
Abstract
Yeast cells transformed with high-copy number plasmids comprising a green fluorescent protein (GFP)-encoding gene optimized for yeast under the control of the new DIN7 or PLM2 and the established RNR2 and RAD54 promoters were used to assess the genotoxic potential of chemical compounds. The activity of potential DNA-damaging agents was investigated by genotoxicity assays and by OxoPlate assay in the presence of various test compounds. The fluorescence signal generated by GFP in response to DNA damage was related to the different concentrations of analytes and the analyte-dependent GFP synthesis. The use of distinct DNA damage-inducible promoters presents alternative genotoxicity testing strategies by selective induction of promoters in response to DNA damage. The new DIN7 and PLM2 systems show higher sensitivity than the RNR2 and RAD54 systems in detecting 4-nitroquinoline-N-oxide and actinomycin D. Both DIN7 and PLM2 systems are able to detect camptothecin while RNR2 and RAD54 systems are not. Automated laboratory systems with assay performance on 384-well microplates provide for cost-effective high-throughput screening of DNA-damaging agents, reducing compound consumption to about 53% as compared with existing eukaryotic genotoxicity bioassays.
Introduction
In the recent years, Vietnam has been experiencing fast industrialization, urbanization, and population growth leading to an increased demand for agricultural and industrial production and health care services. However, these growing problems also contribute to environmental pollution, particularly to outbreaks of foodborne and waterborne diseases caused by pathogens, 1 antibiotics, 2 heavy metals, 3 toxins, 4 and so on resulting from contaminated food, antibiotic residues in food, untreated or inadequately treated wastewater disposal from households, and effluents from hospitals or factories in industrial zones. The harmful chemicals daily released into the environment cause bioaccumulation, biomagnification, or acute toxic effects for exposed people. Thus, early detection of those hazardous substances is of great current interest in food safety and quality control and environmental monitoring in Vietnam.
Conventional and advanced physicochemical analyses were used to identify toxic compounds. 5,6 These methods mainly provide analytical information (ie, identity and quantification of known chemicals), rather than functional information regarding the possible biological effects of contaminants (cytotoxic or genotoxic) and their potential short- or long-term impact on the ecosystem, that is, on organisms, including humans. Additionally, when harmful substances are new, unknown, or not yet deposited in available databases, they cannot be identified by these approaches.
Therefore, a large number of biosensors using whole cells instead of individual molecules (DNA and proteins) have been developed for cytotoxicity, genotoxicity, and environmental monitoring. Some of these biosensors are yeast based and applied to investigate the activity of genotoxins or chemical substances 7,8 and for drug screening. 9 Approximately 40% of yeast and human genes share conserved sequences; thus, fundamental biochemical pathways and cellular processes are similar between yeast and humans as are DNA repair mechanisms. 10 Yeast cells possess mitochondria and a cell nucleus, into which the chromosomal DNA is packaged in a highly structured form, analogous to that found in mammalian cells, thus substantially differing from bacteria. Accordingly, yeast provides an adequate platform overcoming many of the limitations of bacterial assay systems and further can reduce the need for expensive mammalian cell culture and animal testing.
Many yeast genes are inducible by DNA damage, such as RAD54, RNR2, DIN7, PLM2, POL30, THI4, and so on, or involved in the regulation of multidrug resistance, such as PDR1, QDR2, and so on (Saccharomyces Genome Database [SGD] accessed via http://www.yeastgenome.org). RAD54 is known to be involved in the repair of double-stranded breaks 11 and induced above a constitutive level by ultraviolet (UV), 12 ionizing radiation, and alkylating agents. 7 RNR2 is also involved in DNA damage repair including DNA lesions induced by alkylating agents, 7 gamma and ionizing radiation, and hydroxyurea. 13,14 Like RAD54 and RNR2, DIN7 (DNA damage inducible) and PLM2 (PLasmid Maintenance) belong to a group of DNA damage repair genes. The role of DIN7 in DNA repair and replication as well as its role in modulating the stability and metabolism of mitochondrial DNA (mtDNA) had been described and characterized previously. 15,16 Of particular interest is its protective function for mtDNA, which may be particularly susceptible to reactive oxygen species (ROS), for which DIN7 can provide additional sensitivity. 15 -17 PLM2 had been reported to be involved in the maintenance of the 2μ plasmid. 18 Furthermore, PLM2 is also involved in cell cycle control and is required for the activation of the factors involved in DNA replication and repair. 19,20 DIN7 and PLM2, like RNR2, RAD51, RAD54, and DUN1, also belong to the DNA Damage Signature cluster involved in homologous recombination and DNA damage repair, including repair of double-strand breaks (DSBs), in response to DNA damaging agents 20 and had been shown to be upregulated in the telomerase deletion response induced by methyl methanesulfonate (MMS) and ionizing radiation. 21
Therefore, the promoter regions of some of these, in particular RAD54 and RNR2, have been used as biosensing elements to construct reporter plasmids and to develop yeast-based biosensors for investigating and screening a broad range of chemical compounds including genotoxins, cytotoxins, and antibiotics. 7,8 Many studies indicate that the expression of DIN7 and PLM2 is also induced in response to ROS and DNA-damaging agents. 17,22 We used the 2 RAD54-GFP and RNR2-GFP reporter plasmids 7 as reference systems and further created new reporter plasmids containing a promoter of the damage-inducible genes DIN7 and PLM2 in order to determine whether the promoters of DIN7 and PLM2 may also be used as biosensing components, like RAD54 or RNR2. This is done with the intention of establishing new reporter plasmids and to subsequently develop alternative toxicity monitoring systems with the aim of contributing to the development of yeast-based biosensors for cytotoxicity and genotoxicity or for large-scale analysis of environmental samples. These reporter plasmids were transferred into Saccharomyces cerevisiae cells that were subsequently used to investigate the genotoxic potential of standard chemical compounds and drugs by genotoxicity assays performed on 384-well microplates. In addition, the short- or long-term biological impact of potentially DNA-damaging agents on cellular respiration was assessed by OxoPlate assay. For a less time-consuming and more accurate process, a set of R packages developed in our earlier studies 23 and modified for suitability in this study as well as Excel macros were applied to execute all steps, including liquid handling and pipetting, measurements, data processing and analyzing, of experiments in a fully automated and continuous manner without the need for user interactions.
Materials and Methods
Plasmids, Strains, and Chemicals
Two reference reporter plasmids, RAD54-GFP pWDH444 and RNR2-GFP pUMGP5 plasmids, 7 and S cerevisiae strain Y486 (also known as FF 18984, MATa leu2-3,112 ura3-52, lys2-1, his7-1) were obtained from Stefan Wölfl (Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Germany).
All test compounds used for our genotoxicity assays were purchased from Sigma-Aldrich (Taufkirchen, Germany): hydrogen peroxide solution (CAS No. 7722-84-1), N-nitroso-N-methylurea (NNNM, CAS No. 684-93-5), 4-nitroquinoline-N-oxide (4-NQO, CAS No. 56-57-5), camptothecin (CAS No. 7689-03-4), actinomycin D (CAS No. 50-76-0), netropsin dihydrochloride (CAS No. 1438-30-8), bleomycin sulfate (CAS No. 9041-93-4), menadione (CAS No. 58-27-5), and 1 positive genotoxicity control, MMS (CAS No. 66-27-3). Other chemicals or substances used in this study were purchased from Merck (Darmstadt, Germany) and Fisher Scientific (Germany).
