
Abstract
Tenofovir disoproxil fumarate (TDF) is a prodrug of tenofovir that exhibits activity against HIV and hepatitis B. The goals of this study were to evaluate the molecular mechanism of TDF-induced toxicity in mice after 13 weeks of daily oral administration (50-1000 mg/kg) by correlating transcriptional changes with plasma drug levels and traditional toxicology end points. Plasma levels and systemic exposure of tenofovir increased less than dose proportionally and were similar on days 1 and 91. No overt toxicity was observed following the completion of TDF administration. The kidneys of TDF-treated mice were histopathologically normal. This result is consistent with the genomic microarray results, which showed no significant differences in kidney transcriptional levels between TDF-treated animals and controls. In liver, after 4 and 13 weeks, cytomegaly was observed in mice treated with 1000 mg/kg of TDF, but mice recovered from this effect following cessation of administration. Analysis of liver transcripts on day 91 reported elevated levels of Cdkn1a in TDF-treated animals compared with controls, which may have contributed to the inhibition of liver cell cycle progression.
Introduction
Tenofovir disoproxil fumarate (TDF, Figure 1), a prodrug of tenofovir, has been widely used for long-term treatment of HIV and chronic hepatitis B (CHB) infections in adult patients. 1,2 Tenofovir is an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate and acts by inhibiting viral reverse transcriptase. 1 Its favorable pharmacokinetic profile and intracellular drug level in lymphoid tissue allow for a once-daily oral dose regimen. 3 -5 Compared with other HIV treatments, TDF’s oral dosing route, more compliant dosing schedule, and reported better-tolerated safety profile have propelled it to become a first-line treatment for HIV. 6 Tenofovir disoproxil fumarate is also frequently used in combination with other antiretroviral drugs. Tenofovir’s primary route of elimination is through the urine, where it is excreted largely via glomerular filtration and proximal tubular secretion. 7,8
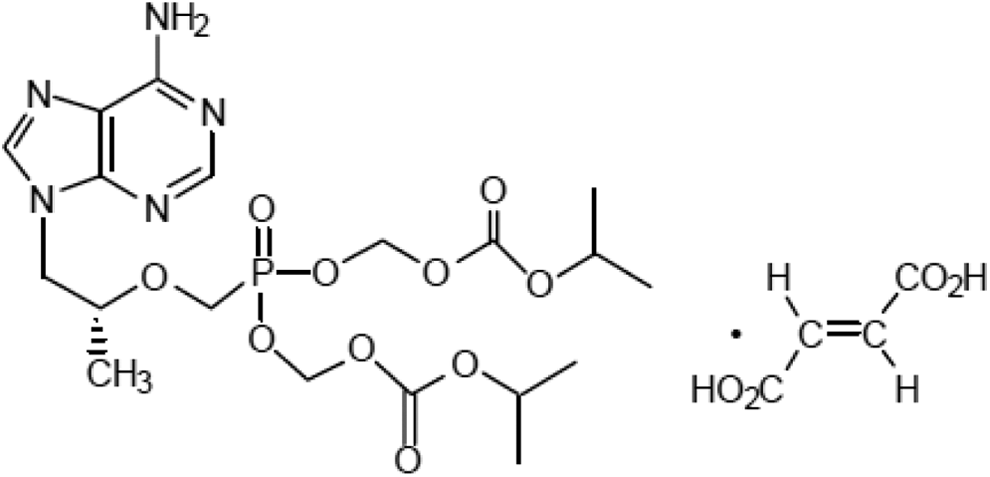
Structure of tenofovir disoproxil fumarate (TDF).
