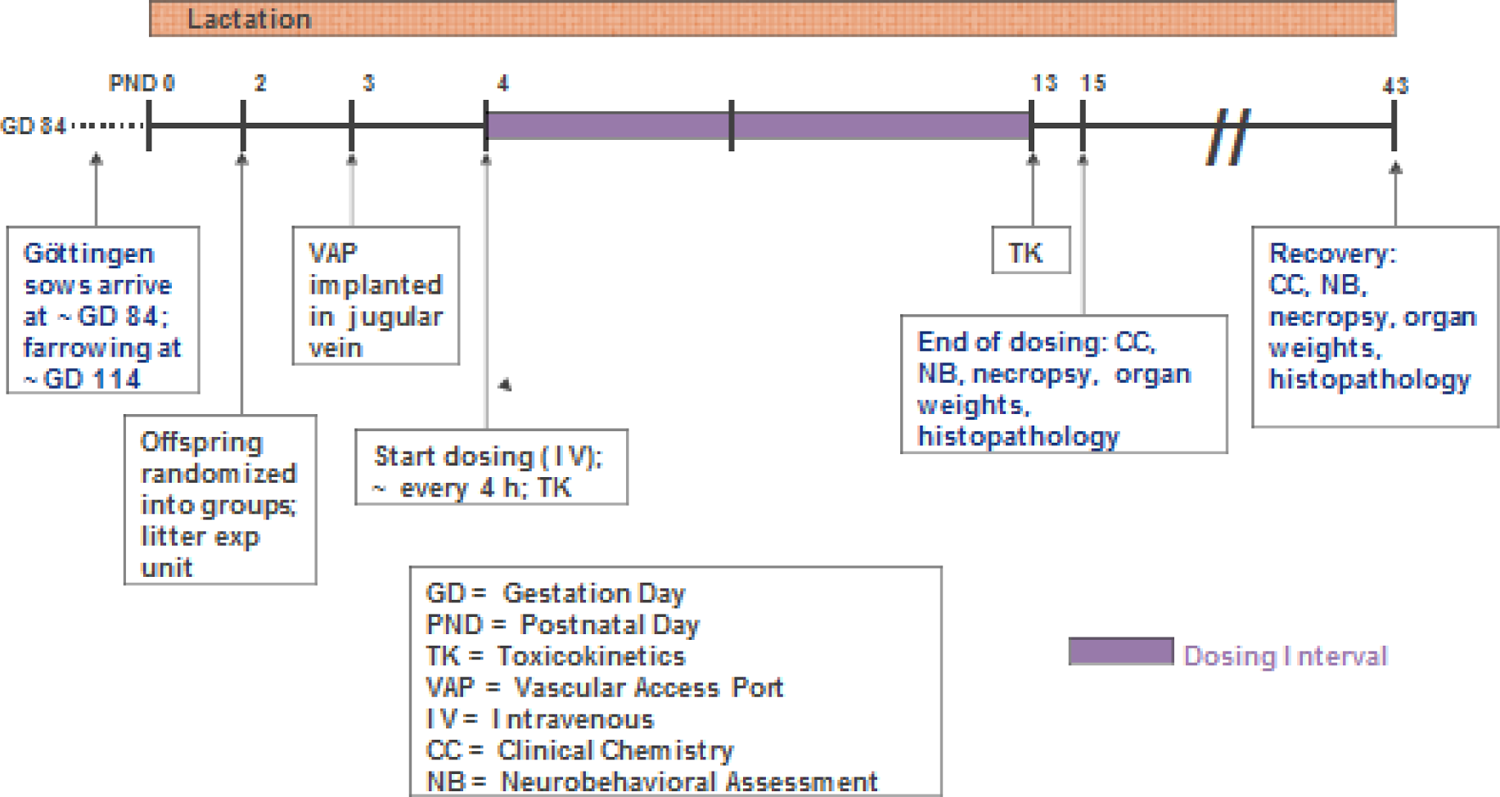
Example of juvenile toxicology study design in minipigs. 27
Both species have organ-specific preferences. For example, dogs are preferred for lung development studies using inhalation routes, while minipigs are suitable models for studying gastrointestinal system development. Both species provide flexibility and potential advantage to evaluate and study the entire development until sexual maturity in a single study. As a potential shortcoming, neurobehavioral testing options are rather limited in dogs and minipigs. However, some limited behavioral evaluation can be performed in the dog at selected times (Figure 9).