Construction of New Promoter-Reporter Plasmids
The 2 reference reporter plasmids, RAD54-GFP pWDH444 and RNR2-GFP pUMGP5, were fully known and well characterized. 7 The RNR2-GFP pUMGP5 plasmid that carries a few genes including a gene encoding green fluorescent protein (GFP) optimized for yeast (yEGFP) under the control of the RNR2 promoter, a gene bearing resistance to kanamycin (KanMX3), and a gene conferring uracil prototrophy (URA3), was utilized as a source for creating 2 new reporter plasmids, DIN7-GFP pUMGP5 and PLM2-GFP pUMGP5, by replacement of the RNR2 promoter region at BamHI and PacI positions.
The primer pairs listed in Table 1 were used to amplify DIN7 and PLM2 promoter regions by polymerase chain reaction (PCR) of genomic DNA extracted from the FF 18984 strain (Yeast DNA Extraction Kit; Thermo Scientific, Dreieich, Germany). The PCR comprises 100 ng of genomic DNA, 0.5 µmol/L (each) forward and reverse primers (Integrated DNA Technologies, USA), 200 µmol/L deoxynucleotide triphosphates (New England Biolabs, NEB, Frankfurt, Germany), 1× standard Taq reaction buffer (NEB), 1.25 U Taq DNA polymerase (NEB), and ddH2O in a total volume of 50 µL. The amplification was performed as follows: 94°C for 1 minute; 30 cycles at 94°C for 30 seconds, at 55°C for 30 seconds, and at 72°C for 1 minute; and 72°C for 4 minutes (Mastercycler pro, Eppendorf). The PCR products were examined by 1% agarose gel electrophoresis analysis (Bio-Rad, Munich, Germany) and purified (QIAquick Spin PCR Purification Kit; Qiagen, Hilden, Germany).
Primers Used for Construction of 2 New Promoter-Reporter Plasmids, DIN7-GFP pUMGP5, and PLM2-GFP pUMGP5.a

Abbreviations: PCR, polymerase chain reaction.
aThe underlined and bold bases are the restriction sites of BamHI (GGATCC) and PacI (TTAATTAA) incorporated in forward (F) and reverse (R) primers, respectively. The extra bases upstream of the restriction sites are for improvement in cutting efficiency.
The purified PCR products with the expected size were ligated into pJET1.2 cloning vector (CloneJET PCR Cloning Kit; Thermo Scientific) and cloned in Escherichia coli-competent cells (DH5α; Invitrogen, Darmstadt, Germany) by the conventional KCM (KCl, CaCl2, and MgCl2) transformation method. Only the cells able to propagate were used for plasmid extraction (QIAprep Spin Miniprep Kit; Qiagen). Subsequently, the concentration of the purified plasmids was determined (NanoDrop 2000; Thermo Scientific) and the sequencing primers included in the kit (CloneJET PCR Cloning Kit) were used to sequence the cloned inserts (ABI Prism 3100 Genetic Analyzer).
The insert, DIN7 or PLM2 promoter, and nicked GFP pUMGP5 plasmid were achieved after single digestion of the pJET1.2 plasmid containing DIN7 or PLM2 promoter and the RNR2-GFP pUMGP5 plasmid with each enzyme, BamHI and PacI (NEB), respectively, separation in 1% agarose gel, and extraction and purification (QIAquick Gel Extraction Kit; Qiagen). Prior to ligation reaction, the digested GFP pUMGP5 plasmid was dephosphorylated by antarctic phosphatase (NEB) for preventing recircularization. Each insert was joined by ligation (T4 DNA ligase, NEB) into digested GFP pUMGP5 plasmid forming new DIN7-GFP pUMGP5 and PLM2-GFP pUMGP5 plasmids. These enzymes were all used and inactivated (if necessary) according to the instruction of the manufacturer (NEB). Again newly formed plasmids were transformed in homemade chemically competent cells of E coli DH5α. The transformants were selected by plating on LB agar (Miller's LB broth base; Invitrogen) supplemented with kanamycin (100 µg/mL). The ligation products were then extracted, purified, and digested with BamHI and PacI, and the digests of ligation were checked by separation in agarose gel same as that mentioned previously.
Genotoxicity Assay
Preparation of yeast cultures
The individual plasmids were transformed into yeast cells (wild-type Y486) using the LiAc/SS carrier DNA/PEG method developed by Gietz and Schiestl. 24 The transformants and recombinant plasmids were maintained during cell growth and division by further selection for uracil prototrophy in SD/-Ura agar (Clontech, TaKaRa, France). Hence, the strains transformed with RAD54-GFP pWDH444, RNR2-GFP pUMGP5, DIN7-GFP pUMGP5, and PLM2-GFP pUMGP5 plasmids were designated as RAD54, RNR2, DIN7, and PLM2 strains or systems, respectively. Every yeast strain was then cultured in SD/-Ura medium and incubated overnight. The overnight culture was transferred into fresh SD/-Ura medium and incubated at 30°C with shaking until mid-log phase, subsequently diluted to optical density measured at 600 nm (OD600) = 0.4 in F1-Ura medium. Basically, the composition of F1-Ura medium used in this study is the same as described by Afanassiev et al. 7 It was however alternatively prepared in a total final volume of 1 L as follows: 3.13 g (NH4)2SO4, 2 g KH2PO4, 0.27 g MgSO4, 0.09 g CaCl2·2H2O, 0.1 g NaCl, 0.01 mg H3BO3, 0.01 mg CuSO4·5H2O, 0.01 mg KI, 0.05 mg FeCl3·6H2O, 0.07 mg ZnSO4·7H2O, 0.1 g leucine, 0.1 g histidine, 0.1 g lysine, 31 mg myo-inositol, 14 mg thiamine-HCl, 4 mg pyridoxine-HCl, 4 mg calcium pantothenate, 0.3 mg biotin, and 20 g glucose. Aliquots of 1 mL of yeast cultures (OD600 = 0.4) were applied into 1.5 mL tubes.
Preparation of test compounds in aqueous solution
From the published reports 7,8 a set of test substances was selected and prepared in stock solutions as follows: 8 mmol/L hydrogen peroxide (272.08 µg/mL), 80 µmol/L NNNM (8.2464 µg/mL), 80 µmol/L netropsin dihydrochloride (40.2712 µg/mL), 80 µmol/L menadione (13.7744 µg/mL), 80 µmol/L actinomycin D (100.4336 µg/mL), 8 µmol/L 4-NQO (1.5212 µg/mL), 8 µmol/L bleomycin sulfate (11.3244 µg/mL), 8 µmol/L camptothecin (2.7868 µg/mL), and 0.1 mmol/L MMS (11.01 µg/mL) as the positive genotoxicity control were diluted in F1-Ura medium plus 4% DMSO (v/v). Aliquots of yeast cultures and stock solutions of substances were brought to the workstation of the Tecan robotics (Genesis RSP-150 Liquid Handling System; Tecan, Austria) for genotoxicity assay.
