
Abstract
Toxicogenomic technologies query the genome, transcriptome, proteome, and the epigenome in a variety of toxicological conditions. Due to practical considerations related to the dynamic range of the assays, sensitivity, cost, and technological limitations, transcriptomic approaches are predominantly used in toxicogenomics. Toxicogenomics is being used to understand the mechanisms of toxicity and carcinogenicity, evaluate the translational relevance of toxicological responses from in vivo and in vitro models, and identify predictive biomarkers of disease and exposure. In this session, a brief overview of various transcriptomic technologies and practical considerations related to experimental design was provided. The advantages of gene network analyses to define mechanisms were also discussed. An assessment of the utility of toxicogenomic technologies in the environmental and pharmaceutical space showed that these technologies are being increasingly used to gain mechanistic insights and determining the translational relevance of adverse findings. Within the environmental toxicology area, there is a broader regulatory consideration of benchmark doses derived from toxicogenomics data. In contrast, these approaches are mainly used for internal decision-making in pharmaceutical development. Finally, the development and application of toxicogenomic signatures for prediction of apical endpoints of regulatory concern continues to be area of intense research.
A Primer on Toxicogenomics Approaches
Toxicogenomics is the application of genomics approaches to understand toxicological responses in biological systems. These genomics approaches may be based on probing the genome, transcriptome, epigenome, proteome, and metabolome and involve a variety of technologies including hybridization, next-generation sequencing, mass spectrometry, nuclear magnetic resonance, and chromatography, among others. These technologies provide a global snapshot of molecular alterations in a single analysis and have been called omics with an appropriate prefix based on the approach being used, that is, transcriptomics, proteomics, and metabolomics. Due to limitations in the respective technologies, dynamic range of the target molecules, and cost, much of the toxicogenomics data are based on gene expression. 45 Since the advent of this subdiscipline, there have been much hype and hope for this technology to revolutionize the field of toxicology and chemical carcinogenesis. Some of the main goals or expectations from this technology are the reduced use of animal testing, prediction of long-term effects from short-term animal studies or in vitro studies, biomarker discovery, and understanding the mechanisms of toxicity or carcinogenesis to establish the translational relevance of animal studies. In this article, we will discuss the current state on the utility of these technologies and the potential evolution in this space.
The central premise for toxicogenomic studies is that gene expression alterations precede the phenotype (toxicity or carcinogenicity). This premise is the foundation for many types of toxicogenomic experimental designs including class discovery, class comparison, and class prediction. 38 While much of the older technologies to identify gene expression alterations were limited by throughput, the advent of cDNA-based and oligonucleotide-based microarray technology has opened the possibility to scan the global gene expression changes in a single experiment. 43 Newer technological advancements related to next-generation sequencing such as RNA sequencing (RNA-Seq) have allowed for a more comprehensive assessment of the transcriptome including the non-coding genes. Over the last two decades, the microarray platforms as well as the respective bioinformatics pipelines have matured and produce technically reproducible results. Several iterations of the MicroArray/Sequencing Quality Control (MAQC/SEQC) consortia have demonstrated the reproducibility of the data using both the microarray and RNA-Seq technologies. 61
The platform of choice for toxicogenomic studies depends on the objectives of the study along with resource considerations. If the objective of the study is to compare the transcriptomic signatures to legacy compounds where data were generated using microarray technology, then selection of a similar microarray platform is a rational choice. However, if the goal is to discover novel transcripts or biomarkers, gene fusions, splice variants, and allele-specific information, then RNA-Seq approach is a better choice. Reduced sequencing costs and mature bioinformatics pipelines have increased the accessibility of RNA-Seq technologies. The next frontier in this space is single cell sequencing and spatial transcriptomics that provide more detailed mechanistic insights previously unavailable from bulk transcriptomic approaches. While these genome-wide transcriptomic approaches provide a comprehensive insight into the molecular changes, it may not be a suitable choice for certain experiments. For example, if the goal is to screen large number of chemicals (100s to 1000s) for biological activity, then targeted approaches such as the S1500+ 34 or L1000 55 are usually preferred due to resource considerations. These targeted panels are based on expert curation of landmark genes to represent the overall transcriptomic signal in the data. Recent studies have shown that these targeted approaches provide sufficient pathway level concordance with the global transcriptomic approaches. 7
Toxicogenomics is a multidisciplinary area that requires a collective expertise related to toxicology, pathology, and bioinformatics disciplines. The contribution of pathologists is especially important in determining the adverse responses in the animal study and for identifying appropriate sample collection for anchoring the phenotype to the gene expression changes. Based on the goals of the toxicogenomic project, some of the sample collection considerations include consistent site/subsite of tissue collection (i.e. same liver lobe or same subsite in the brain), lesion and adjacent normal tissue, randomization of tissue collection across treatment and control groups, controlling for circadian rhythm alterations, fasting status, and time of exposure (toxicokinetic effects). 45 In addition, collection of tissues for appropriate biological and technical controls is important to increase the confidence in the toxicogenomics data. As pathology is the main endpoint in most guideline animal toxicity and carcinogenicity studies, toxicogenomic studies are usually a retrospective consideration in these cases. Due to the high costs associated with repeating animal studies to gain mechanistic insights, it is prudent to include sample collection protocols for potential toxicogenomic studies in routine animal studies. These additional steps incur minimal expense but provide significant value in derisking strategies that require determination of the mechanisms and/or translational relevance of an apical endpoint of regulatory relevance. Some of the considerations include interim target organ collection from 3 to 6 animals/group to gain temporal insights into the progression of the molecular alterations. Ideally, the samples should be fresh frozen or collected in OCT (optimal cutting temperature compound) to maintain the spatial information. In the absence of interim sample collection or freezing facilities, representative target organs may be collected in RNAlater, Paxgene system, or ethanol-based fixatives during terminal sacrifice. At the very least, examination of transcriptomic alterations at a single time point (usually the terminal end point) helps to formulate mechanistic hypotheses that may be addressed in short-term animal studies or targeted in vitro studies.