When using the NHP model, a distinction between juvenile and neonatal animals is mandatory, since time-mated NHPs are not readily available from animal suppliers. Although technically feasible, it is not recommended to generate these animals on-site in a breeding program, since litter size in macaques predominantly consists of a single offspring and in consideration of the limited numbers, there is limited control of gender distribution. Neonatal NHP studies have been done in exceptional cases only. 28 Owing to transport restrictions and animal welfare consideration, infants without mothers should be generally older than 12 months for transport to the test site, but occasionally 9 to 12 months may be acceptable. The NHP model has been used in several juvenile studies of varying designs. 29 Typically, study initiation is feasible at the age of 10 to 12 months. Prior to the initiation of a juvenile toxicology study in NHP, many prestudy activities require much advance planning (Figure 13). Except for lung and bone, the development of most other organ systems generally compares well in cynomolgus monkeys and humans (Table 5). In contrast to the dog and minipig, validated neurobehavioral test batteries including testing for learning and memory are available for NHP. 31
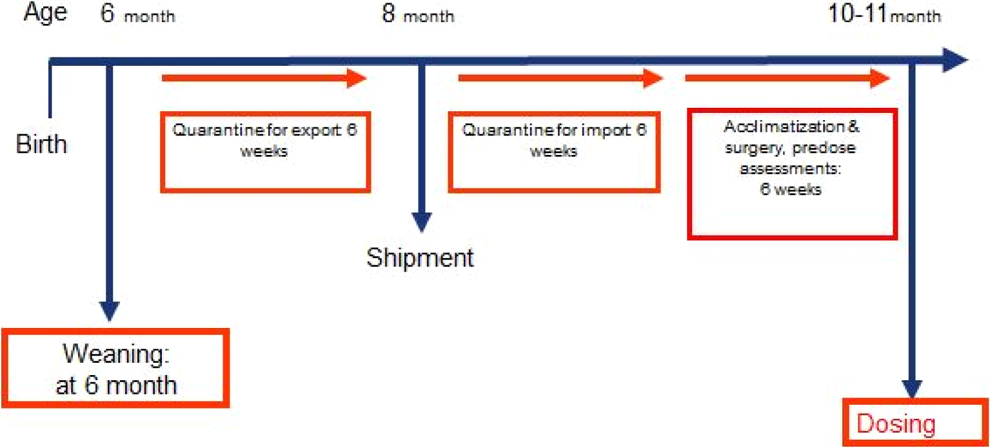
Prestudy activities for a juvenile toxicology study in nonhuman primates. 29

aThe right column represents the recommendation for the minimum age of macaques for studying potential organ system effects.
bBone age at birth is more advanced in macaques than that in humans. For the skeletal system, differences also depend on which bone is being studied.
cSystems with major functional differences compared to human postnatal development.