Assay execution by Tecan Robotics
The assay was performed in 384-well microplate (24 × 16 well format; Greiner Bio-one, Germany) with black walls and transparent flat bottoms. First, 8-tip robotic arm controlled by Tecan Gemini software aspirates 8 test substances from prepared stock solutions and dispenses 70 µL of that into alternate 4 wells (columnwise) in triplicate. Next, it fills the rest of wells with 35 µL of F1-Ura medium. The serial 1:2 dilution was implemented in that the 8-tip robotic arm took 35 µL from wells filled with stock solutions putting it into adjacent wells filled with F1-Ura medium only, mixed 3 times, then transferring 35 µL into the next wells (columnwise). After making 3 serial dilutions, the 35 µL of the last diluted solutions were discarded in the waste station. The nontreated (NT) wells containing only 35 µL of the medium were designated as the control. Subsequently, every 35 µL of prepared yeast cultures was added to both treated and NT wells to reach a total final volume of 70 µL for each well and an initial OD600 of about 0.2. This process was implemented in the same way for the positive genotoxicity control (MMS) and negative control (untransformed yeast cells). In addition, for the process controls, other wells were filled with 70 µL of medium or 70 µL of test compounds to verify microbial contamination or chemical absorbance/fluorescence, respectively. Each well was pipetted in triplicate for determining mean values, standard deviation (SD), and the assay was repeated at least 3 times for reproducibility. Finally, whole microplates were sealed with breathable membrane (Diversified Biotech, USA) and transferred into the microplate reader (Safire 2 Reader; Tecan) by robotic gripper. The plate was incubated in this reader for 24 hours at 30°C with agitation. The measurements were carried out at the time points 8, 16, and 24 hours during incubation.
Data acquisition and analysis
The genotoxic potential of test compounds was evaluated by fluorescence signals generated by GFP and yeast culture absorbance. The fluorescence intensity was measured at 485 nm (excitation wavelength) and 535 nm (emission wavelength); the absorbance (OD) at 600 nm (OD600) by microplate reader controlled by XFLUOR4 SAFIRE II software (Xfluor Excel macros, Version: V 4.62n for Safire 2 Reader; Tecan). Fluorescence data were divided by absorbance data giving specific fluorescence. For normalization, the specific fluorescence of treated wells was then divided by that of NT wells (controls equal to 1) giving GFP fold induction.
The threshold definitions as described in a previous report 8 were applied for conclusions regarding negative or positive genotoxicity data. According to that the expression of GFP was induced upon treatment of test compounds as both the GFP fold induction and the OD600 must be greater than 1.3 and 0.3, respectively, giving positive genotoxicity data. Conversely, negative genotoxicity data are concluded as GFP fold induction of less than 1.3 threshold. To avoid ambiguous genotoxicity results, a supplementary condition was used in this study: The raw fluorescence intensities of treated wells were taken into account in calculation of specific fluorescence and normalized data only if they were higher than those of NT wells. When the raw intensities of treated wells are equal or less than those of NT wells, after being divided by low denominators, that is, low OD600 values but higher than 0.3 (OD600 of treated wells) and normalized, they could produce false-positive genotoxicity results. Toward those means, a set of Excel macros (written in Visual Basic Editor) was developed to automatically rearrange raw data from plate format to time points, check additional conditions, and remove the outliers from raw data before further calculation and normalization. These macros were also used to determine mean and SD values of probes from multiple wells. The genotoxicity threshold and calculated GFP fold induction together with SD values of measurements were represented by bar graphs.
In fact, the individual steps of pipetting liquid, transferring plates, measuring fluorescence as controlled by manufacturer software work separately and independently. To alleviate these tasks and save time, some of the R-packages (R scripts written in the R programming language) developed in our previous study 23 were modified and applied to carry out all these steps automatically. Except for preparation of stock solutions and sealing plates with membrane, a set of modified R scripts controls both the robot and the reader by translating the instructions into machine commands, executing liquid handling and measurements, collecting the data, and processing it without user interaction. The data were saved either in file.RData or exported to Microsoft Excel for conventional processing and analysis by the Excel macros.
Data comparison
The systems bearing RAD54- and RNR2-GFP reporter gene constructs 7 and assays employing RNR2-GFP reporter construct alone 8 have been shown to be able to identify a wide range of substances including genotoxins, antibiotics, and so on with higher sensitivity and specificity than other conventional tests such as Ames, SOS, S9, mouse lymphoma assay, and so on. 7,8 The results obtained in this study are compared to those from previous work with the above RAD54- and RNR2-GFP reporter gene constructs.
OxoPlate Assay
In a first effort, the additional OxoPlate assay (PreSens Precision Sensing GmbH, Regensburg, Germany) was designed to determine the effect of test compounds on mitochondrial respiration, through oxygen consumption against cell growth in response to DNA damage. This assay is however not intended to provide analytical information or functional mechanisms. The same experimental design and setup of the genotoxicity assay was applied to the OxoPlate assay, but this assay was performed on 96-well OxoPlates with a round bottom of the manufacturer and each well containing a total volume of 150 µL diluted with SD/-Ura medium. The oxygen consumption (%) together with cell growth (OD600) were monitored every 30 minutes over 24 hours. The measurement setup and data calculation were carried out according to the instructions of the manufacturer as also described in our previous study. 25 This assay was also executed by computer-controlled automated laboratory system, and the data were analyzed by Excel macros.
Results
The role and functional mechanism of 4 DNA damage-inducible genes, DIN7, PLM2, RNR2, and RAD54 whose promoter regions regulate the transcription and expression of the downstream GFP gene are summarized in Table 2. In the present study, the promoter regions of the DIN7 and PLM2 genes were used as biosensing elements to create 2 new reporter systems, the DIN7 and PLM2 yeast strains. These 2 new systems together with 2 reference reporter systems, the RNR2 and RAD54 yeast strains, were used to identify and investigate the genotoxic potential of chemical substances.
Functional Mechanisms of DNA Damage-Inducible Genes Whose Promoters Were Used in this Study.

Abbreviation: dNTP, deoxynucleotide triphosphate.
aSource: http://www.yeastgenome.org
Fluorescence Induction of DIN7 Reporter System
The signal intensity of GFP produced by yeast strains carrying RAD54- and RNR2-GFP constructs has been reported to be directly proportional to increasing concentrations of analytes. 7,8 Similar findings were here also observed when DIN7 strain bearing DIN7-GFP construct was treated with serial dilutions of 8 test compounds (Figure 1). The measured fluorescence intensity is highly dependent on the chemical properties of substances, on their concentrations, and on incubation times.