In the following sections, detailed discussions on toxicogenomic data analyses, regulatory relevance, utility of transcriptomics in environmental toxicology/carcinogenesis and pharmaceutical development, as well as gene network approaches to better understand mechanisms and pathogeneses are presented.
Transcriptomic Approaches to Predict Environmental Hepatocarcinogens
Predicting hepatocarcinogenicity is crucial, as the liver is the most common target organ in rodent carcinogenicity studies, influencing many hazard identification decisions. 22 The liver is also a frequent target of toxicity in humans, especially with drugs. 26 Combining the prediction of hepatocarcinogenicity with mechanistic characterization can enhance the human relevance of cancer findings in rodents and augment safety assessments in various sectors, including industrial, environmental, and pharmaceutical.
The applicability of the models will likely vary across sectors. In the pharmaceutical and agrochemical sectors, which are typically rich in toxicological data due to mandated data generation, transcriptomics-based models are likely to be of greatest use in internal screening and providing supportive data in safety assessment paradigms such as NegCarc.6,54 In areas with limited data, where there is a restricted mandate for data generation, these models offer the possibility of performing predictive hazard identification. Depending on the tolerance for uncertainty, this can be used for chemical classification or as justification for additional studies.
This section of the article addresses the role of toxicogenomics in characterizing environmental chemical carcinogenesis. However, much of the discussion in this section is also relevant to the risk assessment of pharmaceuticals, albeit with some adjustments and discretion. A primary difference between environmental toxicology and pharmaceutical toxicology, as it pertains to carcinogenesis, lies in the balance of risk versus benefit and the choice of exposure. In the realm of pharmaceuticals, a patient weighs the therapeutic benefits of a drug against its known potential side effects. Consequently, they make an informed decision to undergo treatment, fully aware of the potential consequences, such as an increased risk of certain cancers or other toxicities. This is the choice cancer patients make when opting for chemotherapy and radiation treatments. Contrastingly, in the context of environmental chemical carcinogenesis, individuals often do not have a conscious choice regarding their exposure to harmful chemicals. For instance, they might unknowingly consume polycyclic aromatic hydrocarbons present in grilled meat or aflatoxin in Aspergillus-contaminated food. Moreover, these exposures are often uncontrolled, raising concerns about the risks to sensitive subpopulations. In these scenarios, there is typically little to no benefit to counterbalance the risks. Consequently, the tolerance for exposure is significantly lower, leading to the establishment of more conservative and protective allowable exposure levels compared with typical therapeutic exposure levels in the case of pharmacotherapy. As discussed in a later in this section, the quantitative application of toxicogenomics has the potential to identify minimal perturbations linked to carcinogenesis. When integrated with current environmental chemical safety assessment standards, this can help determine minimal risk levels. Such determinations would consider both threshold and non-threshold mechanisms.2,13
Since the advent of toxicogenomics, extensive work has been done in this area, with most studies focusing on rat data. These models range from predicting outright hepatocarcinogenicity to evaluating different modes and mechanisms of action. Currently, there is project support through Health and Environmental Sciences Institute (HESI) that is attempting to formalize the models to facilitate acceptance for regulatory decision-making. 14
At the outset of building and applying a model, it is critical to consider its applicability. The first question the modeler should address is, “Will this test article exhibit substantial elevations in liver cancer at any administered dose level in a cancer bioassay?” If affirmative, it necessitates exploration into the mode or mechanism of action of the carcinogenic response and an evaluation of its potency. Understanding the mechanism is crucial as it sheds light on how the chemical interacts with biological systems to induce cancer, and assessing the potency is vital to determine the degree of response at different dose levels. Conversely, if the test article does not indicate hepatocarcinogenic properties, it raises another pertinent question: “Will it be carcinogenic in any other tissue?” This extends beyond the capabilities of transcriptional approaches described here but is essential to establishing safety. To address this, a meticulous pathology evaluation to detect any adverse morphological alterations in subchronic and 6-month studies is imperative.6,54 In addition, researchers should look for signatures of genotoxicity and cell proliferation in these tissues, as these are indicative of the potential of a substance to damage the DNA in a cell, causing mutations, and to promote the growth of cancer cells, respectively.
The first step in building models is to identify the substrate data to use. While there are several datasets available, the two most used are DrugMatrix 58 and TG-Gates. 28 Both are extensive rat toxicogenomics datasets that collectively evaluated over 700 test articles at different dose levels and durations. Both datasets use Sprague Dawley rats, and the studies are primarily conducted through the oral route via gavage.
