
Abstract
Safety assessment is reported of an orally ingested dihydroquercetin-rich extract (Lavitol) derived from the Dahurian larch tree, used as a food additive and as a dietary supplement ingredient. Dihydroquercetin, a potent antioxidant, is also known as taxifolin. The results of genotoxicity and toxicological tests (Comet assay, micronucleus test in human lymphocytes, chromosomal aberration test, subacute 7-day oral toxicity study, subchronic 90-day toxicology study with histopathologies, and, prenatal and postnatal developmental toxicity studies) on the extract provide further support for the safety of its consumption as a food supplement and food additive.
Keywords
Introduction
The deciduous coniferous Dahurian larch tree, Larix gmelinii Rupr and syn Larix dahurica Turoz (Pinaceae), is indigenous to central Siberia and found growing from the Yenisei Valley in the east of Siberia, west to Kamchatka, Russia. Larix gmelinii is a hybridized Larch species of Larix sibirica Leder 1 and the most northerly growing tree in the world. 2 Compounds found in the heartwood have been studied to determine what role they might play in order to adapt to the extremely stressful environmental conditions the tree is subjected to.
Bioassays have revealed that this tree’s resiliency to stress conditions is likely due to a high concentration of the flavonoid, dihydroquercetin (DHQ; (2R,3R)-2-(3,4-dihydroxyphenyl)-3,5,7-trhydoxy-2, 3-dihydrochromen-4-one), also known as taxifolin. The concentration of DHQ is higher than that found in onions, Douglas fir bark, French maritime bark, or milk thistle, all foods found to contain this compound and studied for their medicinal properties. 3
The finding of a high concentration of DHQ in L. gmelinii. led to the development and manufacture of a DHQ-rich extract, Lavitol (Ametis JSC, Blagoveshchensk, Amur Oblast, Russia), which over a period of more than 10 years secured regulatory approval for use as a food additive and dietary supplement ingredient in Russia. One of the beneficial properties of Lavitol demonstrated in vivo is as an oral radioprotective agent, which has been attributed to its preferential uptake of the compound by the liver to assist in attenuating the damaging effect of radiation exposure. 4,5 Given the recent incidences of significant human exposure to radiation (I-131, Cs-134, and Cs-137), as has been experienced at the Fukushima Nuclear Power Plant in Japan, 6 and nuclear reactors in Chernobyl, Russia, along with the realization that the potential for more such events exist, there is a need to determine whether oral radioprotective agents such as Lavitol are safe to ingest. Experimental evidence indicates taxifolin displays hepatoprotective, cardioprotective, and neuroprotective properties. There is an indication that taxifolin stimulates fibril formation and promotes stabilization of fibrillar forms of collagen. Also, taxifolin inhibits the cellular melanogenesis as effectively as arbutin, one of the most widely used hypopigmenting agents in cosmetics.
Dihydroquercetin is a naturally occurring polyphenol constituent of many plants consumed by humans for thousands of years. It is found in abundance in yellow and red onions, olive oil, fruits (apples, lemon, Citrus aurantium, and tamarind), berries (black currants, and grapes), nuts (pine nuts and Spanish peanuts), grains (sorghum), spices (thyme, oregano, and marjoram), and many other foods. The estimated individual DHQ exposure based on anticipated daily consumption of commonly consumed foods in the United States is 426.24 mg/d. The basis on which a determination was made on December 17, 2009, by an expert panel, that Lavitol is generally recognized as safe in the United States, intended to be added as an antioxidant to those foods defined by the US Food and Drug Act’s Code of Federal Regulation Title 21§170.3(n), at an acceptable daily intake of 300 mg/d, was based in part on the following studies that demonstrated the extract’s safety: 3 in vitro genotoxicity assays including, the Comet assay test to determine any DNA damaging effects, using the single-cell gel electrophoresis assay that measures DNA stand breaks in individual eukaryotic cells, the micronucleus test in human lymphocytes, to assess the potential to cause an increase in the induction of micronuclei in cultured human lymphocytes, and the chromosomal aberration test to assess whether Lavitol displays any mutagenic activity on bone marrow cells and 3 in vivo animal toxicology studies, the subacute 7-day oral toxicity study, subchronic 90-day toxicology study with histopathologies, and prenatal and postnatal developmental toxicity studies. The results of these studies are reported herein.
Materials and Methods
All genotoxicity and toxicological studies were conducted in compliance with good laboratory practice standards. 7 –12 The Comet assay and chromosomal aberration tests were performed at the Zakusov V.V. Research Institute of Pharmacology (RAMN) (Moscow, Russia). The micronucleus test was performed by Huntingdon Life Sciences (Huntingdon Research Centre, Huntingdon, Cambridgeshire, United Kingdom). The 3 in vivo animal toxicology studies were conducted by Amur State Medical University (Blagoveshchensk, Amur Oblast, Russia).
Manufacturing and Characterization
The test substance used in each study is Lavitol (Ametis JSC), an extract of the Dahurian larch tree, provided by Ametis JSC. The raw material is harvested from Dahurian larch trees in Amur Oblast, Russia. The trees are a sustainable and renewable source with annual volumes logged in the region exceeding 1.5 million m3. The heartwood is processed according to a proprietary closed-system technology, performed in hermetically sealed bunkers that are climate controlled for temperature, humidity, O2 content, and ventilation, to meet purity and identity requirements of the finished extract, Lavitol. According to the extract’s specification, the concentration of DHQ is between 90% and 97%, with other flavonoids accounting for 1.8% to 5.0%, and H2O content, 1.5%. Each of the batches complies with the United States Pharmacopeia (USP) 32/NF 27 <467> regulation for levels of residual solvents, class 1, 2 and ethanol, as well as USP regulation for the level of microbiological contaminants and (California’s) Proposition 65 regulations for levels of heavy metals. Although no tests were conducted on the level of mycotoxins and allergens on Lavitol extracts as discussed in the studies below, the hazard analysis critical control system for purity controls requires an absence of mycotoxins in the raw material and finished product. Tests conducted by PhytoLab (Vestenbergsgreuth, Germany) on 10 randomly selected batches showed no aflatoxins in any batches tested. The protein analysis (method 64 LFGN L06.00-t) determined that none of the 10 randomly selected batches of Lavitol contain any protein.
Lavitol is a poorly soluble pale yellow powder, with a slow dissolution rate, insoluble in nonpolar solvents, and soluble in ethanol and water–ethanol solutions, that is stable at room temperature. The test substance’s chemical composition is determined by high-performance liquid chromatography (HPLC)-accredited method 72-08, certified by the Russian Federal Agency for Technical Regulations and Metrology, Moscow. The method utilizes a Milichrome 2 × 75 mm2 A-02, sorbent Pronto SIL 120-5C18 (EcoNova Institute of Chromatography, Novosibirsk, Russia). The analytical length is 290 nm, at 35°C; pressure 20 to 60 atm. The eluent A mass weight of acetic acid in water is 0.1%; eluent B is acetonitrile, with a run time of 27 minutes. Repeat HPLC analyses of commercial batches of Lavitol, according to the prescribed analytical method, using caffeine as a standard, shows the phytochemical composition of the extract to be in the following range for DHQ (taxifolin) of 90.94% to 97.51%, and the balance of ranges of remaining flavonoids to be as follows: aromadendrin (dihydrokaempferol) 1.6% to 3.1%, quercetin 0.0% to 0.2%, naringenin 0.0% to 0.2%, eriodictyol 0.15% to 0.6%, and pinocembrin 0.5% to 3.00%. The exact percentage of the flavonoids in each lot of Lavitol is reported for each study subsequently. It should be noted that DHQ is classified as an antioxidant due to its free radical scavenging properties, given the presence of the o-dihydroxy structure in the B ring, which confers stability, and the 5- and 7-OH groups with 4-oxo function in the A and C rings responsible for maximal radical scavenging potential. 13 Further, the compound has an elimination half-life of 1.3 hours, with a relative bioavailability of 159% (in rat plasma), following oral administration of 50 mg/kg. 14
Comet Assay Test
The test substance was examined in the sensitive Comet assay for the assessment of any DNA damaging effects of Lavitol using the single-cell gel electrophoresis assay, measuring DNA strand breaks in individual eukaryotic cells. The phytochemical composition of Lavitol used in this study was DHQ 97.51%, aromadendrin 1.55%, eriodictyol 0.1%, quercetin 0.15%, naringenin 0.0%, and pinocembrin 0.0%.
Test preparation
Lavitol was administered per os in 1% ethanol at 0.5 mL of solution, allowing for complete dissolution. The reagents methyl methanesulfonate, Tris, NaOH, and disodium EDTA were supplied by Sigma-Aldrich (St Louis, Missouri), in addition to, low-melting temperature agarose, normal-melting temperature agarose, NaCl, dimethyl sulfoxide (DMSO), and Triton X-100 (pancreas). The fluorescent SYBR Green I dye was provided by Invitrogen (Carlsbad, California).
Animals and housing
Twenty pathogen-free CBAxC57Bl/6 male mice used in the study were obtained from the Research Laboratory of Pharmacogenetics, State Zakusov Research Institute of Pharmacology of the Russian Academy of Medical Sciences, Moscow. Animals were 8 to 12 weeks old and weighed 18 to 20 g. Upon arrival in the laboratory they were kept, 10 animals/cage, in polycarbonate cages (940 cm2), at 20°C ± 2°C, at a relative humidity (RH) of 60%, and a 12-hour light/12-hour dark cycle. They were fed a standard rodent laboratory chow (MEST, Moscow, Russia) and provided water ad libitum. After acclimatization, animals were randomly divided into 4 groups with each group (n = 5) caged separately. Bedding consisted of steam-sterilized hardwood chips.
Experimental design
Methanesulfonate was administered intraperitoneally for 3 hours at 40 mg/kg and used as a positive control. A 1% dose of ethanol (Synthesis, Moscow, Russia) was administered 0.1 mL/10 g body weight (bw) per os for 5 days as a negative control. A single dose of 15 and 2000 mg/kg bw of Lavitol was orally administered to males, as shown in Table 1, corresponding to the daily therapeutic dose of 15 mg/kg bw and a subchronic dose that exceeds the therapeutic dose by >100 times that dose. Cytogenic preparations of the femoral bone marrow, blood samples, and liver samples were prepared using the methods of Singh et al, 15 Durnev et al, 16 and Kharbiev. 17 Gel electrophoresis of isolated cells was performed in an alkaline medium. Preparations were stained with SYBR Green 1 for 30 minutes in total darkness. Cytogenic analysis was performed using an epifluorescent microscope Micmed-2 12T model (Lomo, St Petersburg, Russia), with a high-resolution digital camera (model VEC-335, EBC, St Petersburg, Russia), set at 200× magnification. The images of DNA comets were analyzed using CASP software v.1.2.2 (CASPlab, Wroclaw, Poland) using an automatic imaging system that scores the extent of DNA damage. 17
Administration of Tested Compound.
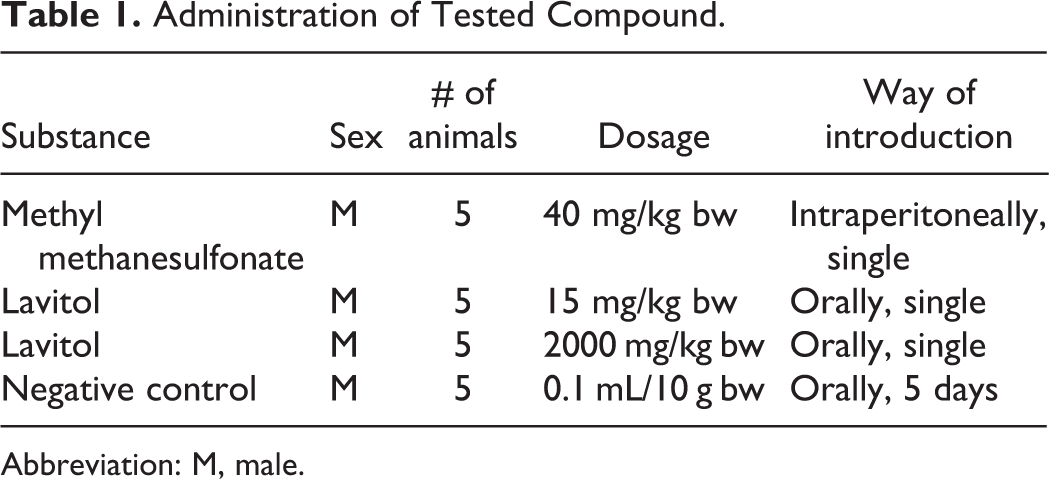
Abbreviation: M, male.
Statistical analysis
At least 100 individual cells for each micropreparation were analyzed for the DNA content in the tail of the DNA comet as an index of DNA damage. Statistical analysis was performed using the analysis of variance (ANOVA) post hoc Dunnet test (Statistica v. 6; StatSoft, Moscow, Russia).
Micronucleus Test in Human Lymphocytes
The micronucleus test was performed according to Organization for Economic Cooperation and Development Guideline 487 to assess the potential of Lavitol to cause an increase in the induction of micronuclei in cultured human lymphocytes in vitro. 18 The test consisted of a preliminary toxicity test and 3 micronucleus tests. The phytochemical composition of Lavitol used in this study was DHQ 97.5%, aromadendrin 1.55%, eriodictyol 0.10%, quercetin 0.15%, naringenin 0.00%, and pinocembrin 0.00%.
Test preparation
Prior to commencing testing, the solubility of the test substance in a vehicle compatible with this test system was assessed. Lavitol was found to be soluble at 304.3 mg/mL in ACS reagent grade DMSO, the vehicle for this study. The highest concentration of Lavitol tested in this study was 304.3 mg/mL in the chosen vehicle, which provided a final concentration of 3043 μg/mL, when dosed at 1% (v/v), in order to test up to 10 mmol/L. The highest concentration in each test was diluted with DMSO to produce a series of lower concentrations. The pH and osmolality of Lavitol in medium were tested at a concentration of 3043 μg/mL. No fluctuations in pH of the medium of more than 1 unit were observed compared with the vehicle control. No fluctuations in osmolality of the medium of more than 50 mOsm/kg were observed compared with the vehicle control.
S9 fraction, prepared from male Sprague-Dawley-derived rats, is dosed with phenobarbital and 5,6-benzoflavone to stimulate mixed-function oxidases in the liver and stored at about −80°C. The S9 mix consists of S9 fraction (10%, v/v), MgCl2 (8 mmol/L), KCl (33 mmol/L), sodium phosphate buffer pH 7.4 (100 mmol/L), glucose-6-phosphate (5 mmol/L), and nicotinamide adenine dinucleotide phosphate (4 mmol/L). The cofactors were filter sterilized with a 0.2-μm nonpyrogenic sterile filter prior to use. The dose levels of S9 mix were selected so as to reach an acceptable toxicity profile per protocol. Dose levels between plus and minus S9 may therefore not be the same as test compounds respond different in either the absence or presence of S9.