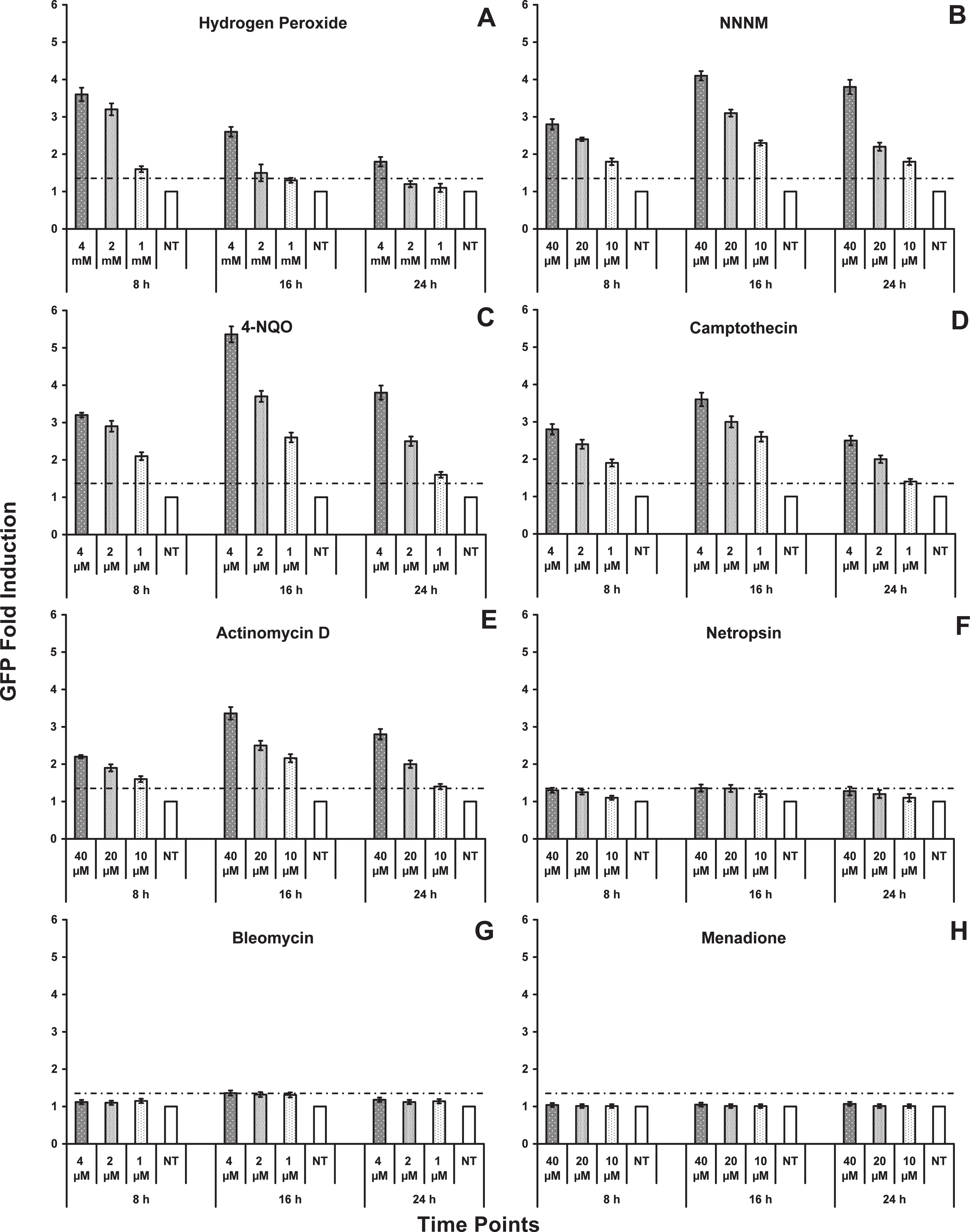
Fluorescence induction in yeast cells transformed with DIN7-GFP constructs in response to DNA damage. Yeast-based biosensor was nontreated (NT, control) or exposed to serial dilution concentrations of hydrogen peroxide (A), N-nitroso-N-methylurea (NNNM; B), 4-NQO, 4-nitroquinoline-N-oxide (4-NQO; C), camptothecin (D), actinomycin D (E), netropsin (F), bleomycin sulfate (G), and menadione (H). The fluorescence intensity of measurements at 8, 16, and 24 hours was compared by caculation of GFP fold induction. The horizontal dashed lines are 1.3-GFP fold induction or genotoxicity threshold. Positive genotoxicity (methyl methanesulfonate [MMS]), negative (untransformed yeast cells), and process controls are not presented here. GFP indicates green fluorescent protein.
Hydrogen peroxide (ROS), NNNM (alkylating agent), 4-NQO (mutagen, carcinogen), camptothecin (cytotoxic quinoline alkaloid), and actinomycin D (antineoplastic antibiotic) all exhibit strong genotoxic effects, since they induce the activity of DIN7 promoter resulting in expression of GFP and thus a more intense fluorescence signal; these substances were assayed at increasing concentrations. Netropsin, bleomycin (antibiotics), and menadione (vitamin K3) showed no genotoxic effect in this system as the fluorescence induction is below the genotoxicity threshold at any treated concentration and time of measurement (Figure 1). In most cases of treatment with DNA-damaging agents, GFP induction tends to increase with incubation time from 8 to 16 hours, reaching a maximum at 16 hours and decreasing after 24 hours incubation (Figure 1). In other words, fluorescence intensity measured at 16 hours is apparently greater than when measured at 8 and 24 hours of incubation. The exception is hydrogen peroxide, where the maximal GFP induction was reached at 8 hours of incubation and then gradually decreased over time (Figure 1). These responses of fluorescence induction were also verified with other reporter systems.
Validation and Evaluation of Fluorescence Signal in Different Reporter Systems
Four reporter systems were exposed to varying serial dilutions of the tested compounds (Table 3). The fluorescence induction of 3 reporter systems, RAD54, RNR2, and PLM2, appears to be similar to that of the DIN7 reporter system in response to potential DNA-damaging agents. Higher concentrations of DNA-damaging agents led to stronger fluorescence signals, that is, higher GFP signals, thus indicating higher level of DNA damage. Also, the highest values of GFP fold induction or the most intense fluorescence signals were observed at 8 hours for hydrogen peroxide and at 16 hours for the other agents except menadione (Table 3).
Validation and Evaluation of Fluorescence Signals in Different Yeast Strains in Response to Serial Dilution Concentrations of Test Compounds.a
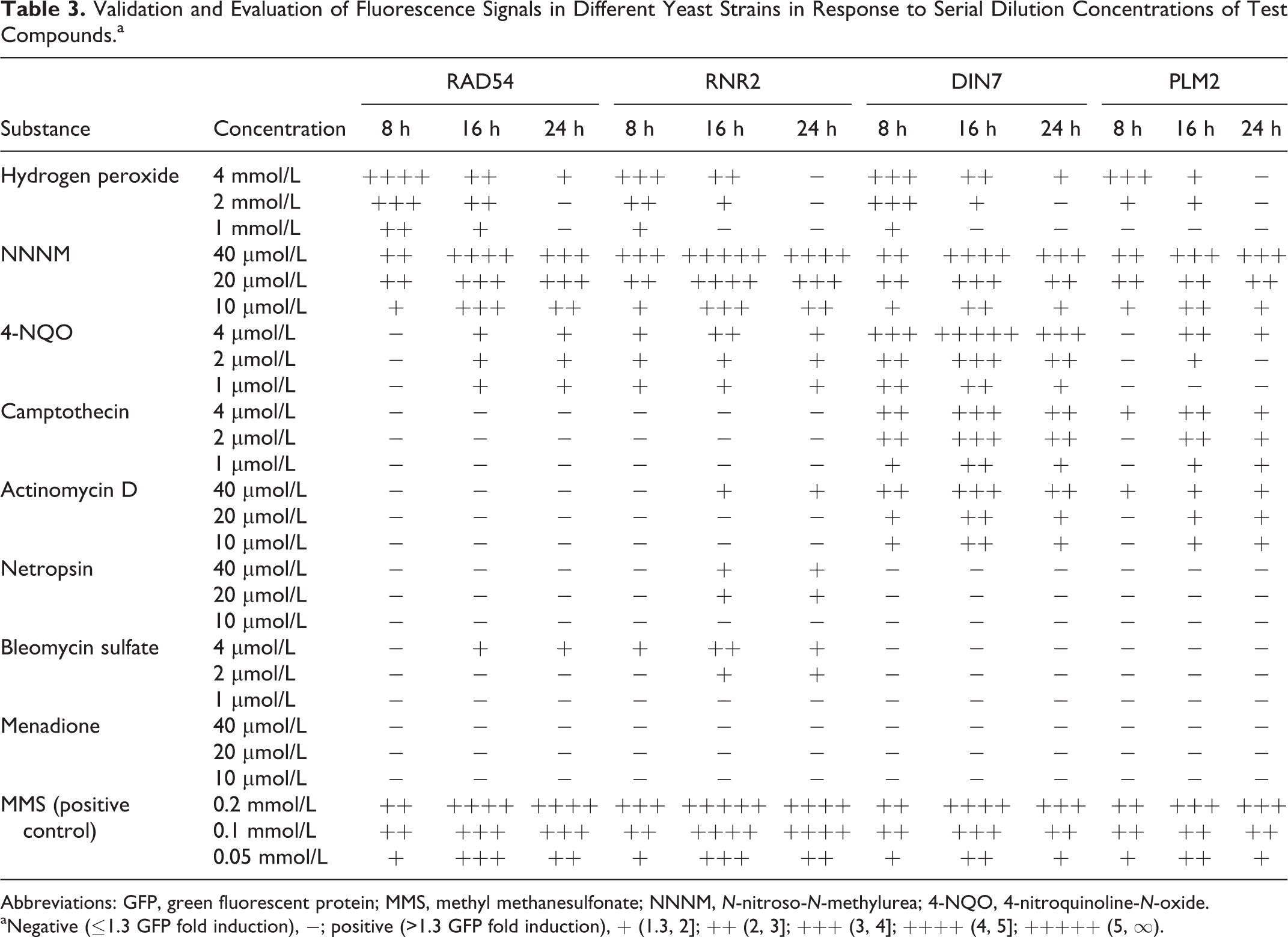
Abbreviations: GFP, green fluorescent protein; MMS, methyl methanesulfonate; NNNM, N-nitroso-N-methylurea; 4-NQO, 4-nitroquinoline-N-oxide.