For biopharmaceuticals, ICH S6(R1) provides the option for an enhanced pre-/post-natal development (ePPND) study comprising delivery and raising infant animals for several months. 32 An ePPND study design in the cynomolgus monkey has gained much attention in the reproductive developmental evaluation of biotherapeutics. The ePPND combines the traditional segmented embryo–fetal development study with the PPND study, where a single cohort of animals is exposed throughout gestation and allowed to give birth naturally. The benefit of ePPND study design evaluates the stages of the traditional 2 study design using fewer animals and assesses the functional consequences of gestational exposure of the molecule. The parameters that could be evaluated include toxicokinetics in mothers and infants, growth, dual-energy X-ray absorptiometry scans for bone density and body mass, neurobehavior, clinical pathology, immunotoxicology, electrocardiography, ophthalmology, and histology (Figure 14). Since neonatal animals will be exposed to the test article via late placental transfer, these studies have been used to address neonatal aspects of juvenile toxicity testing; and under specific circumstances provide a possibility to bridge a juvenile toxicity gap and avoid a separate juvenile toxicity study. Depending on the target organ system, this gap may or may not be relevant. 33
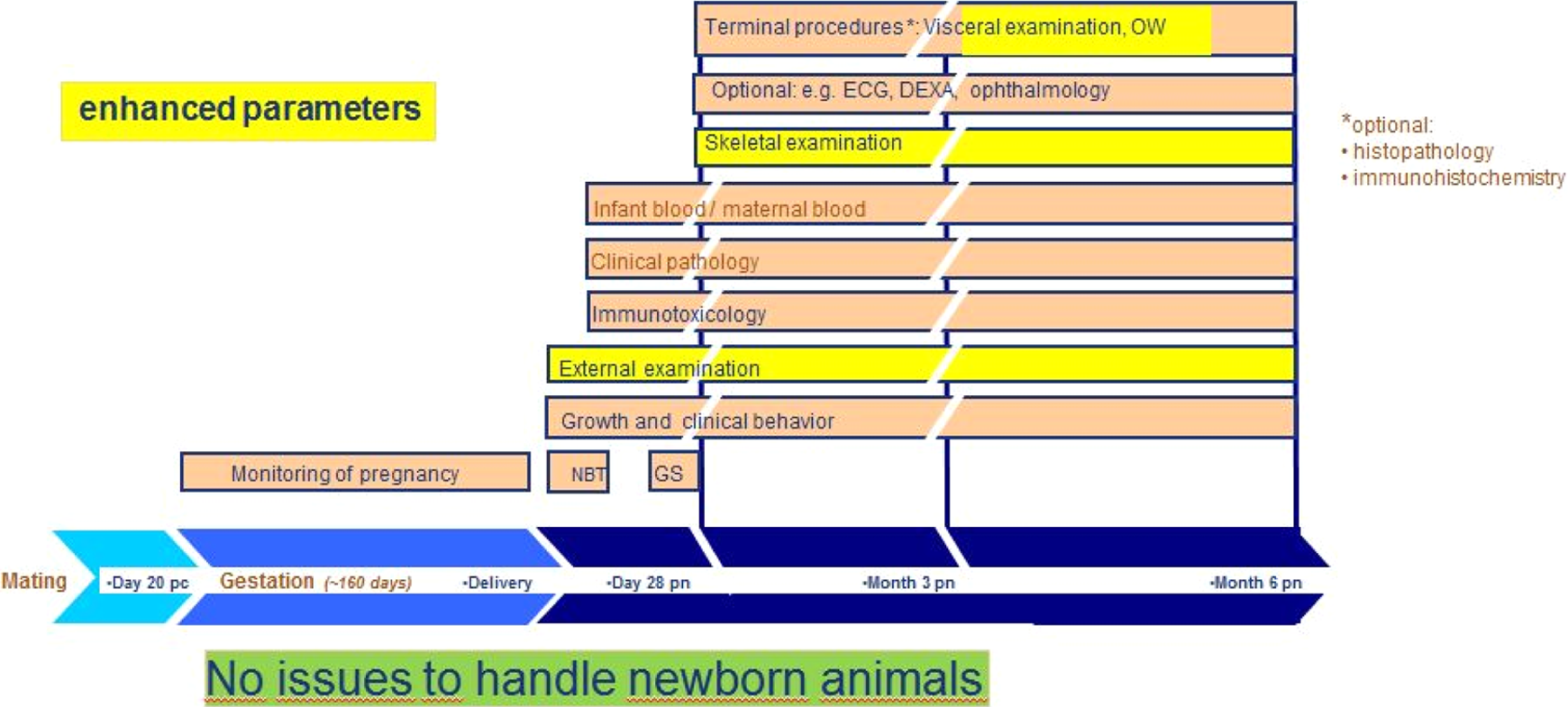
Enhanced pre- and postnatal development (ePPND) study design in nonhuman primates. 32 NBT indicates neurobehavioural battery test; GS, grip strength; ECG, electrocardiography; OWs, organ weights; DEXA, dual-energy X-ray absorptiometry for bone density and body mass; pc, postcoital; pn, postnatal.
Collectively, juvenile toxicity testing is feasible in nonrodent species, and animal size provides opportunities in terms of route of administration and number/frequency of parameter collections otherwise not available in juvenile rodent studies. On the other hand, smaller group sizes can pose statistical challenges, and historical control databases are relatively limited for nonrodent models and the concurrent control group animals will likely serve as a main reference for comparison.