Human blood was collected aseptically from 2 healthy, nonsmoking, adult donors, pooled (in equal volumes from each donor) and diluted with Roswell Park Memorial Institute 1640 tissue culture medium supplemented with 10% fetal calf serum, 0.2 IU/mL sodium heparin, 20 IU/mL penicillin/20 μg/mL streptomycin, and 2.0 mmol/L
Cells were harvested by centrifugation at 500g for 5 minutes. The supernatant was removed and the cell pellet resuspended and treated with a 4-mL hypotonic solution (0.075 mol/L KCl) at 37°C, cultures were then incubated for 3 minutes at 37°C to cause swelling. Cultures were agitated, 4 mL of ice-cold fixative (3:1, v/v methanol:acetic acid) was added slowly onto the culture surface, and the cultures were slowly inverted to mix. The cultures were centrifuged at 500g for 5 minutes. The supernatant was removed, and the cell pellet resuspended. A further 4 mL of fresh fixative was then added and the cells stored at 4°C until slide preparation. Cytokinesis was blocked following mitosis using Cytochalasin B (Cayman Chemical, Ann Arbor, Michigan), the cells harvested, and slides prepared so that binucleate cells could be examined for micronucleus induction. In order to assess the cytotoxicity of Lavitol to cultured human lymphocytes, the cytokinesis-block proliferative index (CBPI) was calculated for cultures treated with the test substance, the vehicle and positive controls. Five Lavitol concentrations were assessed for determination of induction of micronuclei. The highest concentration selected (650 μg/mL) was that which caused a reduction in CBPI equivalent to 55% ± 5% cytotoxicity. Following 3-hour treatment, reductions in CBPI equivalent to 57.8% and 58.0% cytotoxicity were obtained with Lavitol at 650 μg/mL in the absence and presence of S9 mix, respectively. Concentrations of Lavitol selected for micronucleus analysis were 100, 300, 400, 500, and 650 μg/mL in the absence of S9 mix and 100, 350, 450, 600, and 650 in the presence of S9 mix. In the absence of S9 mix following 20-hour treatment, a reduction in CBPI equivalent to 53.5% cytotoxicity was obtained with Lavitol at 200 μg/mL. Concentrations of Lavitol selected for micronucleus analysis were 50, 100, 150, 175, and 200 μg/mL.
Slide preparation and examination
The cultures were centrifuged at 500 g for 5 minutes and the supernatant removed. A homogeneous cell suspension was prepared. Precleaned microscope slides were prepared for each culture by aliquoting the resuspended cells onto the slides and allowing the slides to air dry. Two slides were prepared per culture. The remaining cell cultures were stored at approximately 4°C until slide analysis was complete. Slide staining was performed by rinsing in purified water, staining in acridine orange solution (0.0125 mg/mL) for 4 minutes, washed in purified water for 5 minutes, rinsed in cold tap water for 2 minutes, stored at room temperature protected from light, and immediately prior to scoring, slides were wet mounted with glass coverslips using purified water.
For the microscopic examination of the preliminary test, the prepared slides were examined by fluorescence microscopy. The incidence of mononucleate, binucleate, and polynucleate cells per 500 cells was assessed per culture. The presence of an unusual number of, for example, cells undergoing mitosis, polyploid cells, necrotic cells, and debris was also noted. From these results, concentrations were selected for treatment in the main test. The highest concentration was intended to be that which caused a depression in the CBPI equivalent to 55% ± 5% cytotoxicity (approximately) when compared with the concurrent vehicle control.
For the main test, the prepared slides were examined by fluorescence microscopy. The incidence of mononucleate, binucleate, and polynucleate cells per 500 cells was assessed per culture. The presence of an unusual number of, for example, cells undergoing mitosis, polyploid cells, necrotic cells, and debris was also noted. From these results, concentrations were selected for micronucleus analysis. The highest concentration was intended to be that which caused a depression in the CBPI equivalent to 55% ± 5% cytotoxicity (approximately) when compared with the concurrent vehicle control. Prior to micronucleus analysis, all slides were randomly coded. Interphase cells were examined by fluorescence microscopy and the incidence of micronucleated cells per 1000 binucleate cells per culture was scored where possible. Positive control micronucleus counts are reported for the following treatments only: cyclophosphamide: 5 μg/mL (3 hours, +S9 mix); mitomycin C: 0.2 μg/mL (3 hours, −S9 mix) and 0.1 μg/mL (20 hours, −S9 mix); and colchicine: 0.06 μg/mL (3 hours, −S9 mix) and 0.02 μg/mL (20 hours, −S9 mix). The analyses for micronucleated cells were based on the methods of Fenech and Morley, 19 Fenech, 20 Fenech et al, 21 and Albertini et al. 22
Statistical analysis
The analysis assumed that the replicate was the experimental unit. An arcsine transformation was used to transform the data. Lavitol was compared to control using Williams tests. 23,24 Positive controls were compared to control using t tests. Trend tests were carried out using linear contrasts by group number. These were repeated, removing the top dose group, until there were only 3 groups. Statistical significance was declared at the 0.5 level for all tests. Data were analyzed using SAS 9.1.3 (Cary, North Carolina) and QuaSAR Quantitative Statistics v. 1.4 (Seattle, Washington).
Chromosomal Aberration Test
Lavitol was tested in the in vivo chromosomal aberration assay in compliance with the Guide for Experimental (Preclinical) Study of New Pharmacological Substances (708N, 2010), performed in the Laboratory of Drug Toxicology, Zakusov V.V. Research Institute of Pharmacology (RAMN), Moscow, Russia. The phytochemical composition of Lavitol used in this study is DHQ 93.7%, aromadendrin 1.93%, eriodictyol 0.26%, quercetin 0.37%, naringenin 0.03%, and pinocembrin 0.03%.
Test preparation
Lavitol was administered per os in 1% ethanol at 0.5 mL of solution, which allows for complete dissolution of DHQ (taxifolin). Reagents used in the experiment were supplied by Sigma-Aldrich: methanesulfonate, Tris, NaOH, disodium EDTA, low-melting temperature agarose and normal-melting temperature agarose, NaCl, DMSO, and Triton X-100 (pancreas), and fluorescent dye SYBR Green I (Invitrogen Life Technologies).
Test animals and housing
The Animal Care and Use Committee of the Research Laboratory of Pharmacogenetics, State Zakusov Research Institute, Russian Academy of Medical Sciences, Moscow, approved the study. Pathogen-free CBAxC57Bl/6 mice of both sexes were divided into 2 groups of 25 males and 10 females and were obtained from the Research Laboratory. Animals weighed 18 to 20 g and were kept quarantined in the laboratory’s vivarium under observation for clinical signs of ill health following arrival for 7 days. These animals were used for the DNA-Comet assay and the chromosomal aberration test. Once randomly assigned in groups of 5 animals to either study, 20 for the Comet assay and 15 for the chromosomal aberration test, and given unique identification numbers, the animals were then housed in polycarbonate cages (940 cm2) that included steam-sterilized hardwood chips, maintained at 20°C ± 2°C and a RH of 60%, in a 12-hour light/12-hour dark cycle, while allowed free access to tap water and a standard rodent lab chow (MEST) ad libitum.
Experimental design
Positive controls were administered a single intraperitoneal dose of 20 mg/kg of cyclophosphamide (Sigma-Aldrich). A 1% dose of ethanol (Synthesis) was administered 0.1 mL/10 g bw per os for 5 days as a negative control.
During the first experiment, a single dose of 15 mg/kg bw and 2000 mg/kg bw of Lavitol was orally administered to males, corresponding to the daily therapeutic dose for humans, as shown in Table 1. In the second experiment, Lavitol was administered orally at a dose of 15 mg/kg for 5 consecutive days to both males and females, as shown in Table 2.
Administration of the Tested Compounds.
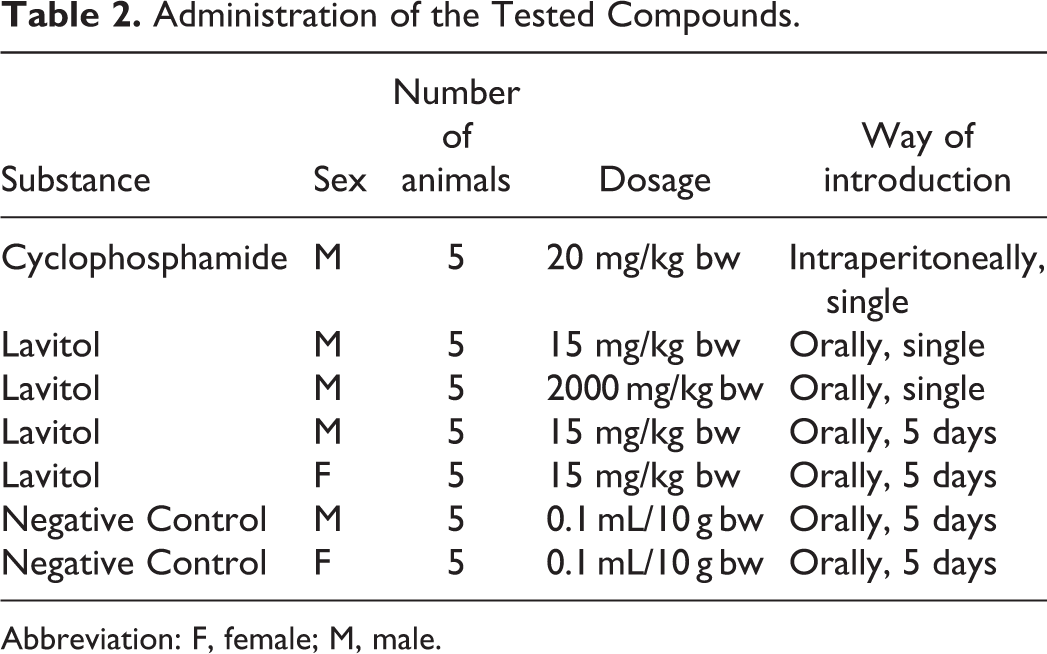
Abbreviation: F, female; M, male.
Cytogenic analysis
The cytogenic preparation of the femoral bone marrow was prepared according to the standard dry-air method. 9,11 For bone marrow metaphase analysis, each dose and week group, including negative controls, consisted of male mice; 2.5 hours before sacrifice, mice were injected intraperitoneally with colchicine (Sigma-Aldrich), at a dose of 4 mg/kg bw. Euthanasia of animals was carried out by cervical dislocation.
Chromosome specimens were prepared from the femoral bone marrow as follows. The femurs of animals were removed, and the femoral marrow cells were flushed out with medium for chromosomal analysis. Bone marrow cell preparations were made by heating up a hypotonic solution of potassium chloride (0.5 mL) and fixative (glacial acetic acid:ethanol, 1:3) to 37°C. All slides were coded and stained with azure-eosin (5 parts of azure [0.1%], 2 parts of eosin [0.1%], and 10 parts of distilled water). The cytogenetic analysis was carried out on a Standart-20 (Carl Zeiss, Gottingen, Germany) microscope with the oil immersion increased 1000×.
Statistical analysis
Results were determined by comparing the “gaps” and ruptures in cells for chromosomal aberrations for both control and experimental groups. Among the experimental group of 5 mice, 100 metaphases were evaluated for each animal. During microscopic analysis, singular and paired chromosomal fragments were evaluated as well as genes and cells with multiple (more than 5 chromosmal aberrations) per cell. Mean standard deviation was calculated for each group of animals. Differences between the control and the experimental groups were analyzed by means of φ-criteria. Statistical analysis was performed using the ANOVA post hoc Dunnet test of the data with Statistica v. 6 (StatSoft, Tulsa, Oklahoma).
Toxicity Studies in vivo
All oral toxicity studies of Lavitol in animals were approved by the Amur State Medical Academy, Blagoveschensk, Russia, in accordance with the Guide for the Care and Use of Laboratory Animals. 25 All studies were conducted on outbred albino rats (Rattus norvegivus).
A previous unpublished acute toxicity study reported nontoxic effects following a single administration of Lavitol by gavage at dose levels of 75, 150, and 1500 mg/kg bw that included gross pathology. 26 In all oral toxicity studies, the positive control groups received a 1% potato starch (Extra calls; Proxima, Novosibirsk, Russia) solution, while the experimental groups received the potato starch in a suspension that included the required amount of Lavitol. One gram of soluble starch was mixed with 20 mL of distilled water, brought to a boil, and then brought to 100 mL by adding cold distilled water to the solution. Lavitol was then mixed with 3 mL of the 1% starch solution. The solution was prepared on a daily basis.
Two stability tests, according to International Conference on Harmonisation Guidelines (1993), were performed (ABC Testing, Tustin, California) on DHQ derived from the Siberian Dahurian larch tree, provided by Ametis JSC, to confirm results reported for DHQ previously by Russian laboratories. For each HPLC (Beckman Coulter) analysis, DHQ was used as the standard. Study methods were performed on 3 batches under normal (25°C ± 2°C; 65% ± 5% RH) and stress (40°C ± 2°C; 75% ± 5% RH) temperature conditions, in finished containers, as well as, in plastic bags. The first stability study was performed for 12 weeks using assay methods ALC114A and AUV203A and showed DHQ to be 94.5% at the end of the study period. The second stability tests were performed for 18 and 30 weeks duration by the same laboratory and showed DHQ to be 97.7% and 97.5%, respectively. Retained samples used for the stability studies are stored for a period of 5 years at the Amur State Medical Academy as well as, Ametis JSC, were microbiological testing is also performed.
The selection of a 1% potato starch solution was based on previous toxicological studies conducted on Lavitol which found that this percentage provided an optimal stable vehicle. 27 When the concentration of the potato starch solution is 1%, the solution remains within a pH range of 7.0 to 8.0. Further experiments showed that the 1% starch solution remained stable whether subjected to temperatures of either 75°C to 95°C for 14 days or 4°C to 25°C for 30 days. A 1% solution (pH 7.0) was prepared daily and shaken vigorously to ensure complete solution homogeneity. The choice of doses for the in vivo experiments was based on the outcome of previous toxicological testing of the Larch extract. 27
To determine whether higher doses of Lavitol given for 7 consecutive days results in toxicity, a subacute 7-day oral toxicity study with Wistar rats was performed, including histopathological examination of the lungs, heart, kidneys, brain, thyroid gland, liver, spleen, stomach, and small and large intestines, following necropsy.
Subacute 7-day oral toxicity study
Upon arrival, animals were housed observed for 14 days in type E, 1032 cm2 polycarbonate cages (MEST), 5 to a cage, by sex, with each animal tail marked by tattooing, maintained at a temperature of 21°C ± −1°C, RH of 50% to 55%, and 12-hour light–dark cycle. All females were nulliparous and nonpregnant. Through the observation period and during the study, heat-treated sawdust was provided as bedding material, cages cleaned daily, purified water provided ad libitum, and a basal diet given daily in accordance with Russian State Standard R 50258-92 that meets the nutritional requirements for the species. Food consumption was recorded daily for each group of animals and quantity of food adjusted for weight of animals.
After the observation period, animals were randomly assigned to treatment groups. All administrations were performed by gavage. Fifty animals (25 female [F] and 25 male [M]) were divided into 4 groups. Group 1 of 20 animals (10 F and 10 M) received 10 000 mg/kg bw of Lavitol daily for 7 days. Group 2 of 10 animals (5 F and 5 M) served as controls, group 3 of 20 animals (10 F and 10 M) were administered 15 000 mg/kg of Lavitol for 7 days, and group 4 of 10 animals (5 F and 5 M) served as controls. Individual doses were calculated based on the body weight of the animal, modified after each weighing. On the day of randomization, the mean weight of males weighed 184.4 g (170.3-200.0 g) and females weighed 165.5 g (150.0-185.5 g). The phytochemical composition of Lavitol was DHQ 90.94%, aromadendrin 2.75%, eriodictyol 1.03%, quercetin 0.63%, naringenin 0.20%, and pinocembrin 0.03%.