aNegative (≤1.3 GFP fold induction), −; positive (>1.3 GFP fold induction), + (1.3, 2]; ++ (2, 3]; +++ (3, 4]; ++++ (4, 5]; +++++ (5, ∞).
In the case of hydrogen peroxide, the RAD54 system shows a slightly higher sensitivity expressed as GFP fold induction than the RNR2, DIN7, and PLM2 systems. The RNR2 system exhibits a slightly higher sensitivity in detecting NNNM as compared with the other 3 systems, whereas DIN7 shows a considerably higher sensitivity than other systems for identifying 4-NQO, camptothecin, and actinomycin D. In respect to specificity, the RAD54 system seems to be nonspecific for camptothecin, actinomycin D, and netropsin, while the RNR2 system showed no specificity for camptothecin. Nevertheless, both DIN7 and PLM2 systems were highly specific to camptothecin and somewhat to actinomycin D but nonspecific in respect to netropsin and bleomycin (Table 3). Also, none of the systems were specific to menadione (negative, Table 3). Regarding the measured signal, the test results summarized in Table 3 indicate that RAD54 and RNR2 systems produce a higher fluorescence signal than DIN7 and PLM2 systems in response to hydrogen peroxide, NNNM, bleomycin sulfate, and MMS (positive control), while the fluorescence signal produced by DIN7 and PLM2 systems was higher than that produced by RAD54 and RNR2 systems in response to 4-NQO, camptothecin, and actinomycin D. In comparison with the 2 reference systems, RAD54 and RNR2, the 2 newly developed reporter systems, DIN7 and PLM2, were slightly less specific and sensitive for peroxide, alkylating agents, or glycopeptide antibiotic but considerably more specific and sensitive for quinoline, quinoline alkaloid, or polypeptide antibiotic.
In reference to fluorescence induction upon treatment with hydrogen peroxide, similar to the effect observed in the DIN7 system, the most intense signal was also observed at 8 hours of incubation in the other 3 systems. In many cases, lower or no fluorescence induction was observed at 16 or 24 hours even upon treatment with high concentrations, for example, 2–4 mmol/L (Figure 1 and Table 3). Probably, treatment with this agent shows a short-term effect, rather than a long-term effect, on cellular activities, including oxygen consumption or mitochondrial activity, of these systems. Our previous study reported that MMS strongly inhibits mitochondrial respiration as a result of mtDNA damage in yeast. 25 Current results from the DIN7 system (Figure 2) indicate the more oxygen being consumed, the higher the cell proliferation rate. Hydrogen peroxide strongly inhibits mitochondrial activity leading to reduced oxygen consumption at the beginning of cultivation (0-6 hours); sometimes the oxygen saturation was in excess of 100% by treatment with the highest concentration (4 mmol/L); afterward mitochondrial function was recovered and cells continued to consume oxygen until all dissolved oxygen in the culture medium was exhausted (Figure 2, panel A). Conversely, NNNM, 4-NQO, camptothecin, and actinomycin D treatments show a long-term effect; they delayed oxygen consumption even longer than hydrogen peroxide. Afterward, depending on the substance concentration, mitochondrial function was either resumed until oxygen in the medium became exhausted or remained inhibited for up to 24 hours of cultivation, that is, no more oxygen was consumed (Figure 2, panels B-E). However, netropsin and bleomycin did not reduce or inhibit oxygen consumption but inhibited cell growth (Figure 2, panels F and G). Menadione, a nutritional and dietary supplement, neither inhibited oxygen consumption nor repressed cell proliferation at all (Figure 2, panel H). The modulation of oxygen consumption of the DIN7 strain and other strains with or without reporter plasmid (wild type) in response to these compounds was about the same in all cases (data not shown). The oxygen consumption of the wild-type strain differed significantly from that of mutant cells defective in genes that control DNA damage and repair checkpoints and mitochondrial biogenesis, like mutants Δrad9 and Δhap4, upon treatment with MMS. 25