Tenofovir-containing regimens have been associated with infrequent renal adverse events during clinical trials and in postmarketing experience. 9 Nephrotoxicity has been primarily characterized as proximal tubulopathy with rare occurrence of Fanconi syndrome. 1,10,11 The exact mechanism of tenofovir-induced nephrotoxicity is not clear but is thought to be related to the accumulation of tenofovir in the proximal tubules. Studies have shown that tenofovir is mainly taken up from the blood into proximal tubule cells by human organic anion transporter 1 (hOAT1) at the basolateral membrane. 12,13 On the other hand, the apical efflux system responsible for transporting tenofovir out into the tubular lumen for excretion is believed to be the multidrug resistance-related protein 4 (MRP-4). 14 It was also shown that tenofovir significantly accumulated in the kidney of the Mrp4 knockout (KO) mice. 15 In regard to hOAT1 involvement, Chinese hamster ovary cells expressing high levels of hOAT1 exhibit greater levels of cytotoxicity following tenofovir exposure compared to cells lacking the transporter. 16
It has been proposed that these elevated tenofovir levels accumulate in the proximal tubule cells, where they interfere with mitochondrial DNA (mtDNA) replication, causing depletion of mtDNA and secondary impairment of its encoded proteins. 17 Furthermore, the direct role of both MRP-4 and OAT1 in transport and efflux of tenofovir in TDF-related renal proximal tubular toxicity was supported by the study conducted by Kohler et al. 18 In that study, renal proximal tubular mtDNA abundance was increased in the MRP-4 KO mice compared with that in the wild-type mice following TDF treatment. In contrast, in the TDF-treated OAT1KO mice, renal proximal tubular mtDNA abundance remained unchanged, suggesting prevention of TDF toxicity due to loss of tenofovir transport into the proximal tubules. However, Biesecker et al. showed that tenofovir did not affect mtDNA content or levels of mitochondrial enzymes in kidney and other tissues. 19
Tenofovir disoproxil fumarate is used as a long-term treatment for HIV and CHB, despite the potential for nephrotoxicity. It is therefore important to better understand the potential mechanisms behind the toxicity associated with TDF. Toxicogenomics uses microarray technology, which provides sensitive and high-throughput data analysis of gene expression in response to treatments, and therefore it can provide valuable insight into mechanisms of toxicity. It may also identify biomarkers of toxicity in response to tenofovir treatment. Microarray toxicogenomic techniques have been used to define potential biomarker gene sets related to nephrotoxicity. 20 For example, we have used toxicogenomic techniques to identify genomic changes associated with pentamethylchoromanol-induced hepatotoxicity. 21 Although toxicogenomics is a powerful tool in understanding the potential mechanisms of toxicity, a more complete picture of response to a drug is built when it is combined with the more traditional toxicology end points, such as clinical chemistry, toxicokinetics (TK), and histopathology. The objectives of this study were to evaluate the molecular mechanism of TDF-induced toxicity, if any, in female BALB/c mice by correlating gene expression changes with plasma drug levels and other traditional toxicology end points after 13 weeks of treatment.
Material and Methods
Animals
Female BALB/c mice (Harlan, Livermore, California), 6 to 8 weeks old, were maintained on Purina Certified Rodent Chow 5002 (Richmond, Indiana) and purified tap water ad libitum in microisolator cages under controlled lighting (12-hour light–dark cycle). All animals were housed 3 to 5 per cage and treated in accordance with a protocol approved by the SRI Institutional Animal Care and Use Committee. Studies were conducted in a facility accredited by the Association for Accreditation and Assessment of Laboratory Animal Care International.
Study Design
Groups of mice were treated daily with 10 mL/kg oral gavage (orally) administration of TDF (Gilead Sciences, Foster City, California) for 1, 28, or 91 days, at doses of 50, 500, or 1000 mg/kg, respectively. Control mice were administrated a similar volume of vehicle, 50 mmol/L trisodium citrate dihydrate (Sigma-Aldrich). Detailed clinical observations were recorded daily for the first week and then weekly thereafter. Body weights were recorded on day 1, once weekly for the duration of the study, and at necropsy. Standard serum chemistry and hematology parameters were assessed at days 92 and 119. Plasma drug levels were determined at 0.5, 2, 6, and 24 hours postdose on days 1 and 91. Mice (7-15 per group) were sacrificed on days 2, 29, or 92 (24 hours after their last dose), while 10 mice per group were sacrificed on day 119 (28 days after their last dose administration). After gross necropsy, organ weights were determined, sections of liver and kidney samples were processed for toxicogenomics assessment, and major organs from mice sacrificed on days 29, 92, and 119 were processed for histopathology.
Clinical Pathology
Standard methods were used to measure hematology and clinical chemistry parameters of the blood collected from the retro-orbital sinus. Blood from 5 mice per group were used for clinical chemistry and the remaining animals in the group (n = 2-9) were used for hematology evaluation.