After administration of the test vehicle for 7 days, twice-daily posttreatment observation period was performed to determine mortality, general state, external appearance, behavior, clinical symptoms, sensory reactivity to auditory, visual and proprioceptive stimuli, and muscle strength by the wire netting method, also known as the invested test. 10 Muscle strength was measured by inside a 40 × 40 cm2 screen. The wire net was tightened and divided into 1 × 1 cm2 segments. The screen is raised 50 cm over a soft surface, and the experimental animals place in the center of the screen, which is slowly inverted 180°. The animal should hold onto the screen for 15 seconds before it falls. Results were recorded. Body weight was recorded daily for each animal during the 14-day period and on the day of necropsy. Hematological laboratory tests were carried out on days 0 and 8. Blood samples were obtained from the tail vein following 14 to 15 hours of fasting, and testing performed at the Veterinary Center of the Far East State University (Blagoveschensk, Russia), which included hemoglobin (Hb), erythrocyte (RBC), hematocrit (HCT), white blood cell (WBC), lymphocytes, eosinophils, monocytes, and basophiles. Biochemical testing included total protein (TP), blood urea, cholesterol, and glucose, all by the photoelectrocolorimetric method and albumin by the photometric method. Urine samples were collected on day 0 and at the end of the administration period on day 8 and included the following parameters: color, transparency, pH, density, proteins, and RBCs.
Following necropsy on day 8 by decapitation, organ weights were determined (as absolute and relative values), and gross pathology was performed including appearance of tissues. Any abnormality was recorded. Tissues were preserved in a 10% solution of buffered formalin for histopathological examination. If no toxic lesions occurred, tissue samples would not be preserved for histopathological examination.
Statistical analysis
Statistical analysis of the differences between the control and the experimental groups was performed using the Student t test. In the case of abnormal distributions, groups would be compared using the Mann-Whitney U test. Depending on the results, pooled or separate variance estimates would be carried out using the 2-sample t test. Statistical significance was set at P < 0.05. Statistical analysis was performed using STATISTISA v. 6 software (StatSoft, Tulsa, Oklahoma).
Subchronic 90-day Toxicology Study
Animals
Ninety-six 2-month-old outbred albino Rattus norvegicus rats (48 M and 48 F) were obtained from the breeding colony of the Far East State University, which maintains historical control data on the strain of animals used in the study. Body weight at the start of the study for males was 208.7 to 239.7 g (mean 224.7 g) and 207.7 to 239.9 g (mean 222.4 g) for females. Females were nulliparous and nonpregnant.
Animals were quarantined for 12 days after arrival, then assigned to either 1 of 3 treatment or control groups. Randomization was by body weight of animals; the individual index of body mass did not deviate from the mean by sex by more than 20%. Each treatment group consisted of 24 animals (M and F). Once assigned, animals were housed in groups of 6 animals per cage, in 1032 cm2 T3S polycarbonate cages (MEST) that contained heat-treated sawdust bedding and cleaned daily. Identification of animals was by the tail tattooing method. Temperature was maintained between 20°C and 22°C at a RH of 50% and 55%, measured daily using a Thermohydrometer TKA-PKM (Ntap TKA, St Petersburg, Russia), and a 12-hour dark/12-hour light cycle maintained. Animal identification was verified whenever removed from or placed back in its cage, as well as, verified prior to dosing and observations.
The animal’s diet contained not less than 13% proteins, or more than 8% fats, or less than 20% carbohydrates, and not more than 10% cellulose: 100 g of feed provided 240 kcal. Distilled water was given ad libitum.
Dosing, route of administration, and observations
Animals were divided into 4 groups and administered either the test material or a control solution daily by intubation as a single dose, at the same time each day to avoid diurnal influences. The individual doses were calculated based on the body weight of the animal and modified after each weigh in. Group assignment of 24 animals of both sexes (12 F and 12 M) was made as follows: 3 experimental groups received 50, 150, or 1500 mg/kg bw of Lavitol each day, while a fourth group served as the control, which received a 1% potato starch solution (Extra Calls, Promixa, Novosibirsk, Russia), and the same basal diet, at a volume equal to the volume received by the experimental groups. The test product was prepared daily before introduction. The required amount of Lavitol was dissolved in the 1% starch to create the solution. One gram of the soluble starch was mixed with 20 mL of distilled water, brought to a boil, then brought to 100 mL by adding cold distilled water to the solution. Lavitol was then mixed with 3 mL of 1% starch solution. This method, prepared daily, allows the test substance to completely dissolve without precipitation prior to administration. The phytochemical composition of Lavitol used in this study was DHQ 92.20%, aromadendrin 2.35%, eriodictyol 0.53%, quercetin 0.26%, naringenin 0.17%, and pinocembrin 0.11%.
All animals were observed daily for clinical signs, with animals observed twice daily for mortality during the study period. Time of onset, intensity, and duration of symptoms, if any, were recorded. Weight of each animal was recorded at the start of the study and 3 times a week on the same days of the week and throughout the course of the study and at termination when relative organ weights were calculated. The group mean of the body weight and percentage of body weight gain were calculated as g/100 g of body weight. The quantity of food consumed by groups consisting of 6 animals of each sex was recorded. The average daily feed and water consumption per head were calculated by registrating the ratio of total volume of consumed feed and water in each cage to a total weight of animals, found in the cage, multiplied by the individual weight of each animal. The registration of average daily feed and water consumption was carried out at least 3 times a week throughout the study period.
Functional observations were conducted 3 times a day for clinical signs of pharmacological and toxicological effects of the test substance. The parameters observed included outward appearance, pose, behavior, and the presence of convulsions, trepidation, abnormal motion, and/or diarrhea. Observations when handling the animals included muscle tone, skin and mucous color, breathing abnormalities, fur state, and lacrimation. Other observations included general mobility, activity, convulsions, abnormal motions, and/or abnormal pose. Observations were recorded daily in individual cards. Ophthalmological examination included state of mucous membranes, presence of normal corneal reflexes, pupil size, and width of palpebral fissure, observed and measured on days 0, 30, 60, and 90 of the study. As part of physiological examinations, and as required by the Ministry of Health’s guidelines, electrocardiogram measures were taken of voltage peaks, intervals, and heart rate monitoring and recorded. During open-field tests conducted on days 0, 30, 60, and 90, the duration of the latent period, horizontal activity, vertical activity, grooming, and number of defecation acts were documented. The detoxification function of the liver function was evaluated by the duration of hexanal-induced sleep, as measured on days 0, 30, 60, and 90 of the study. The rate of latency and duration of hexanal-induced sleep depend on the functional state of the liver. Disturbances in liver detoxification bioactivity results in the induction of the hexanal-induced sleep duration. Thus, to determine whether Lavitol has any negative impact on the liver, 2% aqueous solution of hexanal was administered intraperitonealy based on 80 mg/kg bw to a pair of animals selected from both the tested and the control groups.
Laboratory testing
Laboratory tests were carried out by the Veterinary Center, Far East State University, on days 0, 30, 60, and 90. Blood samples were collected from the tail vein following 14 to 15 hours of fasting. Hematological parameters measured included Hb, RBC, platelet (PLT), WBC, HCT, eosinophils, and monocytes. Biochemical parameters measured included TP, alkaline phosphatase, aspartate aminotransferase (AST), alanine transaminase (ALT), total bilirubin, urea, creatinine, glucose, cholesterol, and triglycerides. Urine samples and urine analyses were collected on all animals on day 0 and on days 30, 60, and 90 of the study and included color, transparency, specific gravity, nitrites, pH, proteins, ketone bodies, glucose, urobilinogen, bilirubin, mucous, and salts.
Necrospy and histopathological examinations
All of the animals were sacrificed on day 91 by decapitation. Complete gross necropsy and histopathological examination were conducted on all animals. Complete gross necropsy included examination of external surfaces, orifices, cranial, thoracic and abdominal cavities, carcass, and all organs.
Macroscopic and microscopic evaluations were conducted on the lymphatic glands, thyroid glands, salivary glands, heart, trachea, lungs and bronchi, esophagus, stomach, brain, small and large intestine, liver, spleen kidneys, adrenal glands, urethra, pancreas, and organs of the reproductive system. The weights of the heart, lungs, liver, kidneys, spleens, testicles, adrenal glands, thymus, and brain were recorded for all surviving animals. Organ weights were recorded as absolute values and relative values as percentage of body weight calculated. Tissue samples of organs from animals were preserved in a 10% solution of buffered neutral formalin. The following organs were subjected to histopathological examination: brain, hypophysis, salivary glands, thyroid glands, thymus, lungs and bronchi, heart, adrenal glands, kidneys, liver, pancreas, stomach, spleen, and lymphatic glands.
Statistical analysis
Statistical analysis of the differences between the control and experimental groups was performed using the Student t test. In the case of abnormal distributions, groups would be compared using the Mann-Whitney U test. Depending on the results, pooled or separate variance estimates would be carried out using the 2-sample t test. Statistical significance was set at P < 0.05. Statistical analysis was performed using STATISTISA v. 6 software (StatSoft, Tulsa, Oklahoma).
Developmental Toxicity Study
Prenatal and postnatal developmental toxicity studies of Lavitol were carried out in the rat (R. norvegicus), supplied by the Far-East State University (Blagoveschchensk, Amur Oblast, Russia), that maintains historical control data on the strain of animals used in this study. Extra animals were ordered to ensure that only healthy animals were enrolled in the study. The phytochemical composition of Lavitol used in this study is DHQ 92.19%, aromadendrin 3.57%, eriodictyol 0.58%, quercetin 0.33%, naringenin 0.17%, and pinocembrin 0.17%.
Animals
Eighty 3-month-old female rats were acclimated in the laboratory upon arrival and observed for 14 days prior to dosing, meeting the maximum age limits on the first day of dosing. Females weighed 199 to 270 g (mean, 231.5 g). After the quarantine period, 1 adult male rat was placed into a cage to cohabitate with 2 females during 2 estrus cycles (2 females:1 male). Mating was carried out over a 13-day period. All female rats were examined for signs of fertilization by taking vaginal smears before quarantine. The fertilization was determined by analyzing vaginal smears. Successful mating was confirmed by the presence of sperm in the vaginal smear. The day of finding spermatozoids in vaginal smear was considered the first day of gestation.
Behavioral and state assessments were performed daily by a veterinary upon arrival and during the acclimation period, including twice daily visual inspection of the animals, to ensure that only healthy animals were enrolled in the study. Identification of animals was achieved by recording each animal’s cage and animal numbers and fur markings. Female animals were housed 2 to a cage, in 720 cm2 T3S polycarbonate cages (MEST), provided heat-treated sawdust bedding, and cleaned daily. Temperature was maintained between 20°C and 22°C at a RH of 50% to 55%, using a Thermohydrometer TKA-PKM20 (NTP TKA) and 12-hour dark/12-hour light cycle. Noise levels were kept below 85 dB, measured by a Voltcraft sound level meter (Kirschau, Germany). Animals were provided certified rat chow (Laboratorsnab, Moscow, Russia), meeting Russian State Standard R 50258-92, and provided purified water ad libitum. Animal identification was verified each time an animal was removed from or placed back in its cage, as well as, verified prior to dosing and observations.
Dosing
Animals were divided into 4 groups and administered either the test material Lavitol or the control daily by gavage. Administrations occurred at the same time each day to avoid diurnal influences. Assignment to groups was made by randomization. The mean weights and weight distributions between groups was determined and varied insignificantly (from 227.1 to 234.7 g).
Group assignments by randomization were as follows: group 1 (n = 20), the control group, received 3 mL of a 1% starch solution; group 2 (n = 20) was administered 75 mg/kg bw of Lavitol during the period of organogenesis—from the 6th to 16th day of gestation; group 3 (n = 20) was given 1500 mg/kg bw of Lavitol; and group 4 (n = 20) received 75 mg/kg bw of Lavitol during the period of implantation, organogenesis, and fetogenesis—from the 1st to the 19th days of gestation. The diet of both experimental and control groups was identical in volume. The individual dose concentration was modified according to the animal’s body weight after each weighing. Lavitol was dissolved and administered in a single dose in a 1% solution of potato starch (Extra calls, Proxima). Although all 4 groups received the 1% potato starch solution, only groups 2 through 4 received the potato starch in a suspension that included the required amount of Lavitol. One gram of soluble starch was mixed with 20 mL of distilled water, brought to a boil, and then brought to 100 mL by adding cold distilled water to the solution. Lavitol was then mixed with 3 mL of the 1% starch solution.
Experimental Phase
Animals were observed 3 times a day, and all observations recorded daily, for clinical signs of pharmacological and/or toxicological effects, including general appearance, behavioral changes, and locomotor activity, starting with the first day of dosing (day 1). Maternal body weight was recorded on day 1, day 8, and day 14, as well as during days 17 to 21 of gestation. All animals were divided into 4 groups of 20 pregnant animals per group. In each group, 10 of the 20 animals survived for birth, 5 were randomly sacrificed on day 18 of gestation, and the remaining 5 randomly sacrificed on day 20 of gestation. The group mean of the body weight and the percentage of body weight gain, as well as feed and water consumption, were calculated and recorded on days 3, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, 36, 39, and 42, of the study. All the offspring were sacrificed after 42 days of observation by decapitation. Male rats were weaned on day 25 (on the 4th week after birth). Male rats of each litter were housed in 720 cm2 cages type T/3C (drinking bowls and dish—polycarbonate; grid and divisor—stainless steel). One cage for 1 litter of each female rat (total 40 cages). No further analyses on dams were conducted within this experiment. All dams were removed from cages with female dams on day 25. Complete physical growth and development of offspring came to an end on days 25 to 30 after birth. The study of emotional–motor behavior of offspring was conducted up to day 45.
Laboratory investigations were carried out at the beginning (before placing males with females together in cages and on the day 20 of gestation). For mating, females were placed in cages with males for 2 astral cycles (2 females:1 male). Three to four days before the expected day of birth, the pregnant females were placed into single cages. Blood draws were performed and analyzed before mating, on the 18th day of gestation or 20th day of gestation, for those animals sacrificed. The first day of pregnancy was the day on which sperm was found in the vaginal smear.
Laboratory testing and physical examinations
All laboratory tests were performed in the Veterinary Center of the Far East State University. Hematological parameters included Hb, RBCs, PLTs, WBCs, eosinophils, basophiles, lymphocytes, and monocytes. Clinical chemistry and urinalysis testing was obtained at before females began mating and on the 20th day of gestation left for delivery and before necropsy on either the 18th or 20th day of gestation, and before necropsy of pups, conducted at the Central Research Laboratory of Amur State Medical Academy. Fetuses were given a gross external examination consisting of an evaluation of the shape of the body, head size, limb extension, and internal and external sexing of pups. Inspection of each fetus included digits, skin, umbilical region, anus and genitalia, the nares, pinna, eyes, and oral cavity. Two-thirds of the fetuses from each litter were subject to skeletal examination, performed by the Department of Embryology, Research Institute of Experimental Medicine (St Petersburg, Russia) using a method reported by Dawson. 17 Skeletons were examined under a stereoscopic microscope and the number of ossification points and skeletal abnormalities were evaluated. One-third of the fetuses from each litter were fixed in Bouin fluid and subject to visceral examination. Visceral examinations were performed using the Wilson method 11 by the same Department of Embryology. Examinations were performed on fetuses in all treatment and control groups and included the facial area of the skull, brain, heart, lungs, bronchi, esophagus, spinal cord, liver, kidneys, intestines, bladder, urethra, and reproductive organs.