Pathologic Considerations in Juvenile Toxicology Studies
The interpretation of histopathology data from juvenile toxicology studies can be challenging. A cross-discipline communication, specifically, knowledge that pathologists can share with toxicologists to enhance the design of juvenile toxicity study and to provide a more comprehensive interpretation of the results can be helpful. Admittedly, pathologists encounter complexity in appropriately interpreting histopathology in juvenile toxicity studies. By understanding these challenges, toxicologists can optimize the study design and enhance the integration of study results.
Three key knowledge points in performing juvenile toxicology studies are (1) understanding interpretation challenges faced by pathologists in juvenile toxicity studies, (2) recognizing when normal looks abnormal during tissue development of animals at various ages, and (3) study design considerations to mitigate these challenges.
When the pathologist performs a microscopic examination of tissues from juvenile animals, there are some unique challenges. This starts with the basic problem of what is “normal” versus “abnormal” in juvenile animals at various ages. Pathologists are most familiar with adult animal tissues, yet tissues from juvenile animals are not just miniature versions of tissues from adult animals. If the microscopic examination is performed at the end of the study as scheduled, the pathologist will use age-matched control tissues for comparison (Figures 15 and 16). The problem heightens when there are early deaths in juvenile toxicity studies which are frequent owing to the procedural challenges in handling young animals. It is not appropriate nor recommended that control animals at the end of the study be used as a baseline comparator of these early death animals. It is preferable to have tissues from age-matched control animals approximately of a similar age as the treated animals that died early. Many of these studies lack concurrent age-matched controls for early death animals. Therefore, the pathologist is expected to be familiar with the normal appearance of all tissues in animals throughout development, from birth through mature adult age, depending on the specifics of the study design. It is important to realize that not all tissues develop at the same rate; thus, the pathologist must be aware of the development of each individual tissue. In addition to this, there are a relatively limited number of references, especially those with images, available in the literature for pathologists to refer to and adequately gain this knowledge. Fortunately, additional valuable references have begun to be published, and selective laboratories have taken the initiative to create their own reference and image libraries.
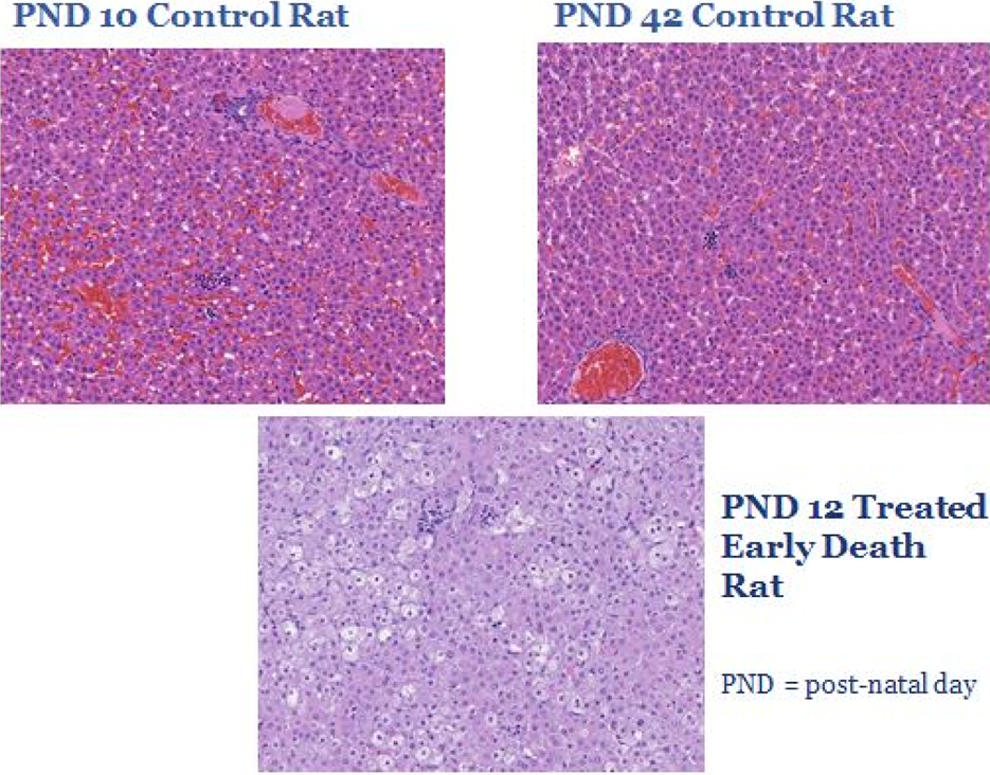
Liver from rat juvenile toxicology study. Comparison between livers from control rats on postnatal day (PND) 10 and PND 42 versus a treated rat that died early on PND 12. The liver has a relatively mature histologic appearance by PND 10 in the rat as observed in the PND 10 control rat shown here. While vacuolation in the liver is normal through approximately PND 3 in the rat, the vacuolated appearance in the PND 12-treated early death rat is not consistent with the normal histologic appearance of the liver at this age; thus, a treatment-related effect is present.