The following hematology parameters were measured using an Advia 120 Analyzer (Bayer HealthCare, Tarrytown, New York): hematocrit, hemoglobin, red blood cell (RBC) count, total white blood cell (WBC) count, absolute and relative differential WBC counts, mean corpuscular hemoglobin, mean corpuscular volume, mean corpuscular hemoglobin concentration, platelet count, mean platelet volume, reticulocyte count (absolute, REA, and percentage, RET), and RBC morphology.
The following clinical chemistry parameters and urine total protein were measured using a Cobas c-501 Analyzer (Roche Diagnostics, Indianapolis, Indiana) and standard methods: aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, triglycerides (TRIs), blood urea nitrogen, creatinine, phosphorus, total bilirubin, sodium, potassium, chloride, cholesterol (CHO), glucose, calcium, total protein, albumin, albumin/globulin ratio, and globulin.
Plasma Concentration of Tenofovir
Blood samples were collected, with EDTA as the anticoagulant, from the retro-orbital sinuses of 3 mice per group per time point. Calibration standards (2.5, 5, 15, 50, 250, 500, 1000, and 2000 ng/mL) were prepared in blank BALB/c mouse plasma, and 50 µL aliquots were processed in parallel with study samples. A 100 µL aliquot of 50 ng/mL indinavir (internal standard) in acetonitrile was added to precipitate plasma proteins. After clarifying by centrifugation, the solvent was removed from the supernatants under vacuum, then the dry residues were reconstituted with 50 µL water containing 0.2% (v/v) formic acid. Samples (10 or 20 µL volume) were analyzed by LC-MS/MS using a Synergi Polar-RP column eluted with a gradient of 0.2% (v/v) formic acid in water and 0.2% (v/v) formic acid in acetonitrile. Tenofovir and internal standard indinavir were detected by multiple reaction monitoring after electrospray ionization in the positive ion mode, using the following transitions: m/z 288.1 → m/z 176.1 for tenofovir and m/z 614.3 → m/z 421.2 for indinavir. The peak area of tenofovir was divided by the peak area of indinavir to obtain a peak area ratio (PAR). Calibration standard curves were generated by performing weighted (1/y) linear regression of the PAR versus tenofovir concentration.
Toxicokinetic Analysis
The measured plasma levels of tenofovir were subjected to noncompartmental TK analysis using WinNonlin software version 5.0 (PharSight Corp., Sunnyvale, California.). The following TK parameters were calculated from the plasma concentration versus time: maximum drug concentration (Cmax), time at which Cmax was observed (Tmax), and area under the plasma drug concentration time curve to the last time point (AUClast).
Histopathology
Microscopic evaluations were performed on the following tissues from all mice tested: right kidney, liver, gross lesions (including tissue masses and abnormal regional lymph nodes), lungs with bronchi, spleen, duodenum, ileum, jejunum, urinary bladder, mesenteric lymph nodes, brain, and heart. Tissues retained in 10% neutral-buffered formalin were subsequently embedded into paraffin, cut approximately 5 micro meter thick, and stained with hematoxylin and eosin. Microscopic examination was performed by a board-certified veterinary pathologist.
Toxicogenomics: Sample Preparation and Gene Expression Analysis
A section (approximately 20-50 mg) of the left kidney and left lobe of the liver from TDF-treated and vehicle control animals (n = 7 per group) was collected for microarray analysis. Total RNA was extracted, processed, labeled, and hybridized to GeneChip Mouse Gene 1.0 ST Arrays (Affymetrix, Santa Clara, California) as described previously. 21 The microarray data were analyzed using GeneSpring GX12.6 software (Agilent Technologies). Per-gene normalization was applied across all of the samples of each experiment to normalize the expression level around the value of 1 (base 2 log transformed to 0). Microarray entities (probsets) were filtered based on their signal intensity so that at least one of the samples had intensity values within the 20 percentile low cutoff and the 100 percentile high cutoff range. The resulting 23 815 and 24 008 probes that qualified (from a total of 28 815 entities on the GeneChip) were selected for subsequent kidney and liver microarray data analysis, respectively.