Litters examined after delivery (lactation day 0) were evaluated for the number and sex of pups, stillbirths, live birth, and the presence of gross anomalies. Thereafter, the day any appearance of primary fur, ear unfolding, incisor eruption, eye opening, testes descent, and vaginal patency was recorded. Live pups were weighed every 3 days, from day 3 to day 42 of the study. In addition, the crania-caudal size of each animal was measured up to the 42nd day to determine whether somatic neural growth was affected. The following sensory-motor reflex changes during the maturation stages were observed or measured in the following chronological sequence: surface-righting reflex (from day 2), negative geotaxis test (from day 5), cliff avoidance (from day 2), open field (from day 8), olfactory (from day 10), pupil (from day 14), auditory (from day 8), muscle tone (from day 15), and open-field motor activity (from day 40).
Clinical chemistry of offspring was carried out on 120 animals (15 of each sex from each group). Blood samples were collected from the tail vein following 14 to 15 hours of fasting and tested (Veterinary Center, Far East State University) and included the following hematological parameters: Hb, RBC, PLT, WBC, HCT, TP, AST, ALT, total bilirubin, urea, and glucose.
Necropsy and histopathology
All offspring, fasted overnight before necropsy, were sacrificed on day 43 by decapitation by trained technicians, supervised by a veterinary pathologist. Complete gross pathology was carried out including examination of external surfaces, orifices, cranial, thoracic and abdominal cavities, and organs, including the carcass of dead or sacrificed animals. Macro- and microscopic evaluation was conducted on the heart, liver, kidneys, spleens, adrenal gland, and testicles. The pathologist was blinded to treatment. Organ weights were recorded as absolute and relative values (% bw) and calculated.
Statistical analysis
Statistical analysis of the differences between the control and experimental groups was performed using the Wilcoxon-Mann-Whitney and Student t test, with significance set at P < 0.05, using STATISTICA v.6 software (StatSoft, Tulsa, Oklahoma).
Results
Comet Assay Test
The results of the effect of Lavitol on the level of DNA damage in marrow cells, liver and blood of experimental animals are presented in Table 3. Administration of ethyl methanesulfonate to animals that served as positive controls was shown to cause a statistically significant increase in DNA damage in all organs and tissues studied. The level of DNA damage in the bone marrow, liver, and peripheral blood of the control animals was 1.8% ± 0.4% DNA in the tail. The DNA damage in marrow cells of animals fed with the test substance at a dose of 15 and 2000 mg/kg bw was 1.1 ± 0.3 and 2.0% ± 0.7% DNA in the tail, respectively. The paired comparison of obtained results with the results from the control group showed no statistically significant changes in the mean or median percentage of tail intensity in either liver, colon, or duodenum of male rats at any treatment level, compared to vehicle control values. No Hedgehog “ghost” cells were observed in any tissue examined.
Effect of Lavitol on DNA Damage in Marrow, Liver, and Blood Cells.
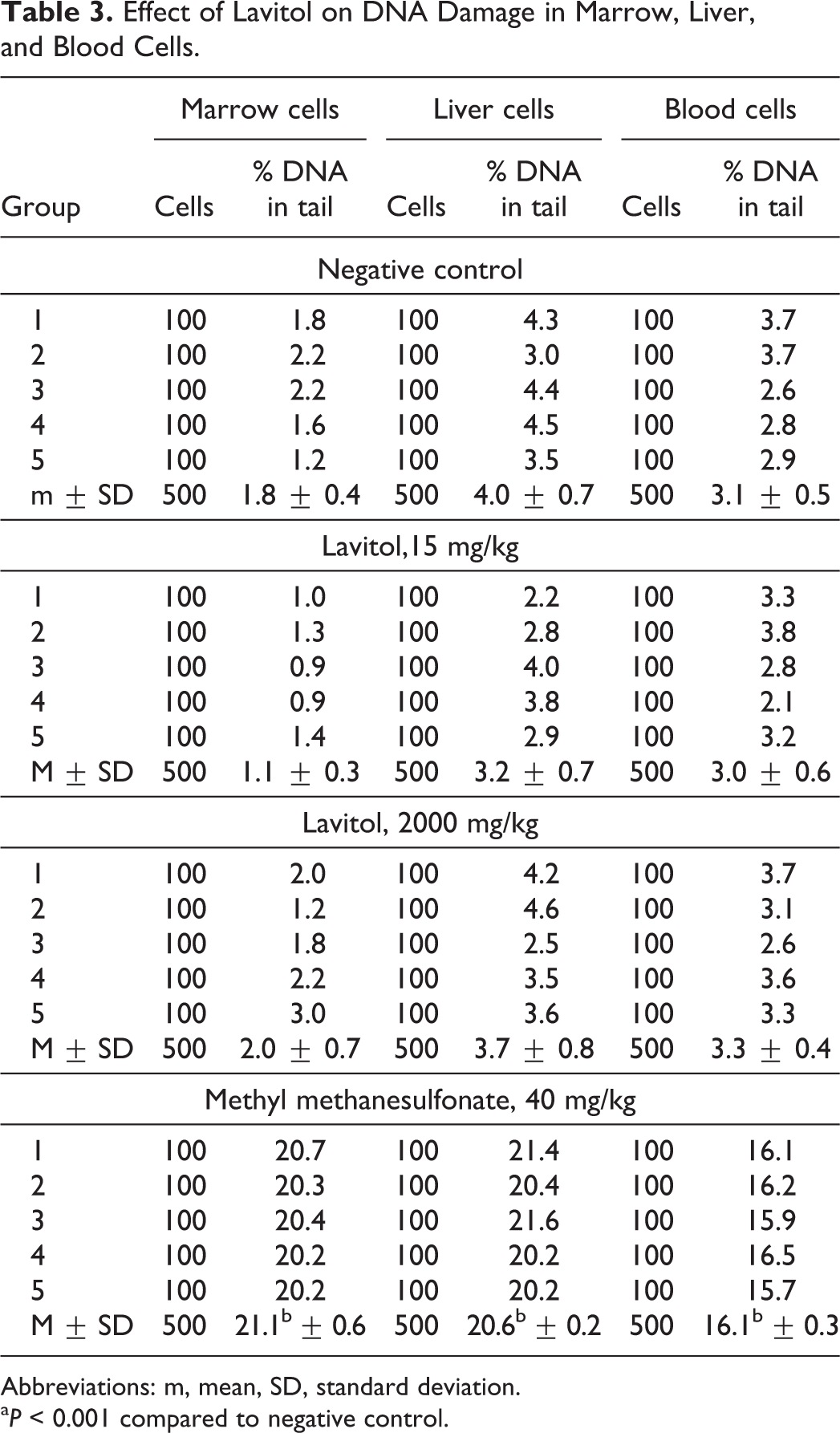
Abbreviations: m, mean, SD, standard deviation.
a P < 0.001 compared to negative control.
The level of DNA damage (strand breaks) in liver cells in animals of the control group was 4.0% ± 0.7%. The level of DNA damage in groups receiving the test substance in a dose 15 and 2000 mg/kg was 3.2 ± 0.7 and 3.7% ± 0.8% DNA in tail, respectively. No statistically significant changes are observed. In blood cells, the level of DNA damages was 3.1% ± 0.5% DNA in the tail. Treatment of animals with Lavitol at a dose of 15 and 2000 mg/kg bw, resulted in a level of DNA damage that was 3.0 ± 0.6 and 3.3% ± 0.4% DNA in the tail, respectively. No statistically significant changes were observed.
In conclusion, the Comet assay study found that a single administration of Lavitol at 15 and 2000 mg/kg did not induce the DNA damage in cells of the bone marrow, liver, and peripheral blood of the experimental animals.
Micronucleus Test in Human Lymphocytes
In both the absence and presence of S9 mix, following 3-hour treatment, and in the absence of S9 mix, following 20 hours treatment, Lavitol did not cause any statistically significant increases in the number of binucleate cells containing micronuclei when compared to the vehicle controls. The positive control compounds (mitomycin C, colchicine, and cyclophosphamide) caused significant increases in the number of binucleate cells containing micronuclei under appropriate conditions, demonstrating the efficacy of the S9 mix and the sensitivity of the test system.
Chromosomal Aberration Test
The results of chromosomal aberration test on frequencies of bone marrow metaphases in mice treated for 2 doses (15 and 2000 mg/kg bw) of Lavitol are summarized in Table 4. The level of chromosomal aberrations in bone marrow cells of male mice in the negative control group was 1.2% ± 0.4%. Administration of 20 mg/kg bw of cyclophosphamide resulted in a statistically significant increase in the level of chromosomal aberrations up to 19.2% ± 1.6% compared to results observed in the negative control group.
Frequencies of Bone Marrow Metaphases in Mice Treated With Lavitol.a
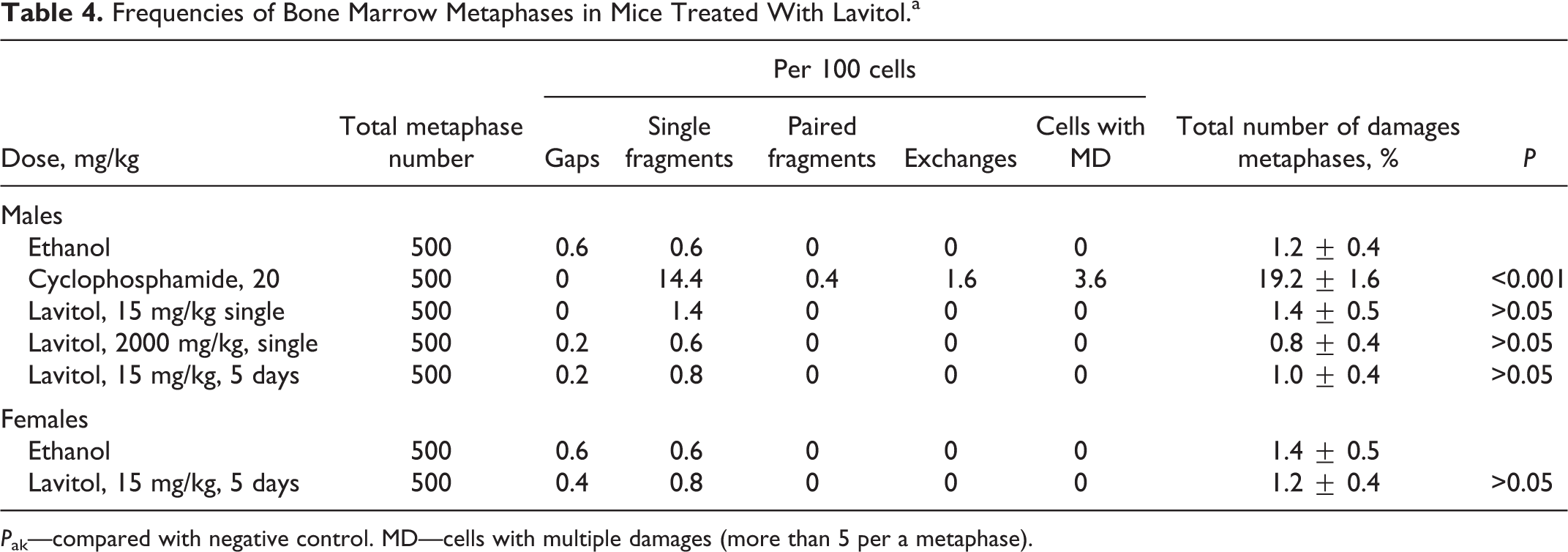
P ak—compared with negative control. MD—cells with multiple damages (more than 5 per a metaphase).
A single administration of Lavitol at 15 and 2000 mg/kg bw resulted in 1.4% ± 0.5% and 0.8% ± 0.4% chromosomal aberrations, respectively. There was no statistically significant difference between the single-treated and negative control groups. Administration of Lavitol for 5 consecutive days at 15 mg/kg bw resulted in 1.0% ± 0.4% of chromosomal aberrations, which was not statistically significant when compared to the corresponding negative control group. The levels of chromosome aberrations in bone marrow cells of female mice in the negative control and the Lavitol groups were 1.4% ± 0.5% and 1.2% ± 0.4%, respectively; while in male mice it was 1.2 ± 0.4 and 1.0 ± 0.4, respectively. Thus, there were no significant differences between the sexes.
Toxicity Studies
Subacute 7-day oral toxicity study
No mortality occurred following single administration of 10 000 or 15 000 mg/kg bw of Lavitol. There were no treatment-related clinical or behavioral effects observed. Quantity of food intake and feed consumption patterns was similar between the control and dose groups. Weight gain during the dosing period of 7 days was comparable for the control and dose groups, as shown in Table 5.
Dynamics of Body Weight, Grams (M ± m).a
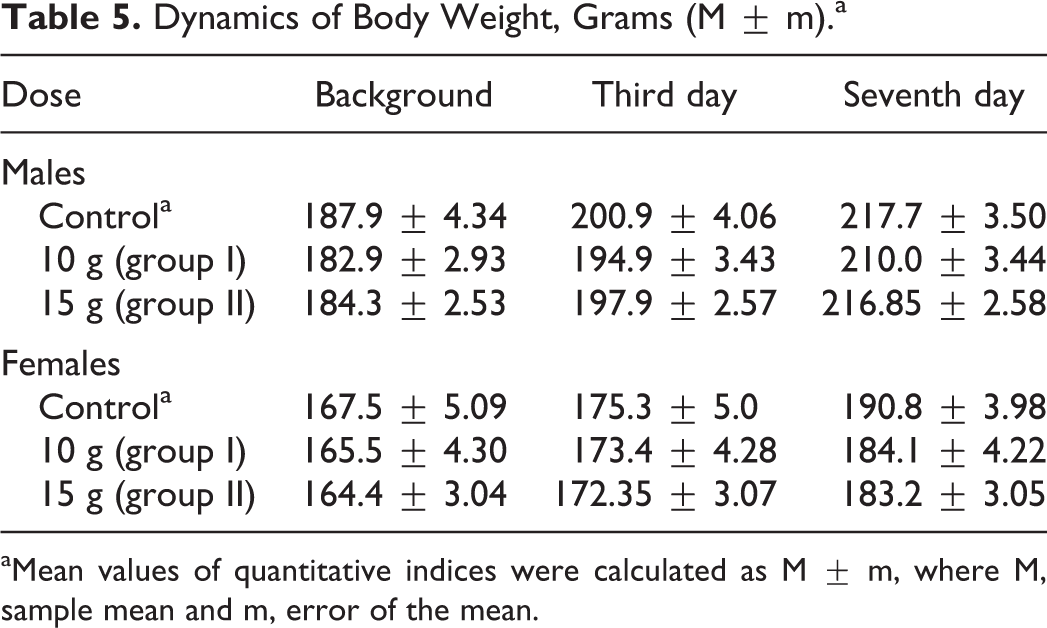
aMean values of quantitative indices were calculated as M ± m, where M, sample mean and m, error of the mean.
Administration of the test substance had no effect on animals’ behavior, skin, and fur condition, or mucous membranes, as the fur was smooth and clear, skin was elastic and mobile, and membranes pale and clear, with no pathological excretions observed. Hematological parameters shown in Table 6, found no significant differences other than decreased RBC values for females at both tested doses on day 8, and significant differences in RBC volumes between the control group of female rats compared to the experimental group of females, as shown in Table 7, which were within normal biological and laboratory historical limits. The RBC volumes in females in the control group shown in Table 8 were higher than historical laboratory limits, which would account for the significant differences observed. As shown in Table 9, significant changes in the values of albumin in males in the 10 000-mg/kg group were seen, yet were within normal biological and laboratory limits. Urine analysis of control and treated animals revealed no abnormalities. No significant difference in organ weights was calculated in either control or treated animals.