After identifying the data substrate, the next step is formulating training and test sets of data. The training set is used to train the model, and the test set, which should be independent of the model training data, is used to independently characterize the accuracy of the model. In the case of hepatocarcinogenesis, three groups of chemicals are typically created in both the training and test sets: non-hepatocarcinogens, genotoxic hepatocarcinogens, and non-genotoxic hepatocarcinogens. If the data were not generated in a manner that parallels the cancer bioassay (strain, dose levels, vehicle, route, etc.), confidently creating these groups can be challenging. If one is using the DrugMatrix and TG-Gates data, several challenges arise because there are several experimental variables that are mismatched. It is often the case that the doses in the short-term toxicogenomics studies far exceed those used in the cancer bioassay. In such cases, it is challenging to identify non-hepatocarcinogenic treatments because high doses of these chemicals may elicit toxicity that, if sustained for the lifetime of the animals, could result in a carcinogenic response. In the case of hepatocarcinogenic chemicals, the high doses used in toxicogenomic studies may elicit a gene expression response where non-genotoxic chemicals may start to elicit cytotoxicity, which can make them manifest expression that is consistent with a genotoxic mode of action. This issue also bleeds over into evaluating specific mechanisms of hepatocarcinogenicity (discussed more below); specifically high doses can elicit a response from a variety of mechanisms (e.g., PPARA, CAR/PXR, Nrf2/oxidative stress), which can make it difficult. Another significant consideration is marginally carcinogenic chemicals or dose levels. If the bioassay parameters (strain, route, vehicle, etc.) were in any way discordant with the toxicogenomic studies the modeler hopes to label, there can be uncertainty as to whether a treatment should be labeled carcinogen or non-carcinogen. Once the various challenges with sample collection and labeling are dealt with, the next step is building the model using machine learning.
There are a variety of algorithms for building high-dimensional models that differentiate between different groups (e.g., non-genotoxic vs genotoxic chemicals). Some of these include decision trees, support vector machines, random forest, deep neural networks, and naïve Bayes. 23 Each identifies descriptive features (e.g., genes), organizes them in a manner to allow multidimensional characterization of patterns in data, and provides a model structure that reports a probability of a sample belonging to a specific class. 66 An important aspect of model generation is the feature reduction process, which avoids overfitting of the training data and increases the generalized applicability of models, that is, maximally accurate prediction of the test data. 37 The primary manner of avoiding overfitting is iterative cycles of modeling where cross-validation is used through a process of splitting the training data into groups for model building and evaluation. 17 After each iteration, the features are ranked, and the least informative features are removed. This is continued until the classification error rates in the cross-validation begin to increase. The final model ends up being the model with the minimum error rate with the smallest number of features. This model is then used to predict the independent test to get a sense of the generalizable accuracy of the model.
Once the models are built, there is significant value in evaluating the underlying features (genes) that inform the model (i.e., explainability). These give the modeler insight into the mechanistic processes that are being captured in the modeling approach and can help to inform risk assessment. Differentiation of modes of action in pathogenesis is crucial to determine human relevance and model extrapolation. 42 Non-hepatocarcinogens at high doses often activate genes and gene sets associated with non-genotoxic hepatocarcinogens; however, typically this activation is much weaker than observed with hepatocarcinogenic doses of non-genotoxic chemicals. A genotoxic mode of action in hepatocarcinogenesis is generally regarded as relevant to humans and is treated with significant caution when performing risk assessments. 25 There are a variety of mechanisms that underlie the genotoxic mode of action, a few of which are described below. Interestingly, most of the different mechanisms tend to elicit a similar response at the transcriptional level, which is the activation of the P53 pathway, with certain prototype response genes being upregulated, including Ccng1, Lama5, Cdkn1a, Mdm2, and Nhej. These will vary a bit depending on the species that are under consideration.
Direct Alkylation/Mutation: Some genotoxic agents, such as certain chemical compounds, can directly bind to DNA molecules, causing mutations. This process is known as alkylation, and it can lead to genetic changes and potential cancer development. 52
Inhibition of Topoisomerase: Topoisomerases are enzymes responsible for regulating DNA supercoiling and preventing tangling during processes like replication and transcription. Inhibitors of topoisomerases by certain drugs and botanicals can disrupt these vital processes, leading to DNA damage and genotoxicity. 49
Microtubule Disruption: Substances that interfere with microtubules, essential components of the cytoskeleton, can disrupt cell division and lead to chromosome segregation errors. This disruption can result in genetic mutations and aneuploidy, a condition characterized by an abnormal number of chromosomes. 13
Nucleotide Synthesis Inhibition: Inhibition of nucleotide synthesis, a process crucial for DNA replication and repair, can result in the incorporation of incorrect nucleotides into DNA strands. 15 This can lead to mutations and genomic instability.
The genes and mechanisms that underlie non-genotoxic hepatocarcinogenicity tend to be more diverse than those observed with genotoxic hepatocarcinogenicity. This is due to the diversity of mechanisms that can elicit a liver cancer response in rodents. These mechanisms consist of activation of certain nuclear receptors (i.e., PPARA, CAR/PXR, ER), PAS domain proteins (i.e., AhR), and other more generalized mechanisms (i.e., cytotoxicity, oxidative stress, inflammation) (Table 1). The mechanisms are described in more detail below.