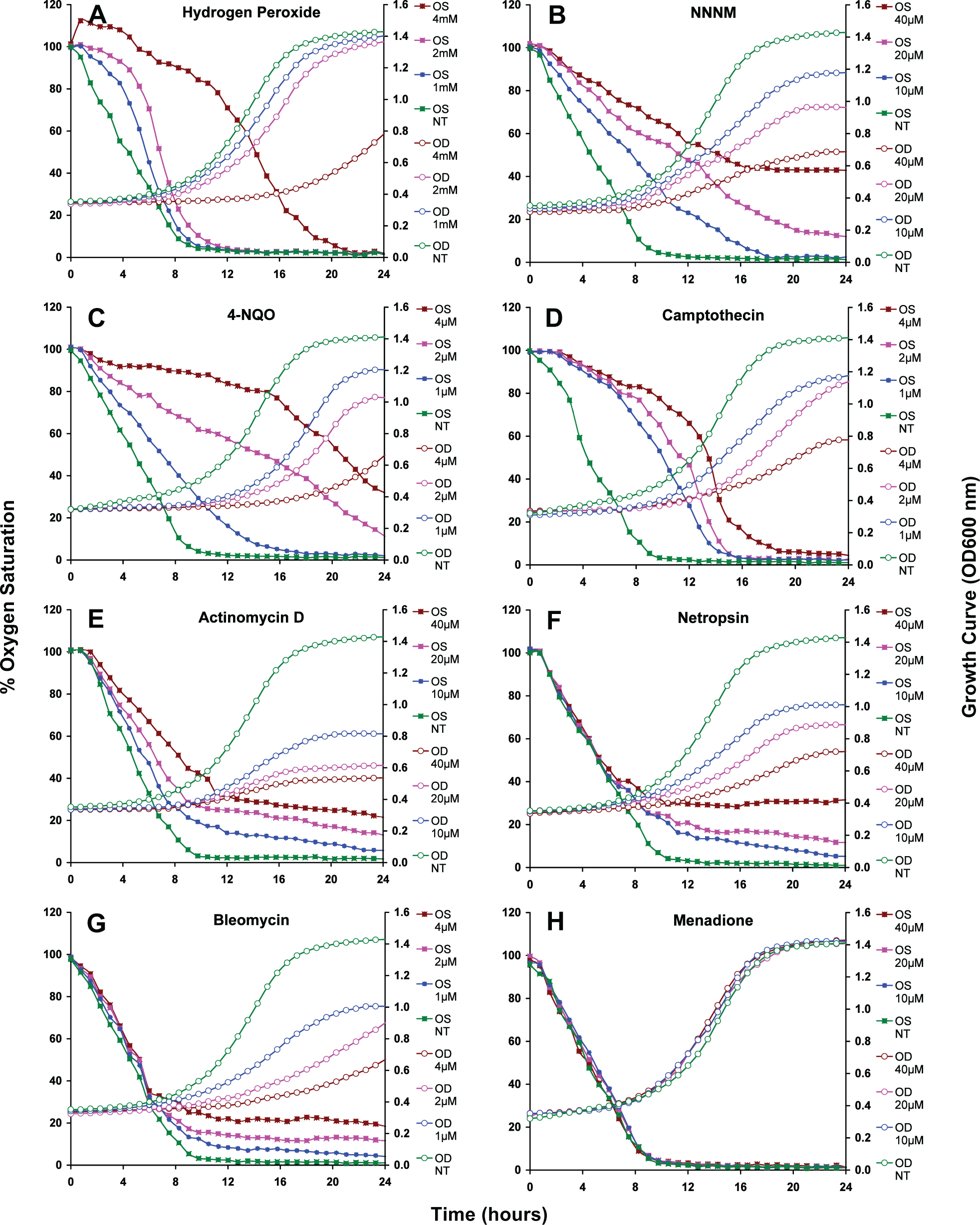
Effect of test compounds on oxygen consumption and cell growth of yeast cells incorporating DIN7-GFP constructs. Oxygen consumption expressed as a percentage of oxygen saturation against cell growth expressed as optical density measured at 600 nm (OD600)values was monitored during 24 hours of cultivation when treated with serial dilutions of hydrogen peroxide (A), N-nitroso-N-methylurea (NNNM; B), 4-NQO, 4-nitroquinoline-N-oxide (4-NQO; C), camptothecin (D), actinomycin D (E), netropsin (F), bleomycin sulfate (G), and menadione (H). The standard deviation values of these measurements were all less than 5% of the calculated values and are thus not presented here. OS indicates oxygen saturation; OD, optical density measured at 600 nm; NT, nontreated control.
Comparison of Test Results
The aim of this study was to develop new reporter systems for alternative testing of DNA-damaging agents. We selected compounds that had not been tested before and others that had been tested at different concentrations. The concentrations were chosen to either overlap or lie inside the ranges of the previously published data sets but must be within the linear range for detection of analytes or for GFP signals produced. In other words, the GFP intensity measured should be directly proportional to increasing concentrations of analytes investigated in this study.
In respect of sensitivity expressed as positive signal (GFP fold induction), the maximum GFP signal was reached when treated with the highest concentrations of analytes, while the minimum signal but greater than genotoxicity threshold (>1.3 GFP fold induction) was obtained when treated with the lowest concentrations (Figure 1 and Table 3). When treated with concentrations outside that range, the positive signal was still induced but not directly proportional to them (data not shown). The GFP signal started to decrease when exposed to levels above the highest concentrations or was less than the genotoxicity threshold when exposed to levels below the lowest concentrations. For example, our previous report showed that the GFP signal started to decrease gradually when exposed to concentrations greater than 0.2 mmol/L MMS or 4 µmol/L camptothecin. 7
The test results are compared with those from previous reports that showed the RAD54 and RNR2 reporter systems to have higher specificity and sensitivity than other conventional tests. 7,8 Regardless to sensitivity, all systems showed positive results (+) in response to a wide range of concentrations of hydrogen peroxide, NNNM, and 4-NQO (Table 4). There are, however, conflicting data regarding certain results obtained here and in the previously established RNR2 and RAD54 systems (–* and +*, or −**), in particular regarding camptothecin, actinomycin D, netropsin, and bleomycin. The DIN7 and PLM2 systems were able to identify these substances though (Table 4). The variation in test results among different test systems will be discussed later.
Comparison of Test Results Obtained From This Study With the Data From Published Reports.a
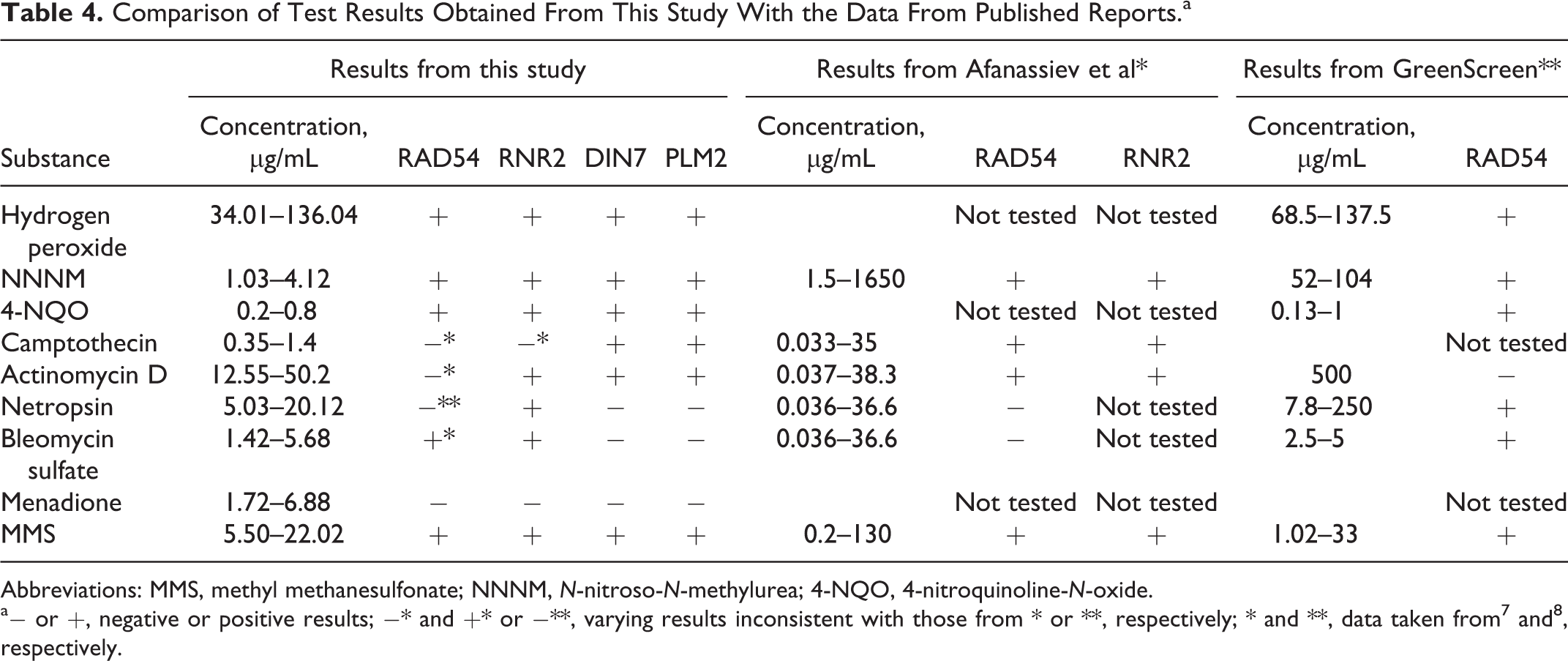
Abbreviations: MMS, methyl methanesulfonate; NNNM, N-nitroso-N-methylurea; 4-NQO, 4-nitroquinoline-N-oxide.