Statistical Analysis and Software
Mean and standard deviation were calculated for body weight, clinical pathology, and organ weight data at each evaluation interval. Body weights, organ weights, and clinical pathology data were evaluated by 1-way analysis of variance (ANOVA), followed by Dunnett test (if the ANOVA was significant). For clinical pathology data, values for parameters that were not within the detection threshold were excluded from the statistical evaluation. For microarray toxicogenomic analysis, t test and Benjamini-Hochberg Multiple Testing Correction were used in statistical analysis.
Results
Toxicity Assessment
Clinical signs
Female BALB/c mice were exposed to 0, 50, 500, or 1000 mg/kg of TDF, once daily via oral gavage, for 1, 28, or 91 days. A subset of mice (10/group) were allowed to recover from any signs of toxicity for 28 days (Day 119) after the discontinuation of the treatment. All animals survived until their scheduled necropsy. A summary of toxicological evaluation following TDF administration in mice is presented in Table 1. Effects in response to the TDF treatment appeared to occur in a dose-dependent manner. In mice treated with either 50 or 500 mg/kg of TDF, 9 of 35 mice exhibited ruffled fur. In mice treated with 1000 mg/kg, 31 of 35 mice exhibited ruffled fur. In addition, in the 1000 mg/kg dose group, 1 mouse exhibited transient incidences of hunched posture, 2 mice exhibited hypoactivity, and 2 mice exhibited gasping. On days 36 and 43, significantly lower mean body weights were observed in mice treated with 1000 mg/kg compared with control animals (P < .05). However, the body weight of the high-dose animals consistently increased over the duration of the study and was comparable with controls for the reminder of the study.
Overview of Toxicology Parameters Following TDF Administration in Mice.a
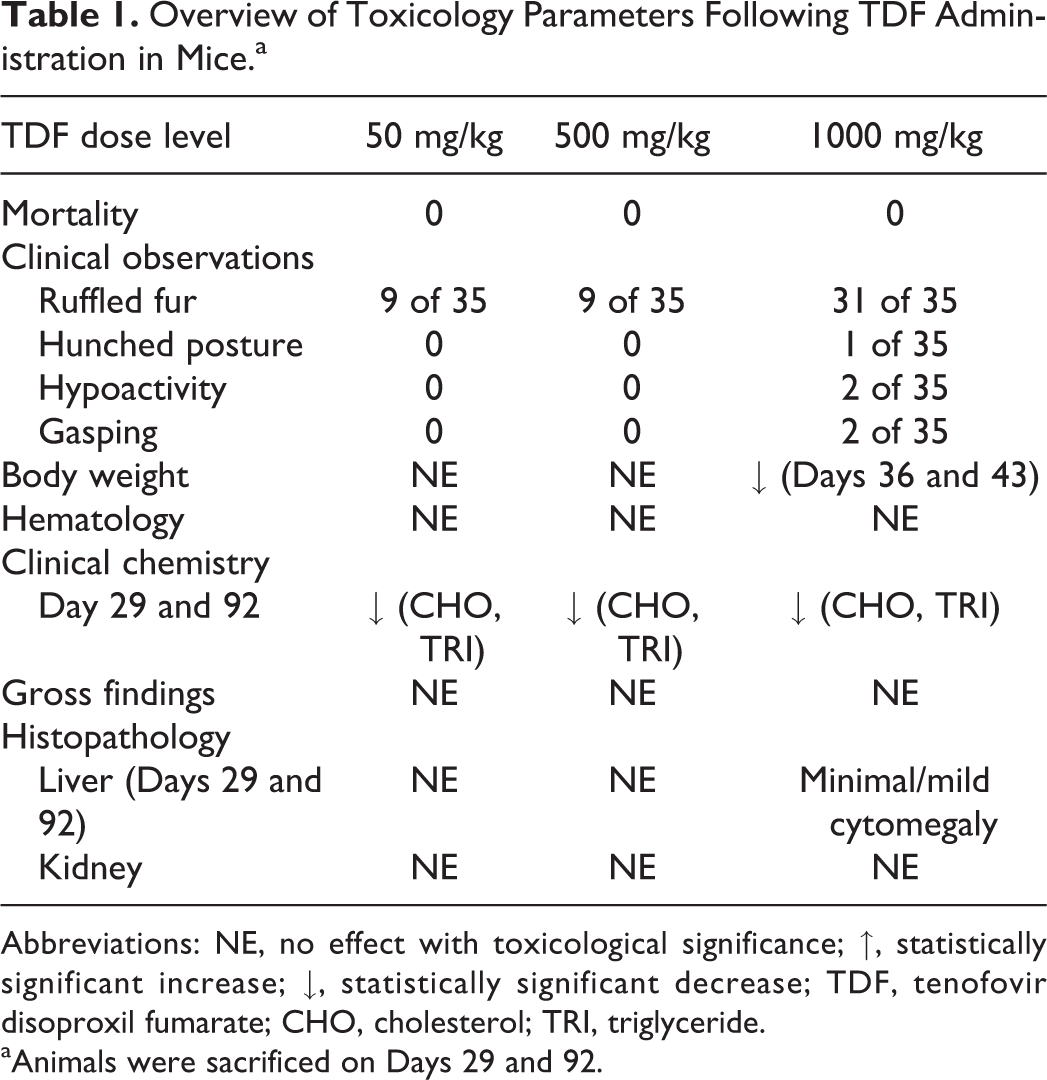
Abbreviations: NE, no effect with toxicological significance; ↑, statistically significant increase; ↓, statistically significant decrease; TDF, tenofovir disoproxil fumarate; CHO, cholesterol; TRI, triglyceride.