Hematological Blood Analysis, Day 8.
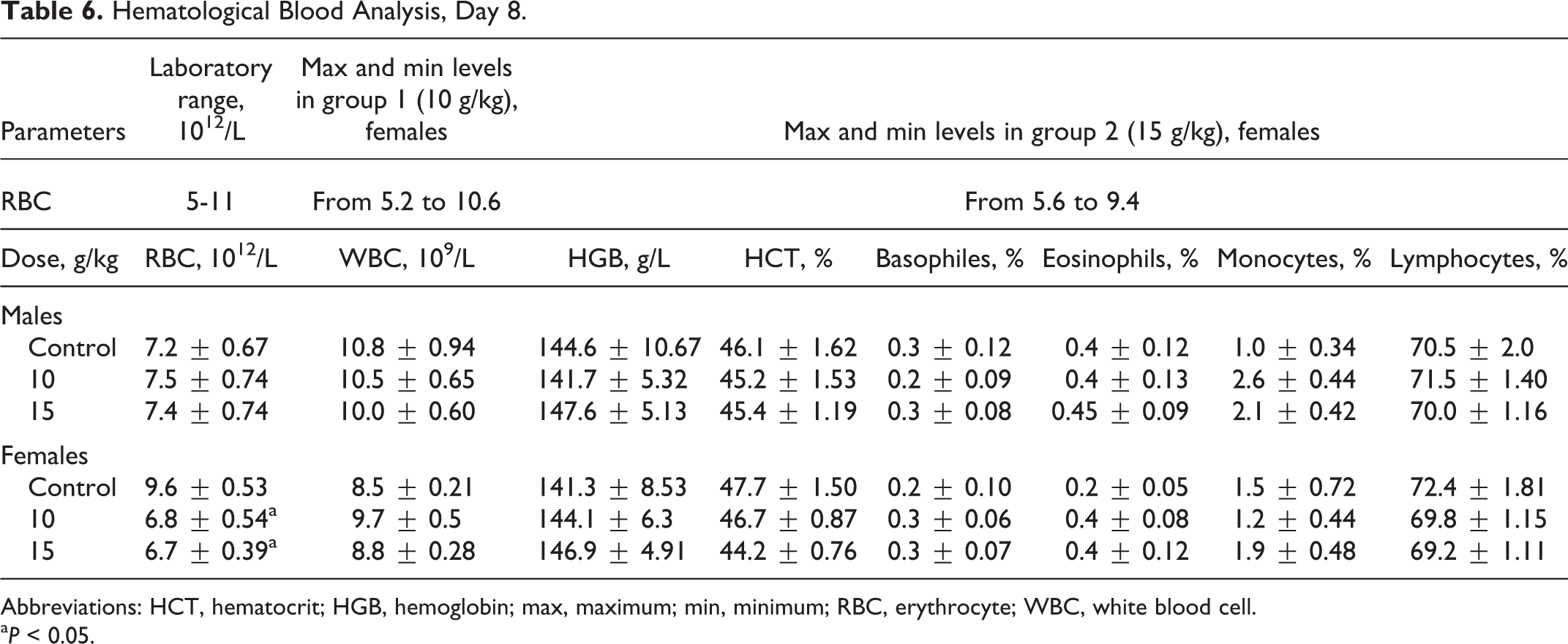
Abbreviations: HCT, hematocrit; HGB, hemoglobin; max, maximum; min, minimum; RBC, erythrocyte; WBC, white blood cell.
a P < 0.05.
Erythrocyte (RBC) Changes in Subacute Toxicity Study.

a P < 0.05.
Erythrocyte (RBC) Changes in Subacute Toxicity (Compare to Control).

a P < 0.05.
Biochemical Blood Analysis, Day 8.
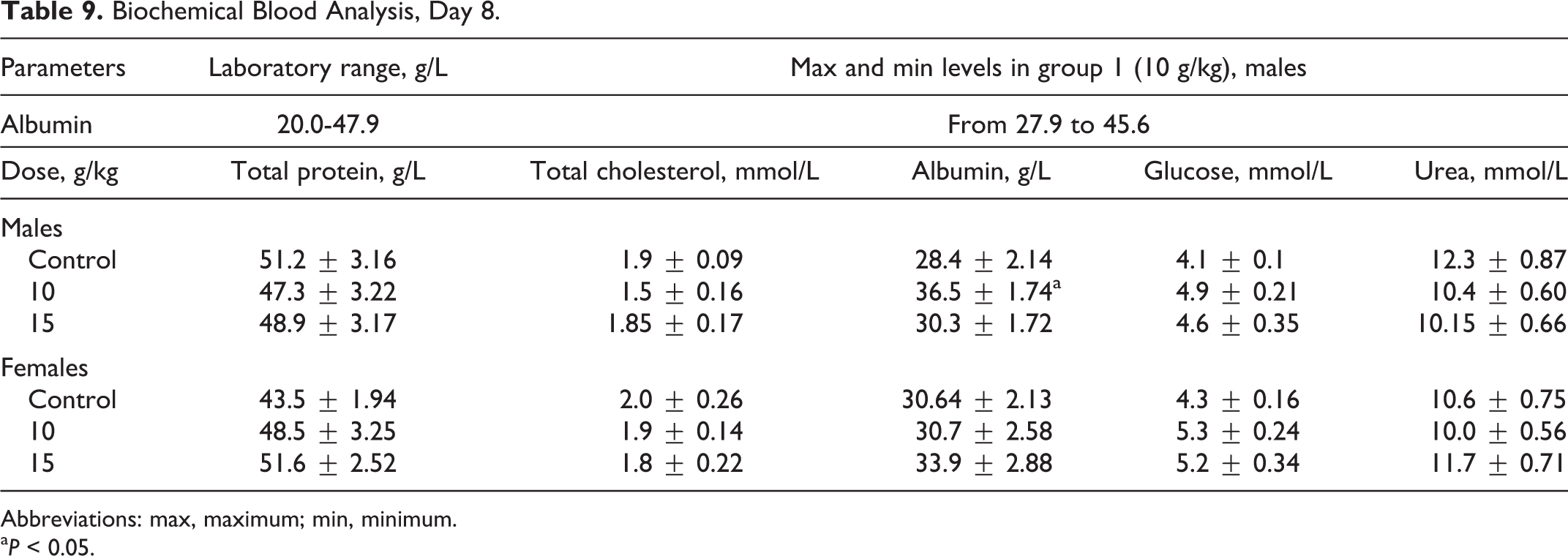
Abbreviations: max, maximum; min, minimum.
a P < 0.05.
Gross pathology revealed no abnormalities. Histopathological examination of the internal organs and tissues of the animals from control and experimental groups revealed single-type morphological changes observed in both males and females from controls groups and different dose groups with similar quality and quantity, considered incidental and physiologically related and not induced by the test substance. No morphological changes induced by the test substance were observed.
Subchronic 90-day toxicology study
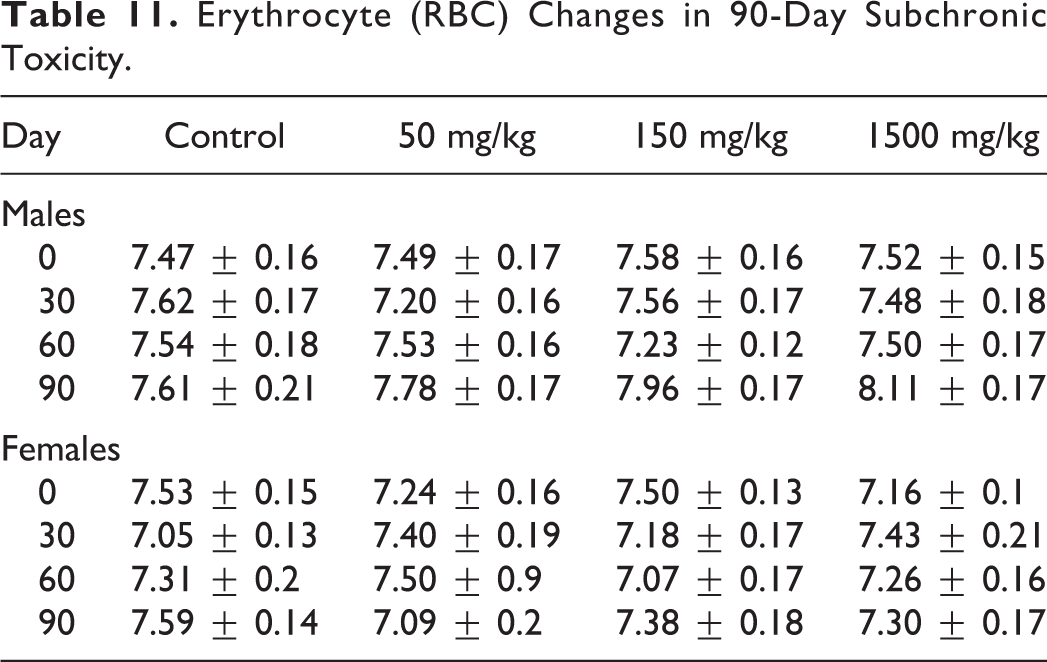
Animals from the control and dose groups exhibited comparable weight gain throughout the 90-day dosing period, as shown in Table 10, and changes in RBC is shown in Table 11. The quantity of food and water consumed by animals in each experimental group during the dosing period was not significantly different than that of the control group.
Body Weight of Rats During Administration of Test Substance (in Grams).
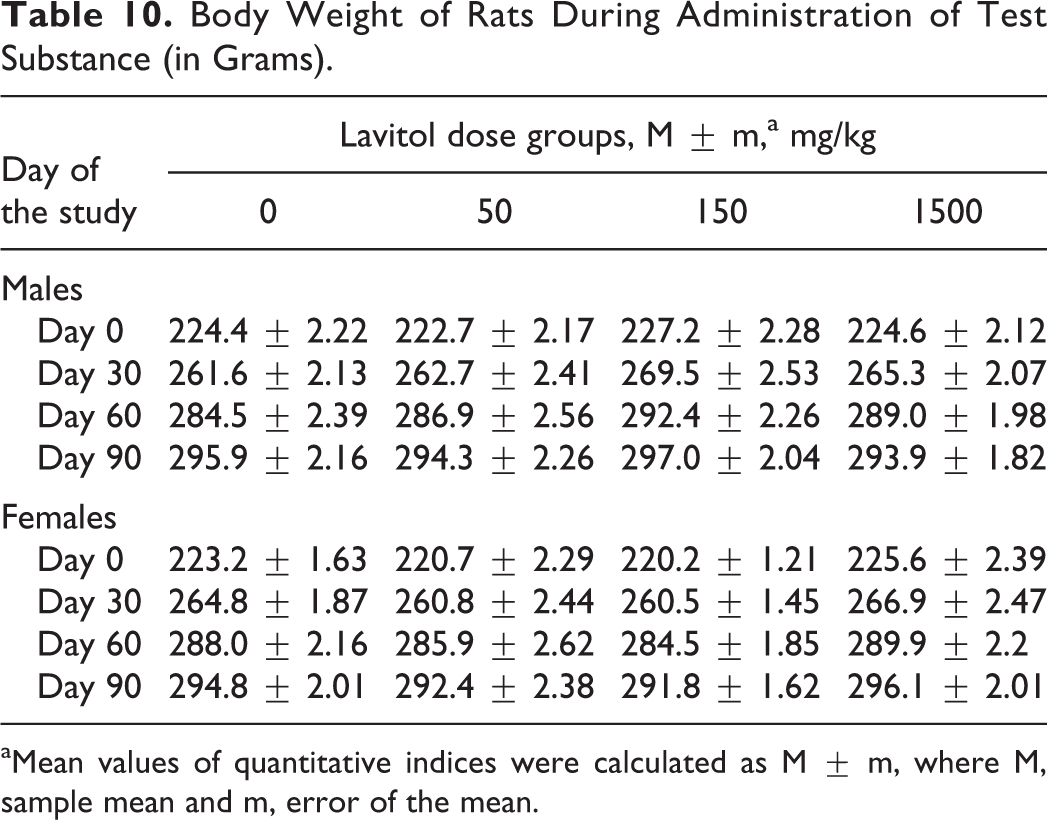
aMean values of quantitative indices were calculated as M ± m, where M, sample mean and m, error of the mean.
Erythrocyte (RBC) Changes in 90-Day Subchronic Toxicity.
No abnormal changes in the skin and fur appearance were observed in any of the experimental groups or controls, other than animals administered 50 and 150 mg/kg that had significantly thicker and fluffier fur compared to the 1500-mg/kg group or controls. No abnormalities in movement or posture, or presence of convulsions or tremors, were observed in any of the groups. During the first month of the study, males in the 150-mg/kg group were more active than males in either of the other 2 experimental groups or controls, while in the third month of the study, males in the 50- and 150-mg/kg group were significantly more active than males in the 1500-mg/kg or control groups. Stool disturbances were observed in all groups during the study, but only significantly lower in both males and females in the 150-mg/kg groups compared to controls. There were no significant differences measured in rectal temperature between the experimental groups and controls. Mucous membranes were normal in all groups. No edema, hyperemia, or pathological excretions were observed in any of the experimental groups. No deviations were seen in the corneal reflex of any animals tested, even at the maximal dose of 1500 mg/kg on pupil size or width of the palpebral fissure.
Physiological examinations, hematology, and biochemistry
No significant changes in heart function or electrophysiology were observed in the 40 animals evaluated at the beginning and at the end of the study on day 0, day 60, and day 90. The auricular (peak value P, interval size P-Q, and ventricular ECG indices [peak T value]) were within normal levels in all tested animals.
Open-field examination data, revealed effects on behavior in some males and females, as follows: males – defecation in the 50 mg dose group at day-30 (P < 0.05), vertical motor activity in the 1500 mg dose group at day-90 (P < 0.05), and looking into holes, in the control group at day-90 (P < 0.05); females – latent period, vertical motor activity, and defecation in 1500 dose group at day-30 (P < 0.05). A statistically significant increase in the latency (time required to leave the center) was observed in females in the 1500-mg/kg group after 30 days compared to day 0. During the same time period, a statistically significant increase in the number of stands (vertical motor activity) and decrease in number of defecations were observed in the same group. A statistically significant decrease in number of stands was observed in males administered 1500 mg/kg for 90 days. Otherwise, no other significant differences between control and experimental groups were seen in other open-field indices.
No statistically significant differences were seen in the duration of hexanal-induced sleep among 40 animals evaluated given Lavitol at any of the dose groups on day 30, 60, and 90, compared to day 0. No statistically differences were found in hematological indices between any of the 96 experimental and control animals for either sex during the study period, the exception of two female control groups or in RBC changes (Table 11), as all indices were within normal values.
Urine analysis of control and treated animals revealed no abnormalities. No significant differences in organ weight between animals of either sex in the control and experimental groups were observed on day 91. Gross pathological examination did not reveal any abnormality within and between the control and experimental groups.