Discussion
The assessment and measurement of harmful substances and pollutants are of crucial importance in food safety management and environmental monitoring in Vietnam. Additionally, the 3R concept (reduction, replacement, and refinement) in toxicology and ecotoxicology promotes the application of genetically modified microorganisms as bioanalytical tools due to the simple cultivation and lack of ethical problems. Thus, 2 newly developed reporter systems, DIN7 and PLM2, and 2 reference systems, RAD54 and RNR2, were used to evaluate the genotoxic potential of 8 chemical compounds by genotoxicity assay.
The performance of the genotoxicity assays here requires only one 384-well microplate (70 µL total volume per well) for testing 8 substances in contrast to the four 96-well microplates (150 µL total volume per well) needed for the same experimental setup and design otherwise. The 384-well format reduces the total amount of required chemicals by 53% when compared to the 96-well format used in previous bioassays for 1 test sample. 7,8
The strongest positive results for detection of hydrogen peroxide (H2O2) were obtained at 8 hours, for the other DNA-damaging agents at 16 hours of incubation. This difference seems to be due to the fact that hydrogen peroxide spontaneously and exothermically decomposes into water and oxygen or is converted by intracellular catalase into the same products so that the percentage of oxygen saturation was slightly decreased or in excess of 100% during lag phase and early log phase (up to 6-8 hours of cultivation) upon treatment with 1 to 2 or 4 mmol/L H2O2, respectively (Figure 2, panel A). Thus, H2O2 caused a strong oxidative damage to DNA, including mtDNA, in the lag and early log-phase that led to stronger positive results observed at 8 hours. Due to the self-decomposition of H2O2 and depending on its concentration, the test results became either weak positive or negative at 16 and 24 hours (Figure 1 and Table 3). Furthermore, treatment with lower concentrations, for example, 0.5 mmol/L H2O2, produced negative signals (<1.3 GFP fold induction) and did not affect oxygen consumption nor cell proliferation at all (data not shown), suggesting that low levels of peroxide cause no oxidative damage but induce the cellular defense system. Even treatment with 1 mmol/L H2O2 only led to oxidative damage during lag phase (0-6 hours), then mitochondrial respiration was recovered and cells continued growing; this could be due to the cellular protective system against oxidative stress (Figure 2, panel A). Also, many studies report that exposure to low H2O2 concentrations (0.5 or <5 mmol/L) enhances tolerance to oxidative stress, induces catalase and glutathione reductase, the 2 key enzymes involved in the cellular antioxidant system, and increases protein production in yeast cells. 26,27 In contrast, other DNA-damaging agents do not self-decompose like H2O2, thus causing long-term damage during incubation. The strongest positive results were therefore monitored at 16 hours, when cells exponentially proliferated in mid-log and log phases (14-18 hours), rather than at 24 hours, when cells slowed down their growth rate in stationary phase (20-24 hours) as a result of nutrition depletion (Figure 2). These findings indicate why a higher intensity of fluorescence signal was observed at 8 hours upon treatment with H2O2 for this study and also agree with the notion that the GFP signal reaches a maximum when measured after 16 hours of incubation upon treatment with other DNA-damaging agents. 7,8
RAD54 promoter drives slightly elevated GFP expression as compared with RNR2, DIN7, and PLM2 promoters upon treatment with H2O2. Being a ROS, H2O2 causes oxidative damage to DNA and induces both single-strand breaks and DSB. 28 Moreover, RAD54, a member of the RAD52 group of DNA repair genes, is involved in the repair of DSBs. 11 RAD54 is induced above a constitutive level by UV, ionizing radiation, 12 or DNA-damaging agents, as for example, MMS. 7 Also, the RNR2 promoter seems to trigger a slightly increased GFP expression as compared with RAD54, DIN7, and PLM2 promoters upon treatment with NNNM. The latter, being an alkylating agent, causes base alterations by transferring its methyl group to nucleobases leading to GC:AT transition mutations. 29 RNR2 is known to play a crucial role in DNA replication 30 and DNA damage repair including DNA lesions. 13 On the other hand, RNR2 induction is related to cell growth and survival, which is clearly increased when exposed to genotoxins, 14 including such alkylating agents as MMS. 7
Interestingly, the GFP expression level driven by the DIN7 promoter is obviously higher than that driven by other promoters upon treatment with 4-NQO, camptothecin, and actinomycin D (Table 3). Since 4-NQO is a potent mutagen and carcinogen mimicking UV-mediated DNA damage, it induces single-strand breaks and base substitutions, mainly G to A transitions, 31 and these DNA lesions should be usually corrected by nucleotide excision repair. Besides that, it is documented that 4-NQO is metabolized into ROS as anion radical metabolites and DNA adducts 32 or causes ROS accumulation in yeast cells. 33 Camptothecin is known to inhibit topoisomerase I that plays a vital role in relieving tension on DNA strands generated during replication. Inhibition of this enzyme by camptothecin may stall DNA replication forks resulting in DSBs. This prevents DNA religation and therefore causes DNA damage leading to cell cycle arrest and programmed cell death, called apoptosis, in yeast cells. 34,35 Actinomycin D is known to inhibit DNA and RNA synthesis by binding DNA duplexes. 36 Although there is still no strong evidence that actinomycin D induces apoptosis in yeast cells, many studies demonstrated that it induces apoptosis in some mammalian cell lines as a result of ROS production. 37,38 On the other hand, oxygen radicals and UV irradiation were shown to induce apoptosis in yeast. 39,40 Our previous research also showed that the high ROS accumulation increases damage to mtDNA leading to inhibition of mitochondrial activity and oxygen consumption upon MMS treatment. 25 Moreover, mitochondrial respiration was strongly inhibited when exposed to 4-NQO, camptothecin, and actinomycin D (Figure 2, panels C-E). This may be due to mtDNA damage as a result of ROS accumulation. Since mtDNA might be more prone to oxidative damage and suffers 3- to 10-fold more damage than nuclear DNA in numerous cell types from yeast, mouse, rats, and humans, it is particularly susceptible to ROS. Enhancement of mtDNA repair should confer protection from cell death, whereas loss of mtDNA repair should promote cell death. 41 Mitochondria are equally important in regulating cell respiration, mtDNA maintenance, ATP biosynthesis, and apoptosis in yeast cells 25,42 as in mammalian cells.