aAnimals were sacrificed on Days 29 and 92.
Clinical pathology
Blood samples were collected from the retro-orbital sinus on days 29, 92, and 119 to evaluate hematology and clinical chemistry parameters. The most noticeable changes are presented in Table 1. Slight decreases in levels of CHO and TRI were recorded across all doses on study days 29 and 92, generally in a dose-dependent fashion, which is most notable (30%-48% decreases) in the animals treated with 1000 mg/kg TDF; however, the changes in these parameters were mostly within the normal reference values and they recovered after discontinuation of treatment. These changes are considered to have no toxicological significance. Slight but statistically significant changes were seen in the hematology parameters following the treatment of TDF. The changes were generally small and within the normal reference range. No differences between experimental and control animals in clinical pathology parameters were reported at the end of the recovery period.
Necropsy and histopathology
No gross findings were observed during necropsy. The only microscopic finding considered related to TDF treatment was cytomegaly in the liver of animals given 1000 mg/kg of TDF at necropsy on study days 29 and 92 (Figure 2). The finding was present minimally or mildly in 7 of 10 and 6 of 15 mice given 1000 mg/kg/d for 28 and 91 days, respectively. This microscopic finding was normalized by the end of the 4-week recovery period, since it was not present in the liver of mice treated with 1000 mg/kg TDF at Day 119 necropsy. Liver cytomegaly was not present in mice given lower doses of tenofovir at 50 or 500 mg/kg/d. No histopathologic changes were identified in the kidneys of any dose group.