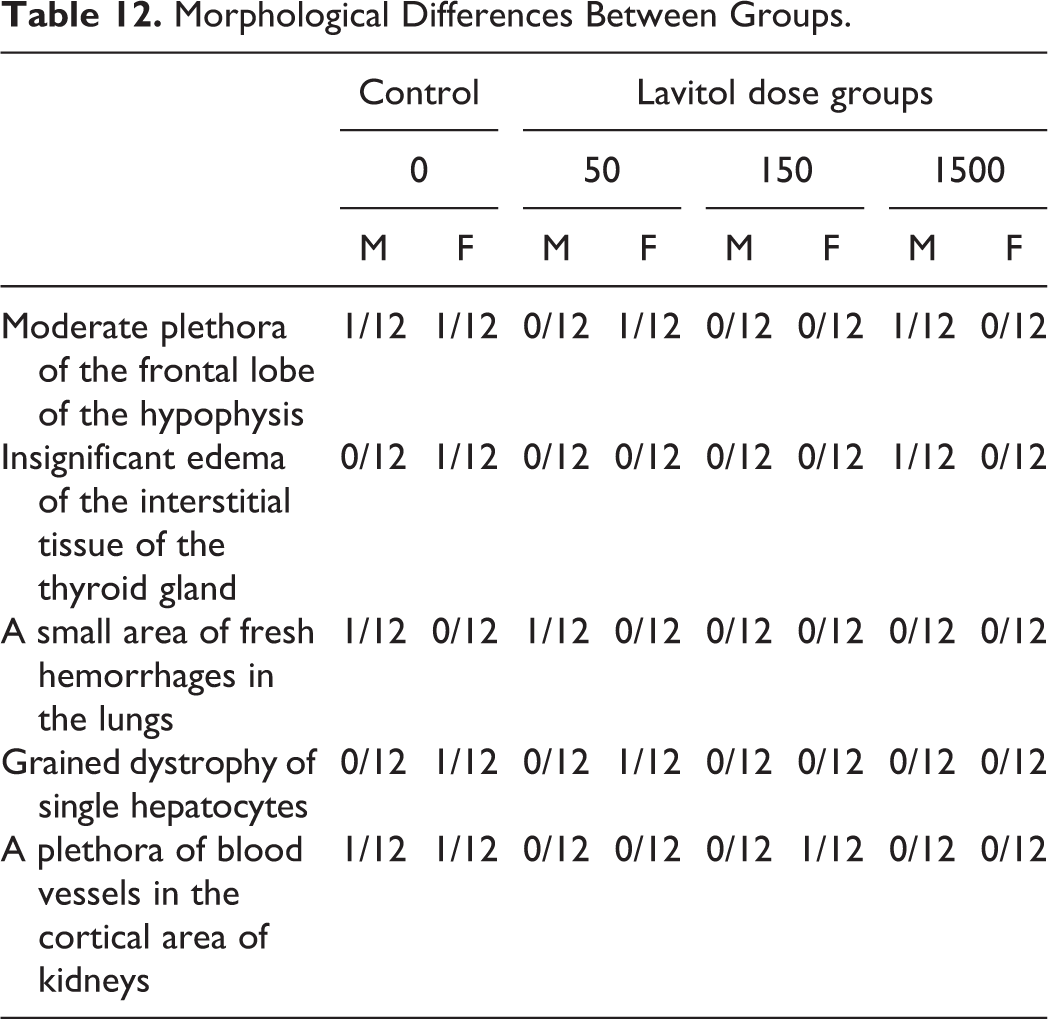
Histopathological examination of internal organs and tissues of the animals in each of the experimental groups did not reveal any significant morphological differences between that of controls, as shown in Table 12. Moderate plethora of the frontal lobe of the hypophysis, insignificant edema of the interstitial tissue of the thyroid gland, a small area of fresh hemorrhages in the lungs, grained dystrophy of single hepatocytes, and a plethora of blood vessels in the cortical area of kidneys observed in the few male and female animals were of similar quality and quantity and hence not considered clinically relevant. Testicles were whitish, of normal size and density. The body of the uterus was of normal density, size, and form. The uterus horns were thin and showed clear to pale mucous membranes. The ovaries were of dark red color, moderate density, usual form, and size.
Morphological Differences Between Groups.
Developmental Toxicity Study
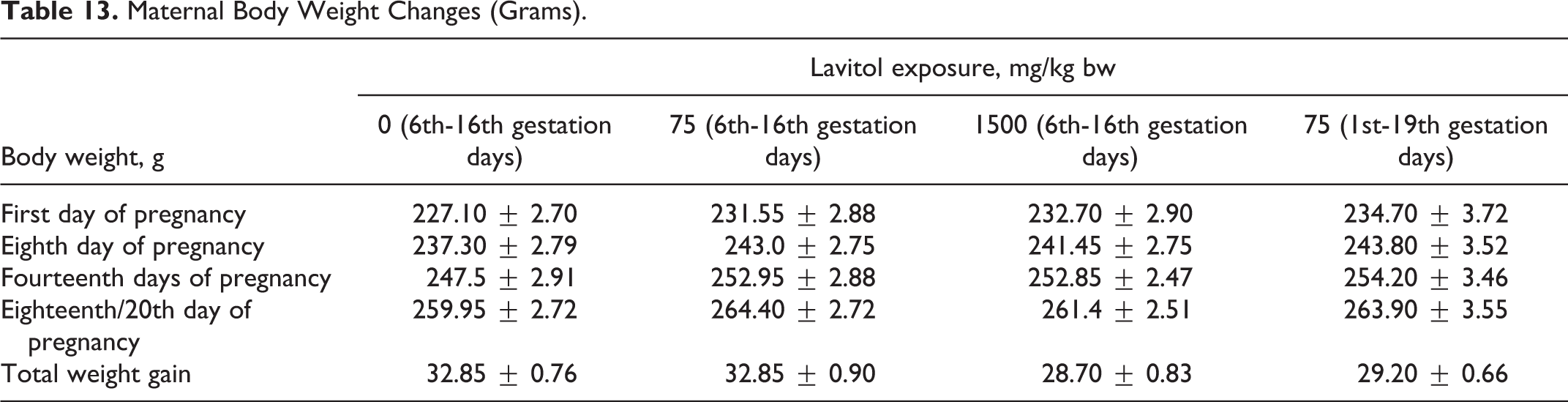
All 4 groups of animals showed no signs of toxicity during the dosing period. No mortality occurred in any of the animals in either the control or experimental groups. Dams in all groups gained weight during gestation during the study. No significant differences in weight occurred in any group. Weight gains in pregnancy ranged from 12.33% (group 3) to 14.47% (group 1). Weights for each group during 4 gestational periods are shown in Table 13. No differences were observed between the control and supplemented groups for appetite, behavior, or activity.
Maternal Body Weight Changes (Grams).
Gross examination of animals during the live phase of the study revealed some incidents in all groups. No abnormalities in lacrimation, breathing, movement, or posture were noted in any animals. Occasional stool disturbances (liquid stool) were recorded for animals in all groups.
Clinical chemistry
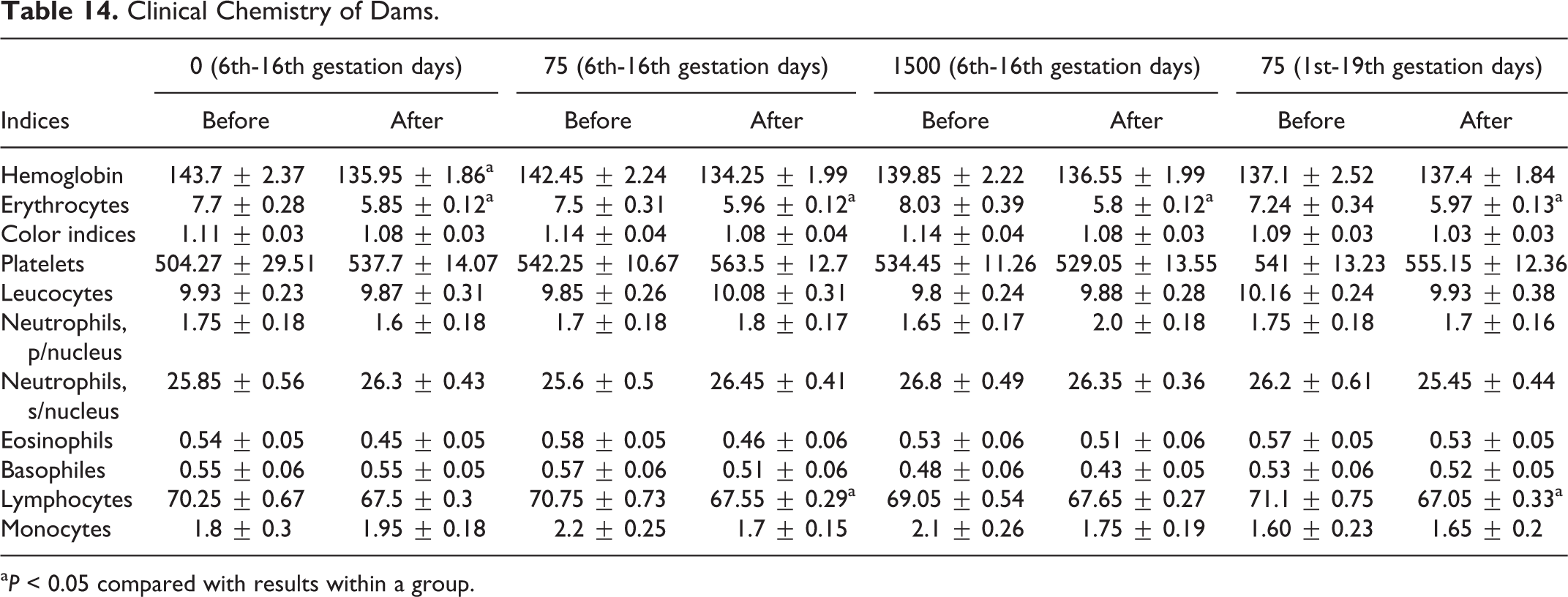
Blood sampling occurred prior to gestation and during the period of gestation on days 18 or 20. No statistical differences were seen between control or experimental groups in clinical chemistries, as indices were within normal values, as shown in Table 14. Differences in hematological parameters of females between treatment and control groups were within normal laboratory limits, with nonstatistical differences of values for treatment and control groups, and nonstatistical differences of values for RBC changes, as shown in Table 15. Clinical chemistry of dams is shown in Table 16. As shown in Table 17, a decrease in RBC volume was observed in all 4 groups at the end of the gestation period, where the volume of RBCs in rats of the control group was 1.1 times lower than historical norms, and groups 2, 3, and 4 were 1.3, 1.4, and 1.2 times lower as well, respectively. The observed decrease in volume of RBC could be considered a common phenomenon during gestation.
Clinical Chemistry of Dams.
a P < 0.05 compared with results within a group.
Hematological Values for Assigned Groups.
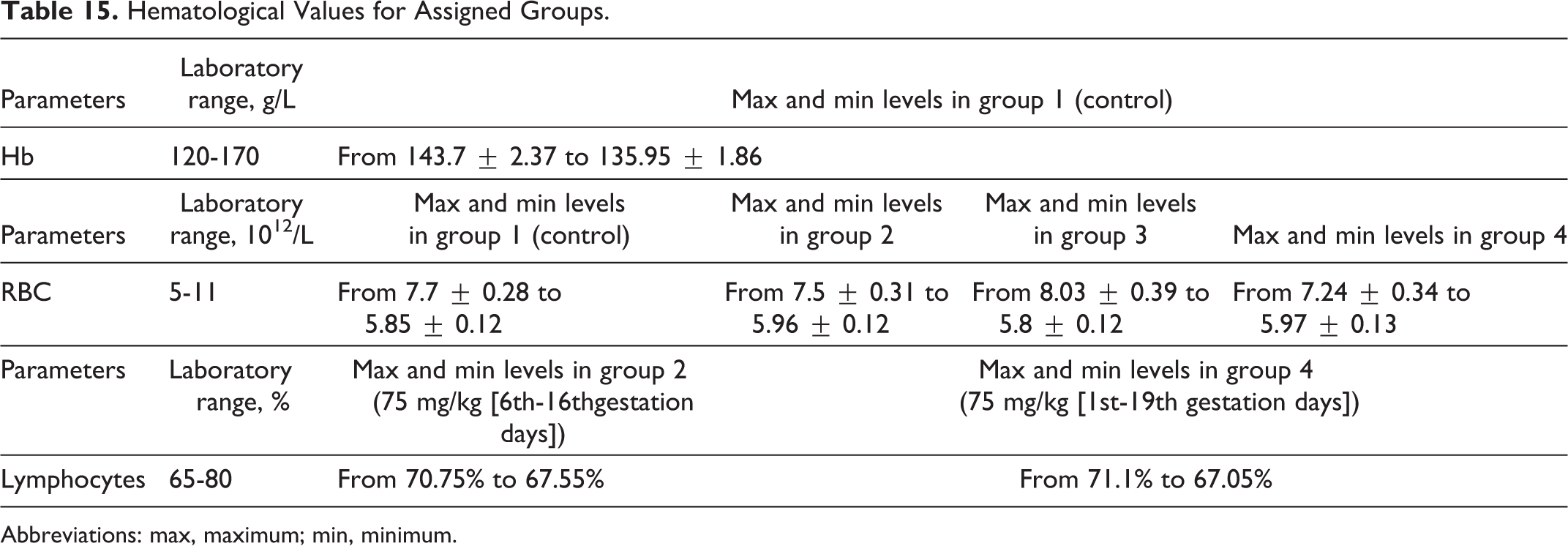
Abbreviations: max, maximum; min, minimum.
Clinical Chemistry of Dams in Developmental Study.
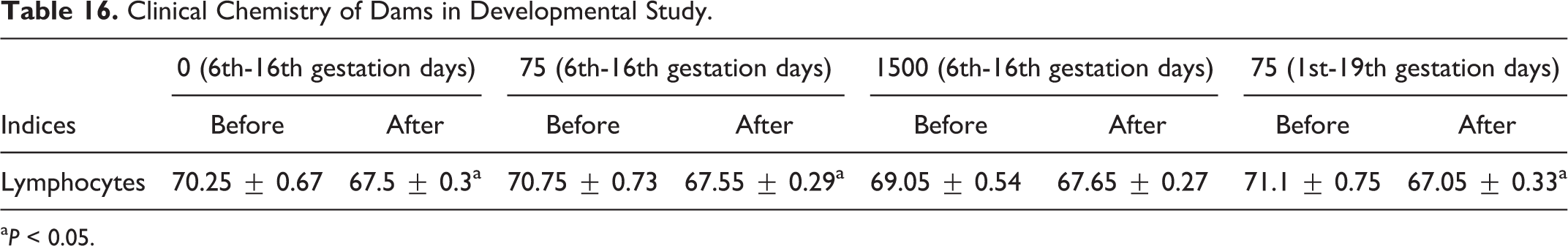
a P < 0.05.
Erythrocyte (RBC) Changes in Developmental Toxicity (Dams, Prior Gestation, and on 18th or 20th Day of Gestation).
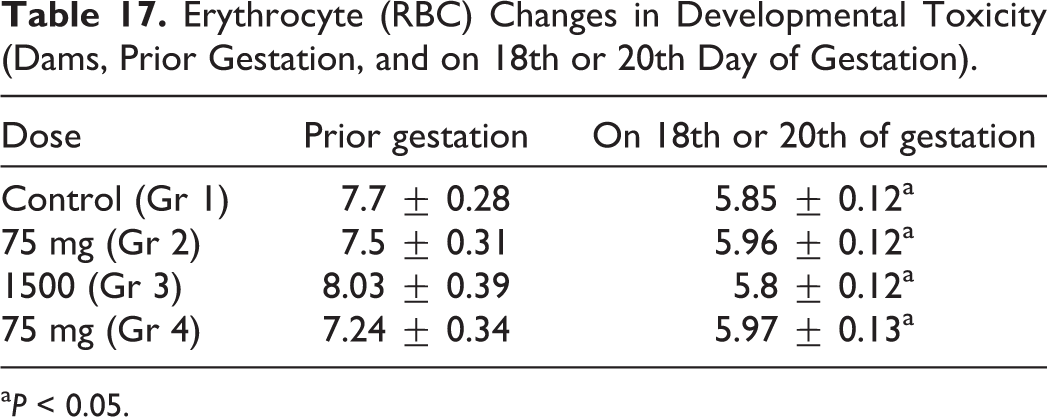
a P < 0.05.