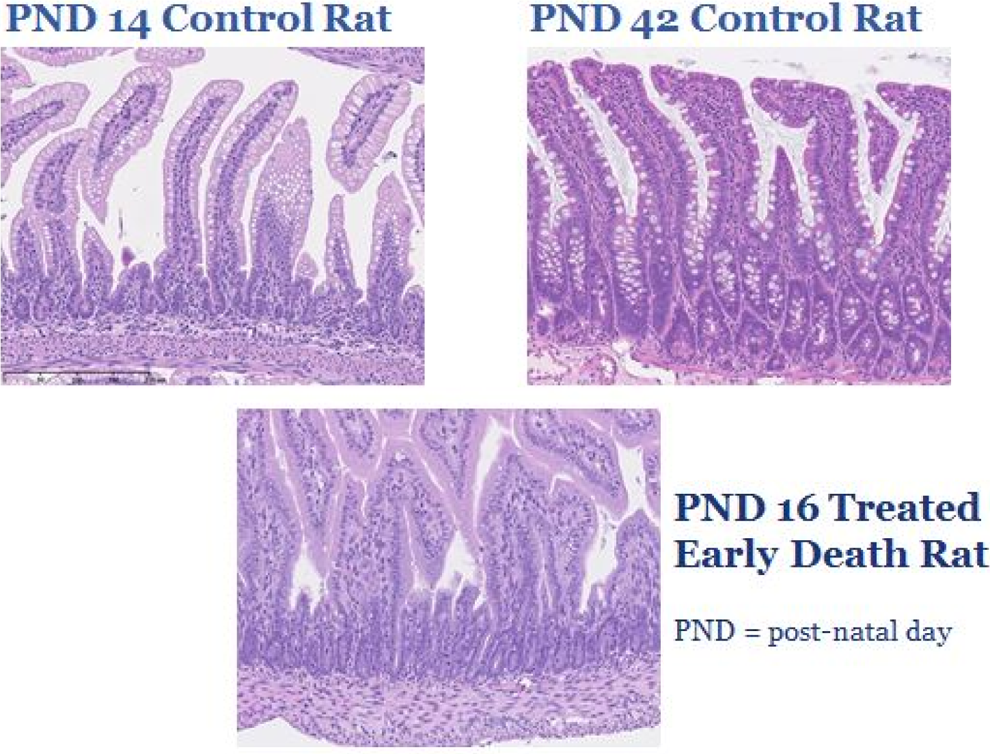
Small intestines from rat juvenile toxicology study. Comparison between the small intestine from control rats at postnatal day (PND) 14 and 42 versus a treated rat that died early on PND 16. The small intestine has vacuolation of the epithelial cells along the villus tips during postnatal development from PND 1 through PND 14, sometimes extending up through PND 24. The vacuoles contain a small amount of eosinophilic protein material and represent protein absorption during nursing. In the mature rat small intestine, represented here at PND 42, the absorption vacuoles are absent and mucous cells are present along the deep villi and crypts. Note the lack of absorption vacuoles and mucous cells in the PND 16-treated early death rat.
In histopathology interpretation in juvenile toxicity studies in particular the tissues from early death animals in the treatment groups on a study when there are no concurrent age-matched controls. In these cases, the pathologist must carefully examine each tissue individually and make an educated decision on the evaluation of the tissue from a list of basic interpretation choices. Do the changes represent (1) normal postnatal development, (2) an effect on development (ie, delayed or precocious development), or (3) a toxicant-related effect which could be either a direct or indirect? Several examples are shown in the micrographs (Figures 15 and 16) to demonstrate this challenge and the importance of knowledge of the microscopic appearance of each tissue throughout development, either through the use of available reference materials or through age-matched controls.