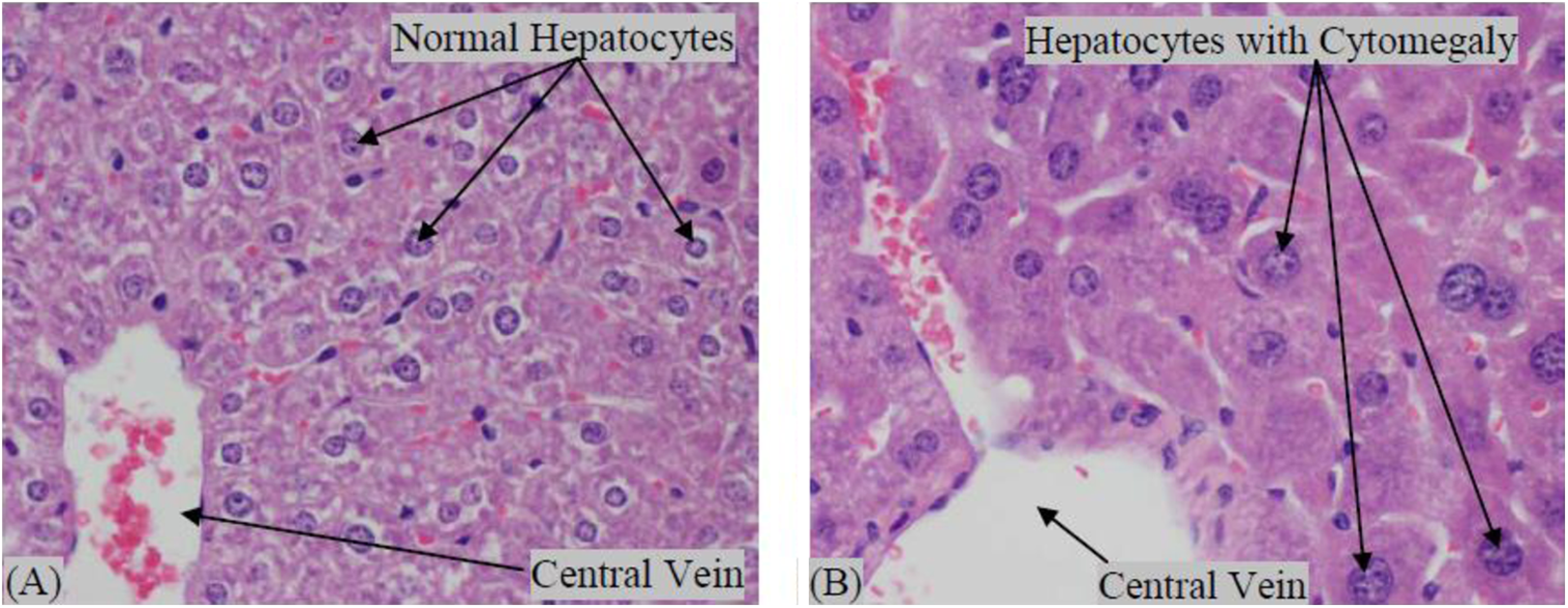
Histopathology of (A) normal vehicle control liver and (B) liver from mouse treated with 1000 mg/kg tenofovir disoproxil fumarate (TDF). In (A), liver tissue has uniform nuclei and normal amount of cytoplasm. In (B), liver tissue with mild cytomegaly of hepatocytes with larger nuclei containing prominent nucleoli.
Toxicokinetic Analysis
We examined the plasma levels of tenofovir in systemic circulation after oral administration of 0, 50, 500, or 1000 mg/kg. Analysis of the TK parameters (Table 2) revealed that an increase in the dose of TDF resulted in a less than dose-proportional increase in exposure based on Cmax and AUClast, with the highest tenofovir exposure observed in the highest dose group tested (6.3 µg/mL and 29.0 h•µg/mL, respectively) after the last dose administration. Tmax was reached 0.5 hour following oral administration of TDF across all dose groups. In addition, no evidence of accumulation was observed between dosing days 1 and 91 for either Cmax or AUC.
Tenofovir Toxicokinetic (TK) Data Following 1 or 91 Days of Exposure.
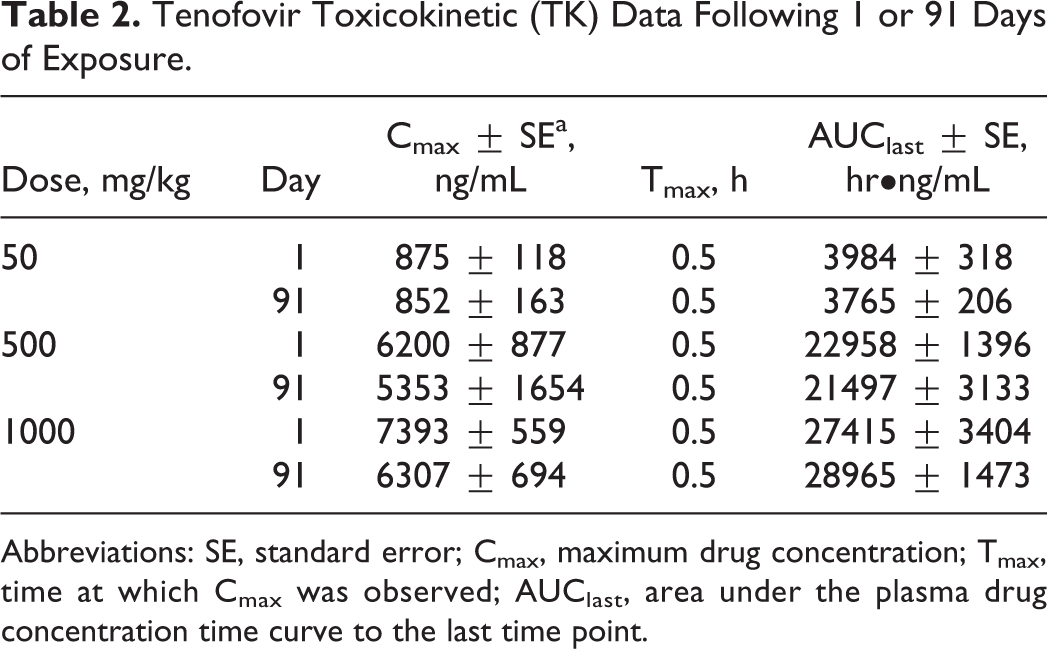
Abbreviations: SE, standard error; Cmax, maximum drug concentration; Tmax, time at which Cmax was observed; AUClast, area under the plasma drug concentration time curve to the last time point.