AhR Activation: Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor involved in the regulation of biological responses to planar aromatic hydrocarbons. 33 Activation of AhR can lead to hepatocarcinogenicity by inducing the expression of cytochrome P450 enzymes, which can convert procarcinogens to carcinogens, leading to DNA damage and mutations in the liver cells, promoting cancer development. Activation of AhR is generally considered to be of carcinogenic relevance to humans. Gene sets that are upregulated by AhR activators are primarily related to xenobiotic metabolism. The most commonly upregulated genes that inform the models are Cyp1a1, Cyp1b1, and Cyp1a2.
CAR/PXR Activation: CAR (Constitutive Androstane Receptor) and PXR (Pregnane X Receptor) are nuclear receptors that regulate the expression of genes involved in drug metabolism and excretion. 68 Activation of CAR/PXR can lead to hepatocarcinogenicity by inducing the expression of enzymes that can lead to increased cell proliferation and decreased apoptosis, creating a conducive environment for the development and progression of liver cancer. CAR/PXR activation is of uncertain carcinogenic relevance to humans; however, there are other human-relevant effects that need to be considered in risk assessment. Gene sets that are upregulated by CAR/PXR activators are primarily related to xenobiotic metabolism, albeit a slightly different subset of xenobiotic metabolism compared to AhR activation. The most commonly upregulated genes that inform the models are Cyp2b1, Ces2c, Cyp3a23/3a1, and Ugt2b1.
PPAR-α Activation: PPAR-α (Peroxisome Proliferator-Activated Receptor Alpha) is a nuclear receptor that regulates lipid metabolism. Activation of PPAR-α can lead to hepatocarcinogenicity by inducing peroxisome proliferation, leading to increased oxidative stress and DNA damage, and by altering lipid metabolism, which can contribute to the development of liver tumors. 59 PPARA activation is not generally considered to be of carcinogenic relevance to humans; however, there are other human-relevant effects that need to be considered in risk assessment. Gene sets that are upregulated by PPAR-α activators are primarily related to fatty acid metabolism. The most commonly upregulated genes that inform the models are Acot1, Cyp4a1, Ehhadh, Cpt1b, and Vnn1.
Estrogen Receptor Activation: Estrogen receptors are involved in the regulation of the reproductive system and secondary sex characteristics. Activation of estrogen receptors in the liver can lead to hepatocarcinogenicity by promoting the growth and proliferation of liver cells, leading to the development of hepatic adenomas and, eventually, hepatocellular carcinomas, particularly in a context of pre-existing liver disease. 11 Activation of ER is generally considered to be of carcinogenic relevance to humans. Gene sets that are upregulated by ER activators are primarily related to estrogen receptor signaling pathways. The most commonly upregulated genes that inform the models are Rbp7, Cited4, Lifr, Orm1, Cyp4a8, and Rgs3.
Cytotoxicity: Persistent chronic cytotoxicity can lead to hepatocarcinogenicity by causing cell death and subsequent compensatory proliferation of surrounding liver cells. 18 This increased cell proliferation can lead to the accumulation of mutations and the development of cancerous cells in the liver. Cytotoxicity is generally considered to be of carcinogenic relevance to humans. Gene sets that are upregulated by cytotoxicity are primarily related to epithelial-mesenchymal transition and wound healing. The most commonly upregulated genes that inform the models are Anxa2, Gpnmb, Tubb6, S100a10, and Trib3.
Oxidative Stress/Nrf2 Activation: Oxidative stress occurs due to an imbalance between the production of reactive oxygen species (ROS) and the ability of the body to counteract or detoxify their harmful effects. It usually occurs secondary to glutathione depletion and is likely closely related to cytotoxicity. 60 Nrf2 (Nuclear factor erythroid 2-related factor 2) is a transcription factor that regulates the expression of antioxidant proteins. 41 Persistent oxidative stress can lead to DNA damage and mutations, leading to hepatocarcinogenicity. Activation of Nrf2 can have a protective role by inducing antioxidant response elements, but if dysregulated, it can also contribute to cancer progression by promoting cell survival and proliferation. Oxidative stress is generally considered to be of carcinogenic relevance to humans. Gene sets that are upregulated by oxidative stress are primarily related to glutathione metabolism and response to reactive oxygen species. The most commonly upregulated genes that inform the models are Hmox1, Nqo1, Akr7a3, Srxn1, and Gsta3.
Inflammation: Chronic inflammation in the liver can lead to hepatocarcinogenicity by promoting a pro-tumorigenic environment. 69 Inflammation can cause DNA damage, stimulate cell proliferation, and inhibit apoptosis, contributing to the accumulation of mutations and the development of cancerous cells. In addition, inflammatory cells can release growth factors and cytokines that further promote tumor development and progression. 70 Inflammation is generally considered to be of carcinogenic relevance to humans. Gene sets that are upregulated by inflammation are primarily related to the innate immune response. The most commonly upregulated genes that inform the models are Lcn2, A2m, Lbp, Tnfrsf21, and Cxcl9.
A summary of various modes of action (MOAs) for rodent hepatocarcinogenicity and their corresponding gene alterations in rodent studies.

It is important to remember that it is rare for there to be solely one mechanism of action for a chemical, although there is nearly always one that is dominant. When using these signatures, it is important to consider the response at different dose levels as well as the genetic background to determine primary versus secondary modes/mechanisms or combined effects.
Regarding the last point above, there have been significant advancements in the application of dose-response modeling of genomic data with the development of software to support data analysis. 39 The application of mode and mechanism signatures in the context of genomic dose response can be employed to understand the effects of chemicals on different hepatocarcinogenic processes and characterize both the effect size and the primary and secondary effects as a function of dose. This type of modeling can inform both human relevance and quantitative risk.