Physiological examinations and fetometrics
There were no cases of spontaneous abortion after administration of Lavitol in either the 75- or 1500-mg/kg groups. Based on the assessments of the animal’s corpora lutea, females in group 4 had a greater ability to be impregnated than any of the other groups; the lowest percentage occurred in the control group, as shown in Table 18.
Fetal Survival After Maternal Exposure to Lavitol.
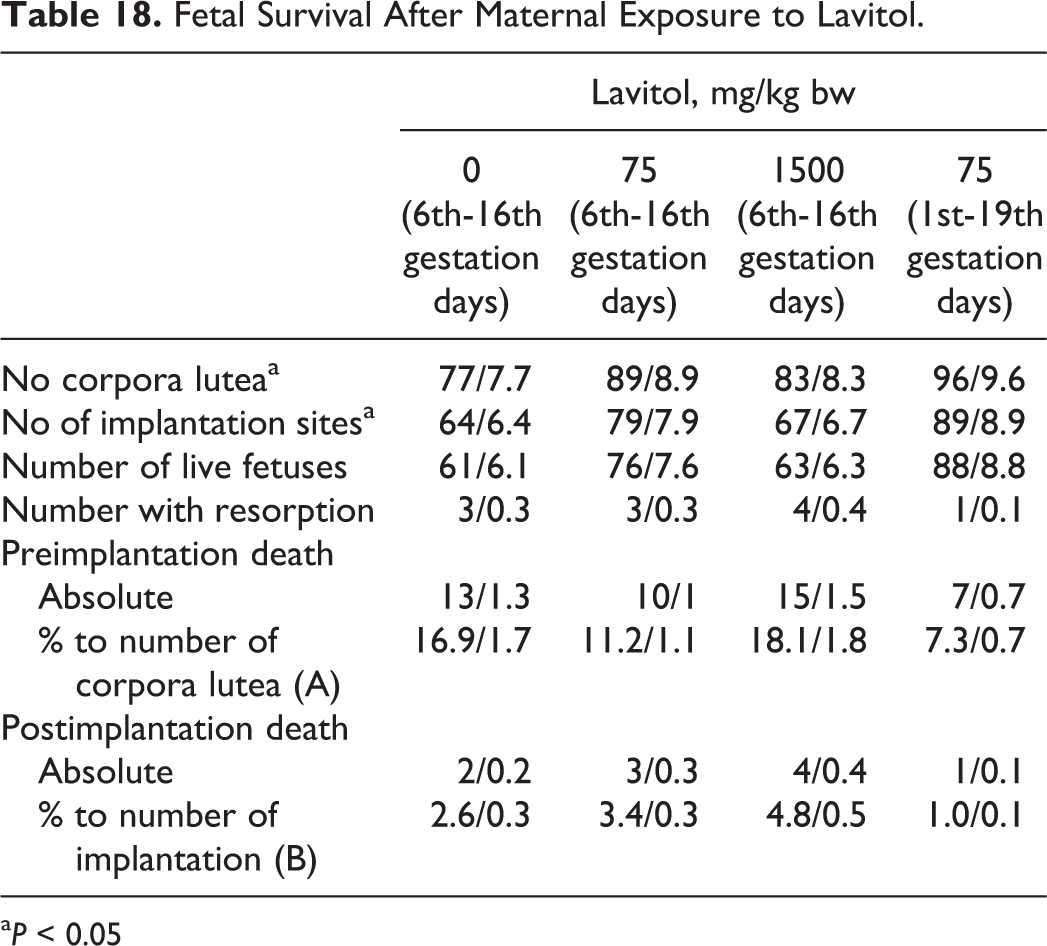
a P < 0.05
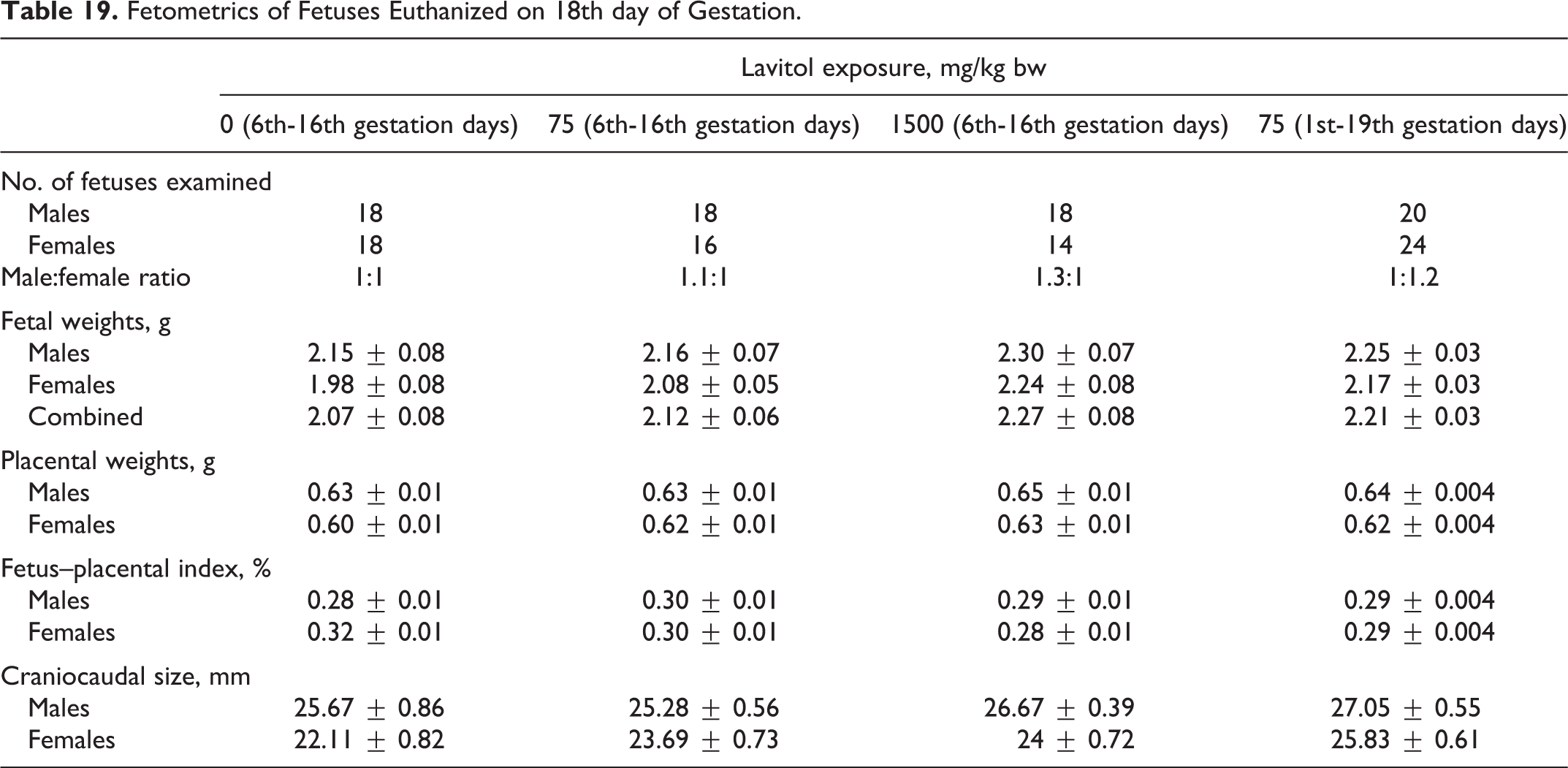
One hundred forty-six fetuses from the dams euthanized on the 18th day of gestation were observed. One hundred forty-one fetuses from the dams euthanized on the 20th day of gestation were observed. The results are shown in Tables 19 and 20. Fetal weight of males on the 18th day of gestation varied from 2.15 ± 0.08 g in group 1 to 2.30 ± 0.07 g in group 3, while fetal weight of females varied from 1.98 ± 0.08 g in group 1 to 2.24 ± 0.08 g in group 3. Placental weight varied from 0.63 ± 0.01 g to 0.65 ± 0.01 g (for males) and from 0.60 ± 0.01 g to 0.63 ± 0.01 g (for females). The craniocaudal size varied from 25.28 ± 0.56 to 27.05 ± 0.55 g (for males) and 22.11 ± 0.82 to 25.83 ± 0.61 (for females). The fetus–placental index varied from 0.28% ± 0.01% to 0.30% ± 0.01% (for males) and from 0.28% ± 0.01% to 0.32% ± 0.01% (for females).
Fetometrics of Fetuses Euthanized on 18th day of Gestation.
Fetometric Indices of Fetuses Euthanized on the 20th Day of Gestation.
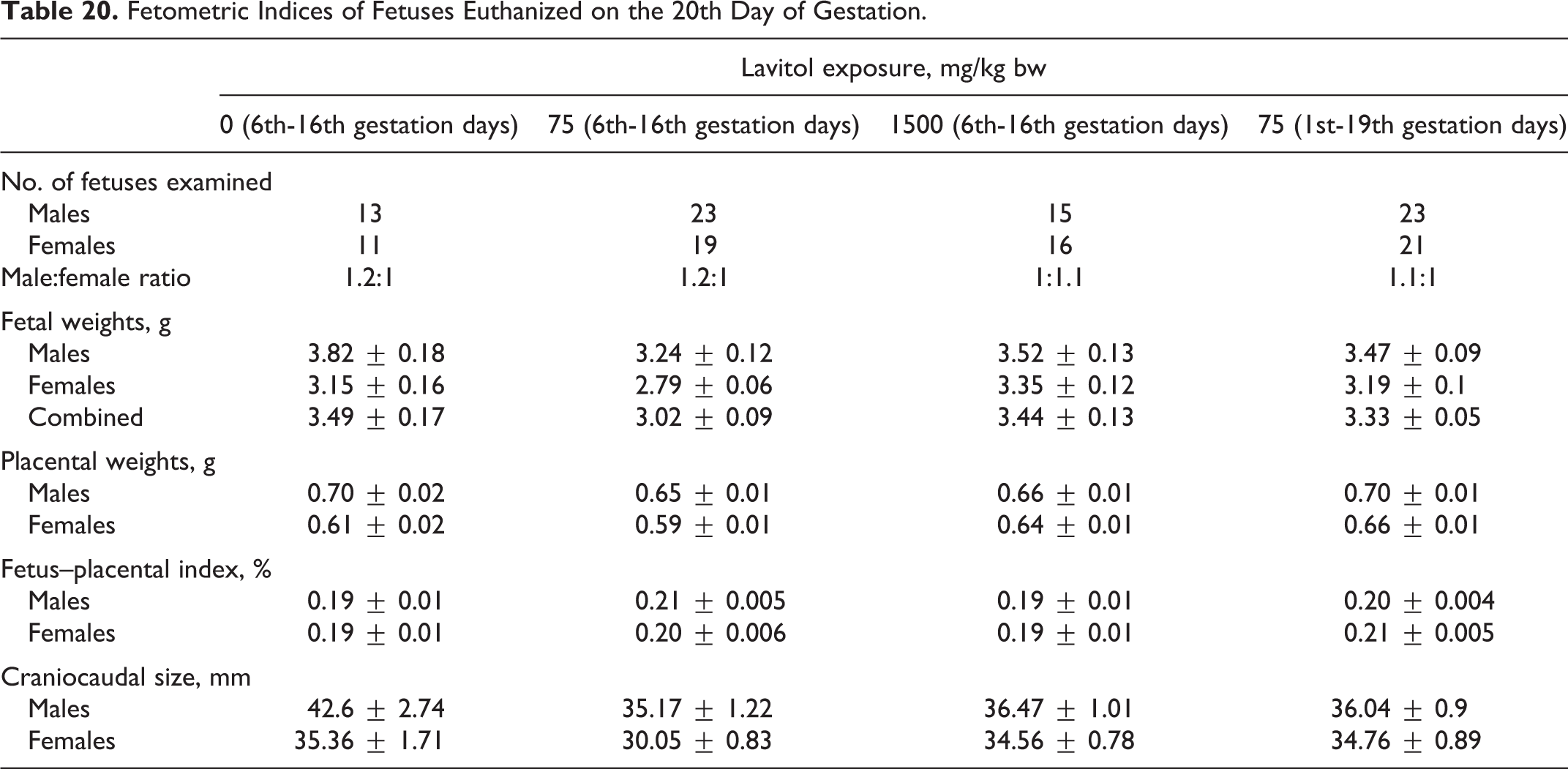
Fetal weight of males euthanized on the 20th day of gestation varied from 3.24 ± 0.12 g in group 2 to 3.82 ± 0.18 g in group 1, while fetal weight of females varied from 2.79 ± 0.06 g in group 2 to 3.35 ± 0.12 g in group 3. Placental weight varied from 0.65 ± 0.01 to 0.70 ± 0.01 g (for males) and from 0.59 ± 0.01 to 0.66 ± 0.01 g (for females). The craniocaudal size varied from 35.17 ± 1.22 to 42.6 ± 2.74 mm (for males) and from 30.05 ± 0.83 to 35.36 ± 1.71 mm (for females).
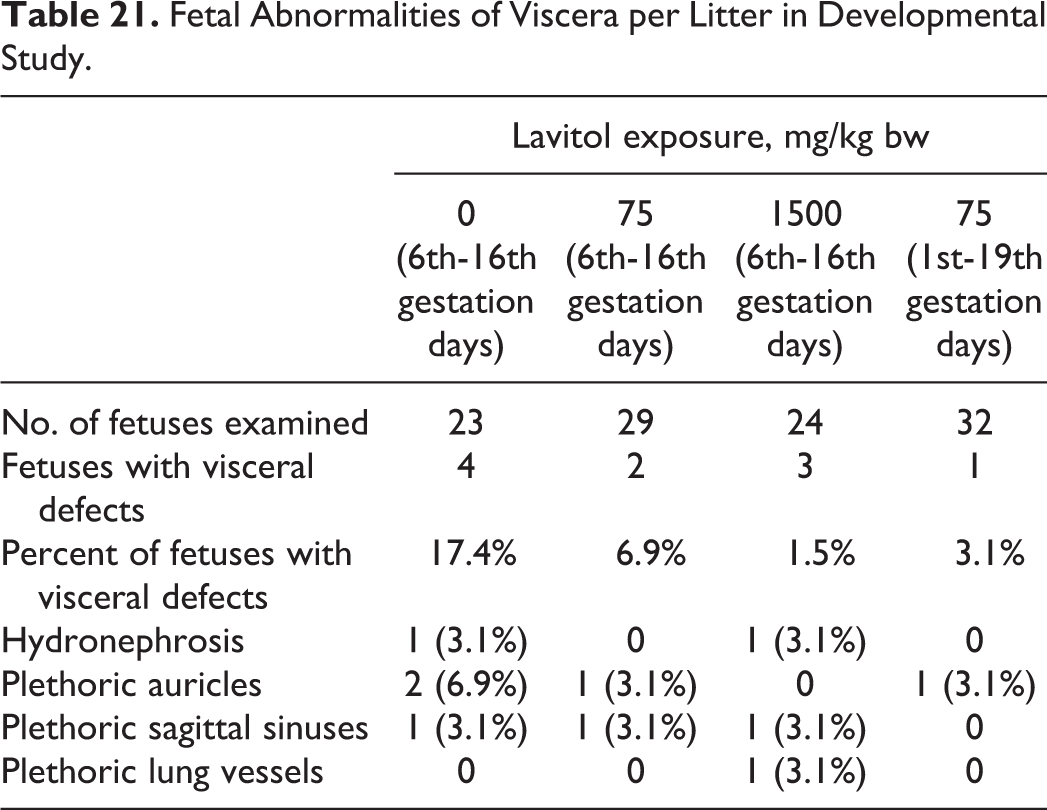
No abnormalities were found during visual observation of 287 fetuses from all groups. Fetal defects of the viscera were seen more so in the control group, as seen in Table 21, than any of the treatment groups.