Toxicogenomics
To analyze the most predominant genomic changes associated with TDF treament, we compared the gene expression profile in the kidneys and livers of the mice in the 1000-mg/kg group with that of the vehicle controls on day 92. Microarray analysis was performed using the Affymetrix GeneChip Mouse Gene 1.0 ST Array. Little differential change was found in gene expression in the kidneys treated with TDF or the vehicle control. A greater than 1.5-fold change was noted only for 4 probes (2 upregulated and 2 downregulated; Table 3). The changes in the kidney were all less than 2-fold.
Genomic Changes in Kidneys of Mice Treated With 1000 mg/kg of TDF for 91 Days.a

Abbreviation: TDF, tenofovir disoproxil fumarate.
a t test P < .05; N = 7.
Microarray analysis in the liver showed 11 genes with at least a 2-fold transcriptional change in the 1000 mg/kg TDF-treated mice compared with the controls on day 92. Six genes were upregulated while 5 genes were downregulated (Table 4A and B). The transcription of Cdkn1a gene, cyclin-dependent kinase inhibitor 1A (p21), in the liver of the 1000-mg/kg dose group was 3.7-fold higher than in the control group.
Genomic Changes in Liver of Mice Treated With 1000 mg/kg of TDF for 91 Days.

Abbreviation: TDF, tenofovir disoproxil fumarate.
Discussion
Since the majority of patients with HIV and CHB will require a prolonged course of therapy, long-term safety is of utmost importance. In the present study, we applied an integrated approach to study TDF-induced toxicity after 13 weeks of daily treatment by evaluating the compound using standard toxicology end points, TK, and differential gene expression of the liver and kidney.
The recommended oral dosage of TDF in adult patients is 300 mg/d, which is 4.29 mg/kg assuming 70 kg of human body weight. The human equivalent dose for mouse study would be 53 mg/kg, and a 10-fold margin would be 530 mg/kg, which is comparable with the mid dose level in this study. Overall, oral treatment with TDF was well tolerated in mice exposed to 50, 500, or 1000 mg/kg for 91 days. Although minimal to mild cytomegaly was detected in the liver of mice treated with 1000 mg/kg of TDF on days 29 and 92, the significance of cytomegaly in liver is not clear and is thought to represent an adaptive process in response to tenofovir exposure. Cytomegaly was not observed on day 119 of evaluation which indicates recovery following cessation of administration. This finding was not observed for mice treated with 50 or 500 mg/kg/d TDF. In line with the observed cytomegaly, a 3.7-fold increase in Cdkn1a transcription was also observed in the 1000-mg/kg dose group from the toxicogenomic data analysis. Cdkn1a, cyclin-dependent kinase inhibitor 1A (also known as p21, Cip1), is involved in the p53-dependent cell cycle G1 phase arrest in response to a variety of stress stimuli. 22,23 The increase in Cdkn1a transcription level may have contributed to the inhibition of cell cycle progression at G1, causing cytomegaly in the liver. It was reported by Aravinthan et al. that high-level expression of p21 is associated with fibrosis and other adverse liver-related outcomes of nonalcohol-related fatty liver disease. 24 The liver cytomegaly observed after treatment with 1000 mg/kg TDF is considered to be of minimal toxicological significance because it has not been reported in clinical use in patients with HIV or in toxicology studies in mice, rats, and dogs, while TDF-related findings in kidney and gastrointestinal tract were reported in ICR CD-1 mice, Sprague Dawley rats, and beagle dogs as reported in the FDA Approval Packages (http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21-356_Viread.cfm).