Transcriptomic Approaches to Predict Hepatotoxicity and Carcinogens in Drug Development
Toxicogenomic or more specifically transcriptomic data are used extensively in the pharmaceutical industry. Here, only the utilization of these data for preclinical safety assessment will be covered. However, it is worth mentioning that most transcriptomic data in pharmaceutical Research and Development (R&D) are used outside of preclinical safety organizations, particularly in Discovery Research for therapeutic target discovery and validation or in clinical development for biomarker development and personalized medicine applications. In addition, the focus of this section will be on applications for in vivo studies, although references will be made to complementary in vitro applications and most principles discussed are applicable to both in vivo and in vitro conditions.
Over two decades ago, as novel technologies and platforms enabling the rapid generation of genome-wide transcriptomic data became available in the form of microarrays, toxicogenomics emerged as a technology with the potential to revolutionize drug discovery and development given that safety is one of the main causes of attrition in drug discovery and development and of market withdrawal. The vision for its application in preclinical toxicology was as a predictive tool for toxicity, thereby decreasing toxicity-related attrition and favorably impacting pharmaceutical R&D productivity. Furthermore, toxicogenomic data were also recognized as useful for providing mechanistic insight. This vision was the basis for substantial efforts by several companies to develop transcriptomic-based predictive biomarkers of toxicity (also referred to as gene expression signatures, classifiers, or models). The underlying assumption of predictive toxicogenomics is that compounds that induce toxicity through similar mechanisms will elicit unique gene expression signatures. Hence, signatures related to a specific toxic mechanism could be derived using the gene expression profiles of well-characterized compounds. To achieve that vision, it was of utmost importance to build databases of gene expression profiles. Consequently, several large repositories of gene expression profiles (mostly from rat liver, but also from other tissues) were generated, such as DrugMatrix 20 or TG-GATES, 28 which are now publicly available. Likewise, some pharmaceutical companies built small-scale databases as initial proof-of-concept. For example, Abbott generated an early database of liver transcriptomic profiles from rats dosed with 15 different hepatotoxic agents complemented by control data to evaluate the potential of the technology. 64
The principle used for gene expression signature development is simple in concept. 4 Briefly, predictive or diagnostic gene expression signatures are generated using a training set of gene expression profiles induced in the tissue of interest by a wide range of structurally and pharmacologically diverse, toxic, and non-toxic compounds. The training set is analyzed using various computational algorithms to develop a predictive model that can classify compounds as toxic or non-toxic in the tissue of interest. This classification can be based on a specific or more general toxic effect. For example, the objective may be to generate signatures for specific histologic changes (e.g., biliary hyperplasia, hepatocyte single cell necrosis, centrilobular hepatocellular hypertrophy) or for more general changes (e.g., hepatotoxicity vs normal). The signature can then be evaluated for its performance in a forward validation step by using a testing set of gene expression profiles distinct from those of the training set.
Multiple gene expression signatures have been published in the literature for various endpoints in diverse tissues, but mostly liver. 45 For example, one of the models developed at Abbott was a quantitative predictive model for rat hepatoxicity. This predictive classifier was developed from a compendium of transcriptomic data generated from liver samples from rat toxicology studies of 3- to 5-day duration and an artificial neural network algorithm. The model classified compounds according to a composite score ranging from 0 to 4, which indicates the probability that the test compound would induce hepatotoxicity in rats upon continued dosing: the lower the score, the higher the probability of being hepatotoxic. 45 A threshold for the binary decision was set at 2.5 based on a receiver operating curve (ROC) analysis. The model was then evaluated with internal projects that were naïve to the training set. For these projects, histopathology and serum chemistry data from rat repeat-dose toxicity studies of at least 2-week duration were considered the gold standard. These data were compared to the prediction based on gene expression profiles generated from livers after 3 to 5 days of dosing at comparable dose levels. The confusion matrix in Table 2 summarizes the excellent performance of the model. This model was used routinely on internal pipeline projects for almost a decade. While most reported signatures were focused on liver endpoints, it is worth noting that robust signatures were also generated in other tissues, such as the kidney. 19
AbbVie rat hepatotoxicity predictive model.