Tissue development is a very dynamic process. There are several histologic changes that occur normally during development and should not be inadvertently mistaken for an abnormal finding. Examples of normal histologic changes during development which may be interpreted as abnormal by pathologists unfamiliar with the changes were presented. These examples, from juvenile rats, included the following normal histologic features: waves of spermatocyte apoptosis in the testes around postnatal days (PND) 15 to 18 (late infantile period) and PND 27 (juvenile period)
46
; “atypical” early tertiary follicles with disorganized granulosa cells in the ovary around PND 10 to 16 (early infantile period)
44,45
; a wave of follicular atresia in the ovarian medulla around PND 26 to 28 (juvenile period; corresponds with falling follicular stimulating hormone [FSH] levels)
44,45
; saccular stage alveolar septae in the lung, which appeared thickened and hypercellular from prior to birth through PND 4 (neonatal period; Figure 17); receptor-mediated endocytosis of protein (vacuoles containing eosinophilic protein globules) in the small intestine from PND 1 to 14 (neonatal and early infantile periods)
45
; and hyperplastic appearance to the gastrointestinal tract from PND 15 to 21 (late infantile period; Figure 18).
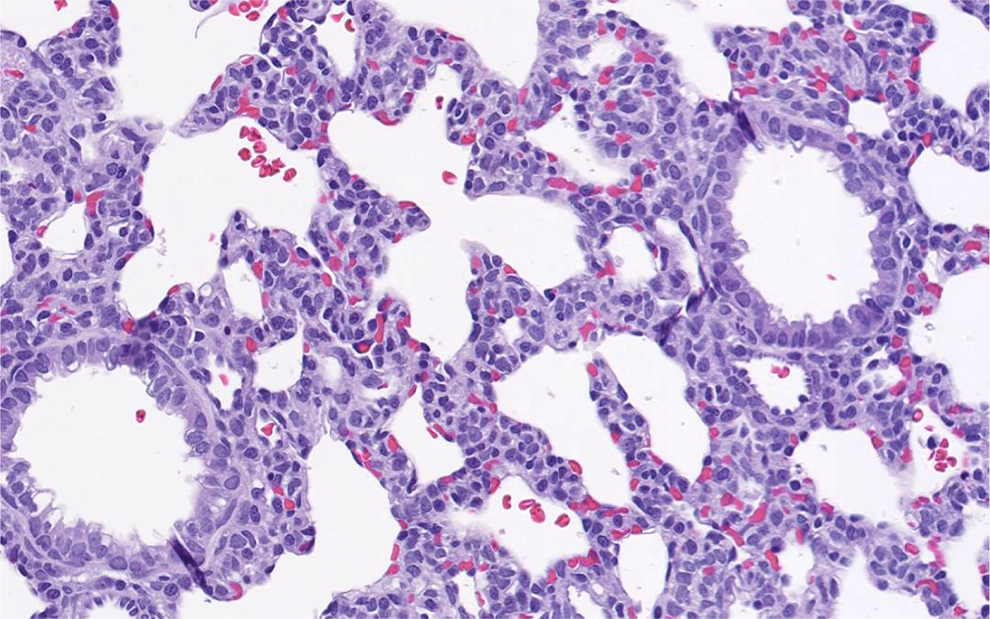
Lung from a juvenile rat. Saccular stage of postnatal lung development in a rat at postnatal day 1.
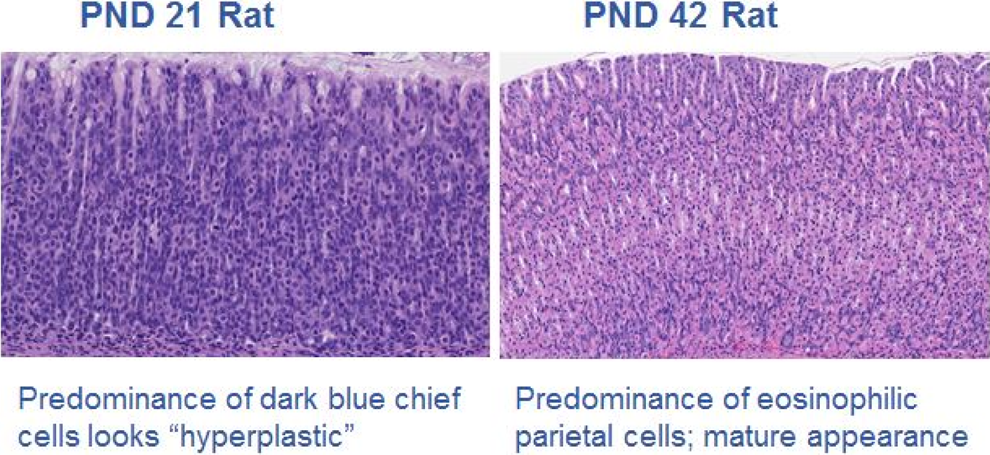
Glandular stomach from a juvenile rat. 41 Predominance of different cell types in the glandular stomach at varying stages of postnatal development in the rat. Dark blue chief cells predominant around postnatal day (PND) 21 in the rat, giving the glandular stomach a “hyperplastic” appearance, while eosinophilic parietal cells predominant in the mature rat.