Interestingly, no nephrotoxicity was observed in BALB/c mice treated with any dose of TDF for 91 days in the present study. In humans, nephrotoxicity is the most notable organ toxicity, albeit with a low incidence of occurrence, associated with TDF treatment. 1,8,10 Nephrotoxicity has also been reported preclinically in rats. Treatment of rats with an oral dose of 100 mg/kg TDF for 8 weeks induced renal toxicity, as evidenced by increased kidney weights, increased proximal tubule diameters, and enlarged mitochondria with disrupted crystal structure. 17 Moreover, TDF-treated rats had reduced mtDNA compared to controls. 17 In another study, nephrotoxicity was observed after treatment with 600 mg/kg of TDF for 5 weeks in Wistar rats. 25 Depletion of the cellular antioxidant was thought to be the cause of oxidative stress and proximal tubule mitochondrial damage. 25 This was supported by the protective role of vitamin E in the tenofovir-induced decreases of renal function in Wistar rats. 26 However, the renal toxicity observed following TDF treatment is not a universal finding. Biesecker et al. evaluated mtDNA content and enzyme activities from the liver, kidney, and muscle from the TDF-treated rats, rhesus monkeys, and woodchucks and reported no effect on mtDNA after oral treatment of TDF. 19
In this study, no renal toxicity was observed in the mice treated with 1000 mg/kg for 91 days. A likely explanation is strain and species difference in either metabolism or toxicologic response to tenofovir. BALB/c is an inbred strain with genetic homogeneity, whereas CD-1 is an outbred stock. Numerous drugs have been shown to be metabolized differently by mice and rats. 27 In addition, TDF is an ester prodrug of the potent antiviral tenofovir. Higher esterase activity in the intestine may lead to a decrease in oral absorption of the prodrug TDF. In fact, TDF showed species-dependent degradation (rat > man > pig) in intestinal homogenates, although mouse was not included in the study. 28 In the present study, the exposure of TFV after oral administration of TDF showed a less than dose-proportional increase based on Cmax and AUC. The mouse AUC plasma levels of tenofovir after 1000 mg/kg dose administration were approximately 8 times greater than those reported in the literature in healthy volunteers. 29
The genomic findings are in alignment with the toxicity data. No significant transcriptional changes were observed in the kidney, and no significant renal toxicity was observed. Moreover, test article-related microscopic cytomegaly noted in the livers of mice treated with 1000 mg/kg of TDF was associated with Cdkn1a transcriptional changes in the same dose group in the same organ. Future studies comparing different strains of mice and with other animal species following TDF exposure would help clarify any specific differences in pharmacokinetics and toxicity in animals.
In summary, chronic treatment with TDF up to a dose of 1000 mg/kg for 91 days did not cause any significant renal toxicity in BALB/c mice, which is consistent with no significant changes in gene expression levels in kidney compared with those in the controls. The findings from this study suggest that the BALB/c mouse may not be the most sensitive animal model to assess TDF-induced nephrotoxicity following chronic treatment.
Footnotes
Acknowledgments
We would like to thank Gilead Sciences (Foster City, CA) for their generous donation of TDF. We also thank Elizabeth Zuo and Michael Eckart at the Stanford University Protein and Nucleic Acid Biotechnology Facility for hybridization and image scans of Affymetrix microarray GeneChips.
Author Contributions
Hanna H. Ng contributed to conception and design, acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy. H. Stock contributed to interpretation, drafted the manuscript, gave final approval, and agreed to be accountable for all aspects of work ensuring itegrity and accuracy; L. Rausch contributed to design, acquisition, and analysis; drafted the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy. D. Bunin contributed to conception, acquisition, analysis, and interpretation; drafted the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy. A. Wang contributed to acquisition, analysis, and interpretation; drafted the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy. S. Brill contributed to acquisition and analysis, drafted the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy. J. Gow contributed to acquisition, analysis, and interpretation; drafted the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy. J. Mirsalis contributed to conception and design and interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of work ensuring itegrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH/NIAID Contract N01-AI-70043 (HHSN266200700043C).