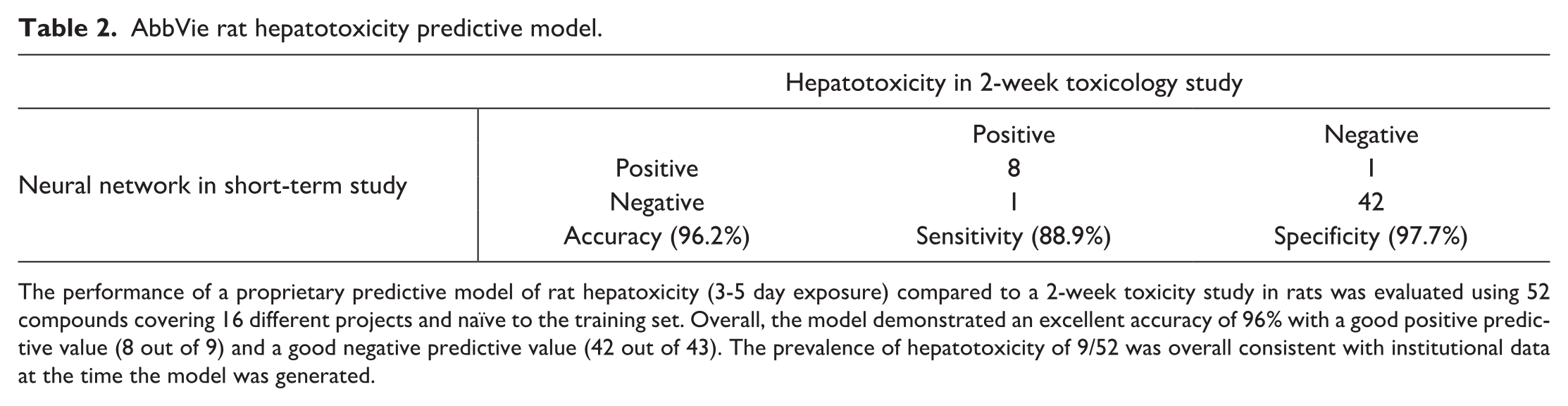
The performance of a proprietary predictive model of rat hepatoxicity (3-5 day exposure) compared to a 2-week toxicity study in rats was evaluated using 52 compounds covering 16 different projects and naïve to the training set. Overall, the model demonstrated an excellent accuracy of 96% with a good positive predictive value (8 out of 9) and a good negative predictive value (42 out of 43). The prevalence of hepatotoxicity of 9/52 was overall consistent with institutional data at the time the model was generated.
Although there were multiple successful examples of the use of toxicogenomics in predictive toxicology, it was also clear that there were some limitations with its use as a predictive tool. First, user experience showed that it was often a more useful tool for mechanistic interrogation. Second, a sufficiently robust database was essential for both building models, and also contextualizing and interpreting the data. Inability to access sufficiently robust toxicogenomic databases limited the ability of multiple preclinical safety organizations to demonstrate meaningful value and hence to more routinely use toxicogenomics. Not surprisingly, organizations that invested in building or buying transcriptomic databases were also the largest users. Third, while a few predictive models were useful, most were not due to multiple challenges. 45 In particular, transcriptomic data are very high-dimensional data (i.e., very high number of features for very few samples). To put in perspective, although relevant current databases are considered quite big, the numbers of samples are still largely insufficient for robust predictive model development: as an example, DrugMatrix contains 600 compounds (i.e., sample category) profiled with Affymetrix microarrays (i.e., >16,000 transcripts, the feature category). These feature/sample imbalance situations are associated with a phenomenon known as overfitting: the model accurately predicts data present in the training set, but the performance of the model fails to generalize against new unseen data. Furthermore, gene expression signatures often face a “black box” problem: they frequently do not offer insight of what contributes to a positive or negative response and often do not have a biological or mechanistic link (i.e., lack of explainability). 29 This lack of transparency results in a lack of trust with the models, especially in preclinical safety organizations where explainability is often required. Finally, toxicogenomics, like most new technologies, faced a hype problem with numerous extravagant and ridiculous claims that negatively affected its ongoing adoption. Despite these challenges, transcriptomic data are still an important part of the toolbox in preclinical safety organizations.
The use of toxicogenomics is diverse across the pharmaceutical industry. In particular, two recent surveys provide data related to the use of the technology, which appears to be more common in toxicology organizations focused on discovery support and/or investigative work. Specifically, a survey of 13 pharmaceutical companies conducted in 2016 indicates that while the technology is not used frequently at the development stage (i.e., GLP studies) or as a predictive tool, it is regularly used for mechanistic investigations. 62 Likewise, another industry survey suggests an increased level of adoption in the last few years. 48 Multiple factors can explain this evolution toward more mechanistic rather than predictive utilization, including: (1) the evolving nature of the pharmaceutical pipelines with the emergence of novel therapeutic modalities and an overall lower proportion of traditional small molecules, (2) the development of novel tools to de-risk problematic chemical matter and of novel platforms such as the microphysiological systems, 3 and (3) the evolution of genomics platforms (e.g., RNA Seq, spatial transcriptomics). In particular, the transition from microarrays to RNA-Seq platforms likely contributed to a change in approaches for several reasons. First, compared to microarrays, toxicity-relevant databases of RNA-Seq data were initially less populated, thereby limiting one’s ability to develop classifiers or even to contextualize the data. Second, gene expression data from RNA-Seq platforms are more quantitative with a much higher dynamic range and with a higher number of differentially expressed genes than data from microarrays. Third, with RNA-Seq, data on the entire transcriptome (including non-coding RNAs such as miRNAs and lncRNAs) are available, hence resulting in richer, but also more complex datasets. Altogether, leveraging institutional historical microarray data in the context of RNA-Seq data proved to be an initial challenge that required the development of novel algorithms and data analysis tools. Nevertheless, experimental evidence supports that both platforms have enough overlap to identify similar transcriptomic dysregulation, although RNA-Seq offers additional insights. 50
Numerous examples demonstrate that toxicogenomics has clear utility when appropriately used in drug discovery and development. For example, during lead optimization, transcriptomic data offer multiple advantages when included in exploratory toxicology studies: (1) gene expression changes induced by treatment with a test article can frequently be detected before morphological and functional changes indicative of toxicity; (2) transcript changes are frequently demonstrating less variability than traditional end points (histopathology and serum chemistry) as illustrated in Figure 1. These two properties (rapid occurrence, low variability) make gene expression profiles a useful complement to traditional endpoints in exploratory toxicology studies by providing higher confidence that compound-related effects are present. Consequently, adding transcriptomic data as an endpoint may enable the use of fewer animals and, hence, the need for lower amounts of test article (which represent a significant benefit during lead optimization programs). Furthermore, transcriptomic changes offer some mechanistic clarity, which can be useful for decision-making. For example, interactions with nuclear receptors can easily and reliably be identified accurately with hepatic gene expression profiles. 4 When used for mechanistic characterization, access to toxicogenomics data helps formulating hypotheses about the mechanism of a toxic change, and this understanding can represent a very efficient way to design a rational strategy to identify a back-up molecule devoid of a specific liability. This was illustrated using an example of a compound series that induced multifocal myocardial necrosis after 4 weeks of dosing in rats (Unpublished data). A transcriptomic analysis demonstrated that this toxicity was associated with a downregulation of genes related to mitochondrial function (e.g., fatty acid degradation, b oxidation and tricarboxylic acid cycle) and an upregulation of genes involved in oxidative stress, suggesting an impact on mitochondrial function. Orthogonal evaluation using complementary mitochondrial functional assays confirmed the hypothesis, resulting in the incorporation of mitochondrial assays early in the lead optimization testing funnel to select a back-up without this liability.