In S cerevisiae, DIN7 is a DNA damage-inducible (DIN) gene involved in the same DNA repair and replication as RAD54 and RNR2 (Table 3), being induced by exposure to oxidative and genotoxic agents. In other words, the product of the DIN7 gene, Din7p, exhibits DNA nuclease activity playing an important role in DNA damage repair. Particularly, it is located inside mitochondria and the level of the nuclease Din7p modulates the stability of the mtDNA. 43 Taken together, the higher GFP expression level induced by the DIN7 promoter can be explained in that, in response to DNA damage, yeast cells struggle with DNA damage caused by 4-NQO, camptothecin, and actinomycin D. They attempt to repair DNA injury and trigger a transcriptional response to detoxify the metabolites of these compounds or induce apoptosis caused by DNA damage as a result of high intracellular ROS accumulation through cellular metabolism.
Netropsin or bleomycin were found to activate only 1 promoter, RNR2, or 2 promoters, RNR2 and RAD54, respectively, and induce weak GFP expression, as the GFP fold induction was below the value of 2.0 in most cases (Table 3). One explanation could be that netropsin binds to the minor groove of double-stranded DNA but it does not induce specific DNA strand cleavage like bis-netropsin analogs that inhibit the binding of topo I to DNA and could alter the DNA structure, 44 while bleomycin induces DNA DSBs and gross chromosomal rearrangements. 45 In addition, RAD and RNR group genes were linked to repair DSBs and mediated cell cycle and replication arrest (Table 2) and induced in the presence of bleomycin, hydroxyurea, MMS, and so on, 7,8 suggesting that the activity of RAD54 and RNR2 in our systems could also be induced by agents that interact with DNA helix-forming DNA adducts or induce DSBs like netropsin and bleomycin. In fact, both of these compounds showed more effect on the inhibition of cell growth than impairment of mitochondrial respiration (Figure 2, panels F and G), that is, they perform their antibiotic effect rather than DNA damage effect. Menadione was not found to activate any promoter to drive GFP expression, since it, as a nutritional additive, showed no damaging effect on yeast DNA, 46 but a cytotoxic effect on yeast cells treated with 0.5 mmol/L menadione 47 was observed, which is more than 10 times greater than the highest concentration of menadione (40 µmol/L) used in this study (Table 3).
In comparison with the published data detected as nongenotoxic or negative (–) and genotoxic or positive results (+), there is agreement that a wide range of concentrations of H2O2, NNNM, and 4-NQO are detected as positive by the 2 similar systems, RAD54 and RNR2, or the 2 new ones, DIN7 and PLM2 (Table 4). There are, however, a number of conflicting data (–* and +*, or −**), which were deduced as negative in this study, but positive in one of other reports and vice versa. This discrepancy could be due to different experimental protocols or setups 7,8 and data analysis and calculation that might also produce false-positive results as mentioned earlier. For example, the negative results of identifying camptothecin and netropsin by RAD54 or RNR2 system in this study could be explained by the fact that the low test concentrations of these compounds were not able to induce the RAD54/RNR2 promoter-driven GFP expression. The positive result of detecting bleomycin is consistent with the findings from GreenScreen assay (Table 4). Bleomycin has been described recently to cause DSBs. 45 RAD genes, including RAD54, are involved in DSBs repair. Thus, when cells were exposed to bleomycin the RAD54 promoter-driven expression of GFP was induced. In the concentrations used, only a weak induction of DIN7 and PLM2 promoter-driven GFP expression below the defined cutoff was observed. These differences may be explained by the different roles of the respective proteins in repair as discussed earlier.
Our yeast-reporter systems, in association with a computer-controlled automated laboratory system, enable cost-effective, rapid, and high-throughput screening of numerous compounds, requiring only small amounts of chemicals. This yeast-based biosensor system can supplement existing analytical methods and is geared toward applications in environmental monitoring, drug discovery and screening, or drug development, where genotoxicity tests are needed to determine the potential genotoxic and mutagenic hazards and risks of drug candidates. It should be noted that detection of genotoxic compounds is only possible when a certain threshold sufficient for triggering damage in yeast strains is reached. Very low levels of genotoxic compounds will not be detected. Thus, the systems are particularly suitable for detecting immediate toxic damage, while at present low levels of contamination that may lead to DNA damage after extended exposure are not accessible with these systems.
Indeed, this genotoxicity assay lends itself to test other compounds with similar properties as well as new substances or agents by replacement of inducible promoter regions of other yeast genes such as FBP1, POL30, and THI4 (SGD). Moreover, these systems could be developed for determining numerous pollutants, for example, polycyclic aromatic hydrocarbons (PAHs), heavy metals, and so on by selection of a specific promoter of regulatory genes induced in response to a specific agent, for example, Pu promoter of xylR gene, Pnah promoter of nahR gene, ars promoter of arsR gene, mer promoter of merR gene, which could be used as biosensing components in the development of whole-cell biosensors for detecting toluene, naphthalene, arsenite, and mercury, respectively. 48 Actually there are a large number of toxic substances that have not been able to be detected by the RAD54 or RNR2 systems. 7,8 Aflatoxin B1 did not induce GFP expression in our systems (unpublished data). Probably, such substances require bioactivation by cytochrome P450s (CYP450) and NADH-P450 oxidoreductase (CPR) to produce genotoxic or mutagenic metabolites. Many studies indicate that besides serving in detoxification, human CYP450 enzymes (CYP 3A4, CYP 2B6, CYP 2C9, CYP 2D6, CYP 2E1, etc) can also biotransform otherwise harmless chemicals or procarcinogens, for example, PAHs, aromatic amines, aflatoxins, into mutagenic and carcinogenic metabolites or carcinogens. 49,50 Human CYP450 and CPR are well documented to successfully be expressed in various yeast species and capable of metabolizing many compounds, drugs, or xenobiotics and only coexpression of both CYP450 and CPR show this effect. 51,52 Based on these studies, our systems should be further developed by incorporating both CYP450 and CPR genes, where CYP450 enzyme would be responsible for converting a specific substrate into the according carcinogenic metabolites that would enable the promoter to drive GFP expression. In consequence, the novel yeast-based biosensor would be able to detect both carcinogens and procarcinogens.
In conclusion, our newly developed systems, DIN7 and PLM2, exhibit higher sensitivity to 4-NQO and actinomycin D than the existing systems, RNR2 and RAD54. Particularly, both DIN7 and PLM2 systems can detect camptothecin within investigated concentrations (1–4 µmol/L), but the RNR2 and RAD54 systems cannot. The 2 new systems, DIN7 and PLM2, together with the 2 reference systems, RAD54 and RNR2, can be used as alternative yeast-based biosensors for identification of a wide range of genotoxins or carcinogens with 53% lower compound consumption than existing eukaryotic genotoxicity bioassays, but they are not able to detect procarcinogens. Thus, further work is aimed to establish novel yeast-based systems that additionally carry CYP450 and CPR genes for determining both carcinogens and procarcinogens.
Footnotes
Acknowledgment
We thank Theodor C. H. Cole for language editing and valuable comments regarding the manuscript.
Author Contributions
V. N. Bui contributed to conception or design, acquisition, drafted the article, critically revised the article, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. T. T. H. Nguyen, T. H. T. Nguyen, T. L. Pham, and T. Y. Hoang contributed to acquisition, critically revised the article, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy. Y. Bettarel, V. T. T. Nguyen, N. M. Nghiem critically revised the article, gave final approval, and Agree to be accountable for all aspects of work ensuring integrity and accuracy. S. Wölfl contributed to conception or design, acquisition, analysis, or interpretation, drafted the article, critically revised the article, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under the grant number 106.16-2012.09.