There are numerous dynamic tissue changes occurring during postnatal development, and each tissue matures at a different rate, so the interpretation of these tissues without appropriate background knowledge, references, and age-matched control tissues can be problematic. To mitigate these challenges, toxicologists and pathologists should collaborate to optimize the study design and to enhance the integration of the study results.
To optimize juvenile toxicity study design, additional control animals (treated or not treated with vehicle) can be included in the study as a separate necropsy cohort to be used as age-matched and sex-matched comparative control when any early deaths that occur during the study. In addition, laboratories performing these juvenile toxicity studies are encouraged to build a strong database of information on juvenile animals that can be readily available for use by toxicologists and pathologists. A historical control database of information should include body weights, clinical pathology, organ weights, histology, and other encompassing data at various ages of development from neonatal to adult. The currently available literature provides some portions of these data. 34 –52
To enhance the integration and interpretation of study results in juvenile toxicity studies, toxicologists should be aware of the challenges faced by pathologists in the interpretation of juvenile tissues. Toxicologists should take the initiative to review the available references and textbooks on juvenile development and consults with the pathologist if a diagnosis is made for a tissue expected to be normal at a certain stage of the developmental process. Collaboration is key in these studies, and the toxicologists and pathologists involved should view themselves as one scientific team and focus on an integrated study package. A well-interpreted and integrated study design and well-interpreted results will benefit the testing laboratories, sponsors, and regulatory agencies. In the end, proactive and collaborative efforts will provide valuable safety assessment of the molecule that will be exposed to pediatric population.
Conclusions
In designing and conducting juvenile toxicology studies, key messages that should be considered are as follows: Juvenile toxicity studies are designed and performed on a case-by-case basis rather than using standardized study protocols. Study design of juvenile toxicity studies needs to cover phases of growth and development of organ systems at risk in the pediatric population. Often the rat is the first-choice species, followed by the dog. Other species are used less frequently but can be justified. Juvenile toxicity studies will involve direct dosing of pups which is technically challenging, and the design of studies are complex. Pathologists must understand “normal” organ development in all tissues at various stages throughout the entire postnatal developmental period. Much background information is needed prior to conducting the juvenile toxicity study. Each study is unique and requires intellectual participation of numerous technical and scientific personnel.
Footnotes
Acknowledgments
Jacqueline Carleer (FAMHP, Brussels, Belgium) and Karen David-Bruno (FDA, Silver Springs, Maryland) also participated in juvenile toxicology continuing education course at the 2015 Annual Meeting for the American College of Toxicology (Summerlin, Nevada) and provided the regulatory perspectives. These are not presented within the confine of this publication, as the regulatory guidance documents are constantly evolving and the forthcoming ICH S11 is expected to harmonize regulatory differences. Authors would like to specially thank Elisa Turner from the American College of Toxicology for providing administrative support; LaRonda Morford of Eli Lilly and Company for guidance on the non-rodent information; Kate Lane of Shire for acting as a liaison for the continuing education course; the American College of Toxicology 2015 Continuing Education Committee led by Patricia Ryan of Medimmune; and the American College of Toxicology 2015 Program Committee led by Hanan Ghantous of the US FDA.
Author Contributions
N. N. Kim and T. Steinbach contributed to conception and design, contributed to interpretation, drafted the manuscript, and critically revised the manuscript. R. M. Parker, G. F. Weinbauer, and A. K. Remick contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.