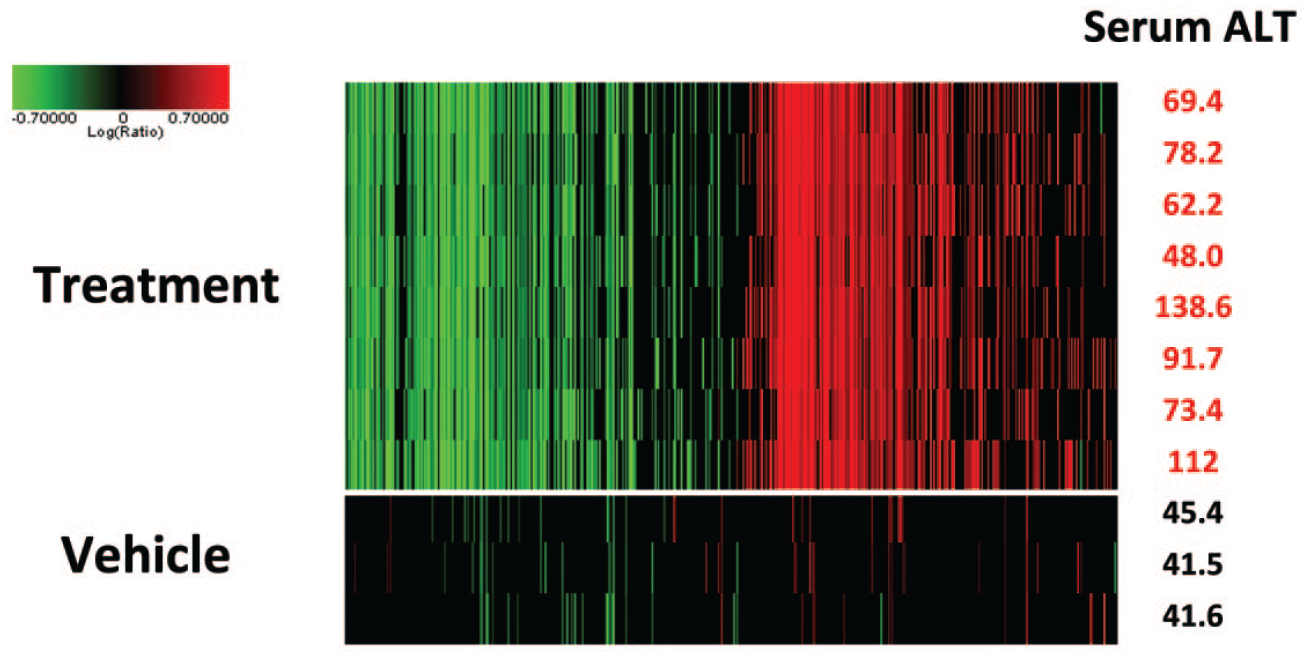
Variability of gene expression profiles compared to serum alanine transaminase (ALT) values. Eight male rats were dosed daily for 3 days with a slightly toxic dose of a tool hepatoxic compound, and 3 male rats were dosed with the vehicle. On the right column are the serum ALT values 24 hours after the last dose when rats were sacrificed. Gene expression profiles were generated using the rat Affymetrix microarray for all rats. Shown here are the genes that were altered at a P value less than .01 and with at least a 2-fold change compared to the three control animal profiles pooled in silico. Note that overall, the gene expression profiles for the rats exposed to the hepatotoxicant show little variability, at least comparable if not better than the serum ALT values. Early in lead optimization efforts, this limited variability enables the use of a low number of animals and hence a much lower quantity of test article, which is clear advantage at a stage where compound amount can be limited, but also from a 3R perspective.