Fetal Abnormalities of Viscera per Litter in Developmental Study.
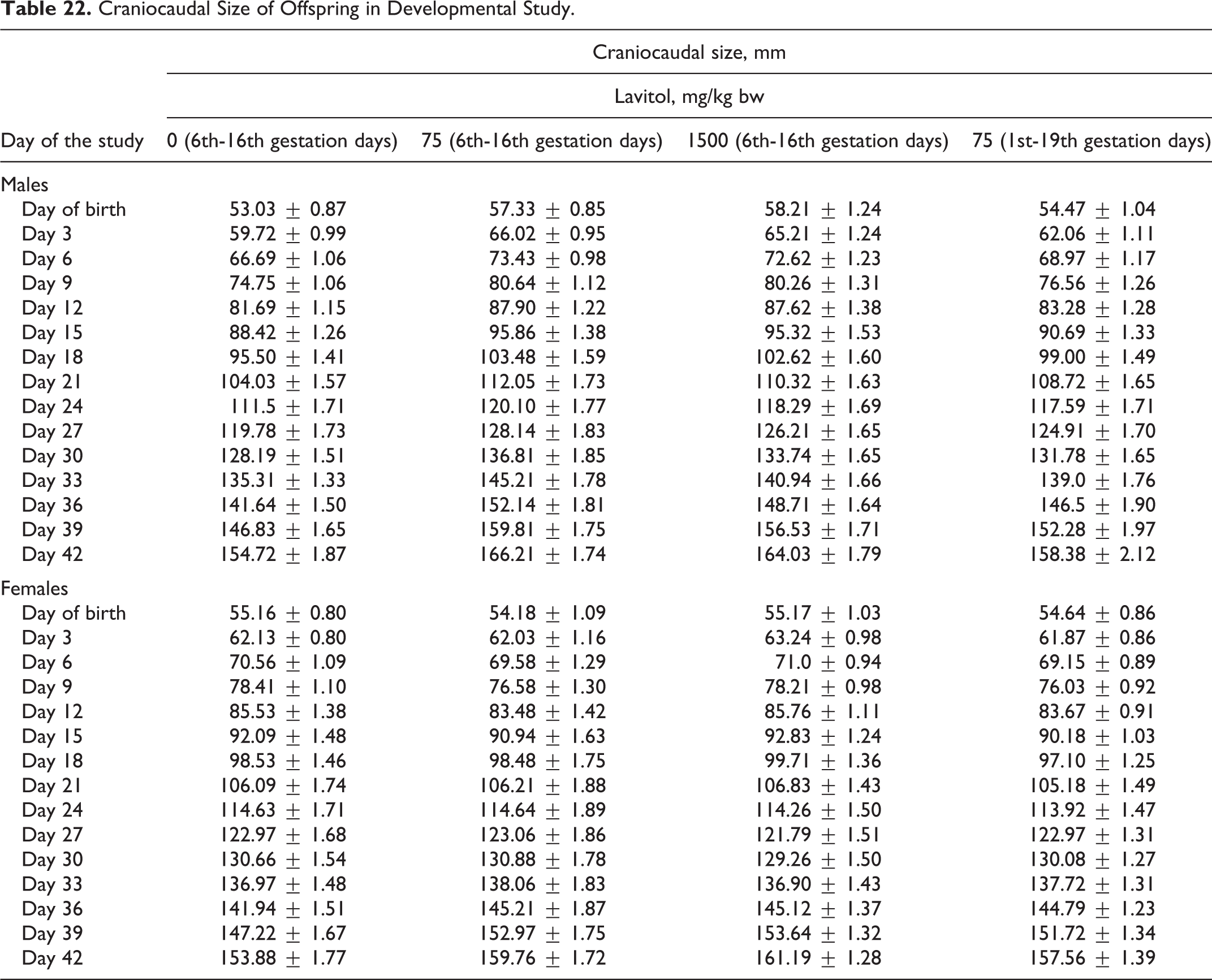
No treatment-related skeletal anomalies were seen in any of the groups as shown in Table 22. Degree of ossification of each bone (other than the skull) was unremarkable in any group.
Craniocaudal Size of Offspring in Developmental Study.
Postnatal development
Postnatal developmental observations of 291 live pups on day 0 and day 42 were carried out. Body weights of newborns in all test groups were within normal limits, varying from 4.72 ± 0.16 g (the mean body weight of newborns in group 4) to 4.89 ± 0.22 g (the mean body weight of newborns in group 2). A steady increase in body weight was observed for all pups. By the 42nd day, body weight of pups varied from 70.36 ± 3.00 to 74.67 ± 2.92 g. In group 1 and group 2, the number of male offspring exceeded that of female offspring: 36 males and 40 males over 32 females and 33 females, respectively. In groups 3 and 4, the number of female offspring exceeded that of male offspring: 42 females and 39 females compared to 34 males and 32 males, respectively. Two dead pups were found in the control group and in group 2, caused by birth trauma in the control group, and the mothers eating the pups following birth. Earflap detachment occurred on the second day in pups from all groups. Hair growth was observed on the 5th day; incisor eruption on the 7th day in the control group and on the 8th day in the treatment groups; and eye opening observed on the 14th day. Testes descent was observed on day 25, while opening of the vagina on day 30 (but on day 29 in group 2).
There was no statistically significant difference in the maturation date of sensory-motor reflexes among the offspring. Observation of emotional–locomotor behavior of 40-day-old pups was unremarkable. The time required to leave the center varied from an average of 5.73 ± 0.12 seconds (group 2) to 7.01 ± 0.22 seconds (group 4). The number of visited squares varied from 21.33 ± 1.23 (group 3) to 27.48 ± 1.76 (group 1); the number of racks varied from 0.51 ± 0.12 (group 1) to 0.72 ± 0.19 (group 3); the number of different types of grooming varied from 0.51 ± 0.06 (group 2) to 0.62 ± 0.07 (group 1); and the number of defecations varied from 0.32 ± 0.06 (group 2) to 0.44 ± 0.07 (group 3).
Necropsy and histopathology
Autopsy of animals revealed no significant pathological changes. Serous membranes of all animals were smooth and shiny. Mucous membranes were pale pink. Heart, liver, kidneys, spleen, organs of the gastrointestinal tract, adrenals, thymus, testicles, and uterus were of normal consistency, color, and size. Postmortem evaluation of constitution, external surfaces, orifices, and mucous membranes as well as thoratic and abdominal cavities and the organs located in the cavities was conducted. The histological examination of the internal organs and tissues of offspring from control and experimental groups did not reveal any significant morphological differences between groups.
Discussion
Genotoxicity Studies
In the micronucleus test in human lymphocytes, Lavitol administered for 3 hours at concentrations of up to 650 μg/mL, in both the absence and presence of S9 mix, and up to 200 μg/mL for 20 hours in the absence of S9 mix only, did not show evidence of causing an increase in the induction of micronuclei in cultured human cells. In the chromosomal aberration test, oral administrations of Lavitol by either a single dose of 2000 mg/kg bw or a repeated dose of 15 mg/kg bw resulted in no cytotoxic effects on bone marrow cells in this test model. Lavitol was not shown in the Comet assay to show any evidence of causing an increase in DNA stand breaks or cytotoxicity in the liver, colon, or duodenum of male rats in this in vivo test procedure.
Toxicological Studies
All male and female animals survived throughout the 7-day dosing period. Animals exhibited comparable body weight gain and food consumption. Hematological, biochemical, and urine analysis and functional battery tests revealed no abnormalities attributable to the treatments. Organ weights at the end of the dosing period were comparable to that of controls. Neither gross pathological or histopathological examinations revealed any toxicological abnormalities in treated animals.
In the subchronic 90-day toxicity study, all male and female animals administered Lavitol orally up to a dose of 1500 mg/kg survived throughout the study. Treated animals experienced no significant changes in weight compared to controls during the study or differences in food consumption. Hematological analyses conducted during and at the end of the study were comparable to that of controls, as were biochemical and urine analyses, in all treated animals. Functional battery observation tests conducted at termination revealed no abnormalities. Organ weights at the end of the dosing phase of the study were comparable to controls. Gross pathological changes in any of the organs were not observed or did histopathological examinations of the organs and tissues reveal any clinically relevant abnormalities. The repeat subchronic toxicology study found no adverse effects of the test substance Lavitol, when administered orally for 90 days up to a dose of 1500 mg/kg bw in male or female rats.
In the developmental toxicology study, dams in all groups survived throughout the dosing administration period. No signs of toxicity were observed in animals during this period. The absence of deaths of pregnant females or spontaneous abortions indicates a healthy duration of pregnancy. Yellow discoloration of the abdomen-covering fur observed in 18 of the 20 animals in group 3 supplemented with 1500 mg/kg of Lavitol starting with day 16 of gestation could be attributed to excretion of Lavitol administered at the highest doses, resulting in discoloration of the fur on the belly.
Although significant decreases in the levels of RBCs were observed in the treatment groups by the end of the study, similar decreases were observed in the control group, while none of the values fell below normal limits.
No statistically significant differences were detected in the number of corpora lutea per dam or implantation sites among groups, or in resorptions, late fetal deaths, nonlive implants, or percent pre- and postimplantation loss, male or female, or combined fetal weights, or male to female fetal ratios. The main fetometric indices indicated that fetuses in all groups corresponded to the gestation period. No abnormalities in internal organs other than singular cases of hemorrhage in the abdominal walls, hydronephrosis, and plethora of atrical and lungs, which were observed in both control and experimental groups. No treatment-related skeletal anomalies were seen in any of the dose groups. In addition, there was no evidence of exposure-related reductions in the ossification of the fetal skeleton. Small differences in the length of ulna and humoral bones in the fetuses of dams from groups 3 and 4 can be explained by differences in craniocaudal measurements as previously discussed.
Lavitol had no effect on litter size, physical development, survival, reflex meansurements, or behavioral variables. Gross pathologies did not reveal any abnormalities nor did histopathological examinations of internal organs and tissues, in animals in the control or experimental groups. Therefore, the present study found that Lavitol exerts no embryotoxic or teratogenic effects on the development of offspring in this mammalian species.
Given the concentration of DHQ in Lavitol, the previously reported studies support the findings of Booth and Deeds. 28 These investigators reported the compound to be nontoxic based on long-term feeding studies performed in albino rats when give a dose of 1% of the test substance in food. They further reported that based on chromatographic analysis of the urine of 2 human volunteers who had ingested 2 g of the compound, the conversion to its 3 metabolites, 3,4-dihydrophenyl-acetic, m-hydroxyphenylacetic, and 3-methoxy-4-hydroxyphenylacetic acids, were the same metabolites that had been seen in rats and rabbits following oral ingestion, in which no toxicity was observed.
In a series of studies carried out by Shkarenkov et al, 27 a single oral dose of “diquertin” (now named Lavitol), which contained the same concentration of DHQ, was administrated at various concentrations orally, intraperitoneally, and by injection into the stomach, at doses 10-fold and 100-fold greater than the recommended dose recommended for humans. In 1 study, doses of 150 and 1500 mg/kg were administered to 80 white mice (18-20 g) and 60 white rats (160-175 g) of both sexes, in a 1% starch solution by intragastric administration and a 5% diquertin alcoholic solution by intraperitoneal administration, respectively, without any mortality or significant adverse effects other than shortness of breath, languor, or cyanosis of skin integuments of auricles and limbs observed in a few mice and rats.
The same authors also reported on a chronic 6-month toxicity study of diquertin given to 48 white male rats (150-165 g), divided into 3 groups of 16 rats each that received either a 1% starch solution serving as controls or 2 experimental groups that received either 150 or 15000 mg/kg of diquertin administered into the stomach. The same investigators also studied the safety of the same preparation in another chronic 6-month study on mongrel dogs (10-16 kg), divided into 2 groups that either received 190 mg/kg/d diquertin in their food or the same diet without the test substance. In both the rat and dog studies, no evidence of diquertin toxicity was shown. Except for slight variations in some functional tests, the safety of the preparation was supported by evaluation and analyses of the animal’s organs and tissues.
In addition, the authors reported in the same article an evaluation of the immunomodulatory properties of diquertin administered to CBA mice. The experimental substance did not affect antibody-forming cells of the spleen or alter the hemagglutinin titer in the serum or result in any delayed-type hypersensitivity or graft versus host reactions. Previous studies reported by Zhanataev et al, 29 that assessed DHQ’s genotoxicity in the chromosome aberration test and the DNA-comet assay, found a lack of evidence of the compound’s genotoxicity. In their studies, DHQ was administered 5 times at 0.15 or 1.5 mg/kg, or in a single dose of 15, 150, or 2000 mg/kg, in 8- to 12-week-old male and female C57B1/6 mice (18-20 g) and found to induce no DNA damage in bone marrow, or blood, liver, and rectal cells. The single administration study at either a dose of 150 or 2000 mg/kg had no effect on the level of chromosome aberrations in mouse bone marrow cells compared to controls. As was seen in our studies of Lavitol, the DHQ in a wide dose range exhibited no clastogenic or DNA-damaging effects in organs or tissues of the animals so studied.
Conclusion
Given the evidence of safety presented herein, and based on increasing use of this extract in foods and supplements, and growing evidence of its application as a radioprotective agent, 4,5 further in vitro and in vivo research into the exact mechanism of DHQ and Lavitol in the prevention, mitigation, and treatment of disease pathology and radioprotection is warranted.
Footnotes
Acknowledgments
The authors are grateful to the technical staff at Huntingdon Life Sciences/Harlan Laboratories in Huntingdon, United Kingdom. To Dr A.K. Zhanataev and A.D. Durney, with the Zakusov Research Institute of Pharmacology, Russian Academy of Medical Sciences, for reviewing and providing assistance in the preparation of the article.
Authors’ Note
Some of the studies in this article were discussed in a poster presented at the 51st Annual Meeting of the Society of Toxicology, San Francisco, California, March 12, 2012 (Toxicological Sciences, 2012;126(Suppl 1):S45).
Author Contributions
A.G. Schauss contributed to conception or design; acquisition, analysis, or interpretation; drafted the article; gave final approval; and agrees to be accountable for all aspects of work ensuring integrity and accuracy. S.S. Tselyico contributed to acquisition, analysis, or interpretation; drafted the article; critically revised the article; gave final approval; and agrees to be accountable for all aspects of work ensuring integrity and accuracy. V.A. Kuznetsova and I. Yegorova contributed to conception or design, critically revised the article, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. VAK is an employee of the sponsor, Ametis JSC. IY is a representative for the sponsor in North America.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Ametis JSC, Blagoveshchensk, Amur Oblast, Russia.