An understanding of gene expression changes can also contextualize some unexpected findings. For example, gene expression analysis provided clarity related to the unexpected plasma glucose and triglyceride lowering effects of inactive experimental Acetyl CoA carboxylase 2 inhibitors through an off-target interactions with PPAR-α. 65 Likewise, the effect of a novel experimental formulation excipient on rat liver and thyroid gland could be rapidly interrogated through liver transcriptomic data, which provided robust evidence that the novel excipient was activating the constitutive active/androstane receptor (CAR), a nuclear receptor that plays a critical role in regulating the activity of hepatic drug metabolizing enzymes. 67
Gene expression data can also be useful as supporting evidence of safety. This is exemplified by the work from the laboratory of one of the authors (E.A.G.B.) that leveraged hepatic gene expression data as a complement to traditional histopathology and serum chemistry endpoints to show that iron deposition in the liver associated with a monoclonal antibody against Repulsive Guidance Molecule C (RGMc) was not associated with genomic, morphological, or functional evidence of toxicologically significant adverse effects on the liver. 5 These data support the safety of using a monoclonal antibody against RGMc as a potential treatment of anemia of chronic disease. Not surprisingly, the utilization of transcriptomic data in support of safety is not as widely published given that there is an inherent bias in publications toward positive findings. The extent of use in the industry for that purpose is not clear. From the perspective of one organization (AbbVie), gene expression data are often used as an additional support for a lack of safety liabilities either in the context of a target evaluation or during the evaluation of exploratory therapeutics.
Gene Network Approaches to Define Mechanisms of Pathogenesis
Supervised models, such as gene signatures, covered elsewhere in this article, have been a major focus in toxicogenomic applications for two decades. In general, these methods use transcriptomic data from sets of chemicals with defined mechanisms of toxicity (the training set) to derive sets of genes that can function as a classifier for new chemicals (the test set). There is no doubt that signature derivation models can identify sets of gene that can be used to classify chemicals with common pharmacological and toxicological properties that have common early effects reflected in gene expression profiles,30,35,40 as suggested at the advent of the toxicogenomic “revolution” in risk assessment. 43 This seems particularly powerful when the pathway leading to an adverse outcome can be clearly linked to an early transcriptional event, and in particular those associated with a ligand-activated transcription factor, for example, PPAR-α. 44
By contrast, pathogenesis of acute and chronic liver injury from many hepatotoxicants is a complex process involving multiple cell types and may require months to years for overt histological evidence of pathology to develop. Given this complexity, one might expect gene expression patterns to differ according to the temporal dynamics for the different cell types and biological processes involved. For example, after acute exposure to a hepatotoxic chemical, gene expression consistent with tissue repair and cell proliferation might be evident while with long-term repeated toxic exposure, activation of stellate cells and deposition of extracellular matrix resulting in fibrosis might be the dominant effect. Moreover, chemicals with different modes of action might be expected to show different patterns of gene expression early while chronic end-stage effects, such as fibrosis, associated with progression to liver failure may have more common histological and gene expression correlates. This complexity complicates supervised approaches to using gene expression patterns to define mechanisms of acute and chronic hepatotoxicity.
Two contemporary survey articles point out that understanding mechanisms of pathogenesis is deemed by practitioners to be among the highest, if not the highest, value output of toxicogenomic analysis.46,62 Interestingly, in the survey of Pettit et al, the same respondents who identified understanding mechanisms as high value also identified extracting that mechanistic information as a significant impediment to realizing that value. This conundrum, that extracting mechanistic information is of high value but high difficulty, suggests that other approaches to toxicogenomics might add value to toxicogenomics analyses if they capture more of the complex biological processes driving the progression of acute injury to tissue degeneration.
Network methods are an attractive approach to modeling biological systems composed of interacting modules of biochemical and biological functions.1,63 Network models capture the modular nature of complex biological systems, including cell and tissues, as sets of highly connected units (modules) of biochemical and biological function. 63 These modules of function are in a dynamic equilibrium under conditions of normal cellular homeostasis and change when cells are forced to adapt to changes in environmental conditions. These modules of function may represent the role of cellular organelles or modular biochemical pathways that work in an integrated manner. For example, cellular organelle functions include the following: mitochondria generate ATP, rough endoplasmic reticulum processes proteins for export, smooth endoplasmic reticulum synthesizes glycogen, lipids, and steroids, and ribosomes assemble proteins. In addition, modular biochemical pathways may include cholesterol biosynthesis and glycolysis that provide substrate for energy production and in the aggregate, the necessary functional building blocks for more complex cellular functions. At a higher level of organization, networks of interactions among different types of cells give rise to complex tissues and organs. These networks, and the modular functions they reflect, may be well defined and connected through biochemical pathways, such as cholesterol biosynthesis, or larger complex networks that reflect the dynamics of cellular function at the level of organelles such as ribosome and mitochondrial biogenesis or DNA replication. At the tissue level, the units of function can be modeled as networks of cell-cell interaction working in concert to achieve a common function.
Assembling the components of a biological network requires that the individual elements (nodes) within the network are connected (edges) by coalescent properties, such as selective protein-protein interactions or protein-substrate interactions, necessary to coordinate formation of modular units of function.8,24 Co-expression in gene networks is a coalescent property of biological systems; genes in the network must change in concert in order to increase or decrease capacity of that modular function. This property of “changing in concert” can be reflected in quantitative measures, such as the Pearson correlation, between two genes in the network providing a value for the connectivity, or edge strength, between the nodes. Examples of network models and the node edge relationship that form the networks can be found in reviews on the application of networks in pharmacology and medicine.1,8
Transcription factors regulate transcriptional networks that coordinate the levels of multiple genes related to a common function. For example, changes in expression of the individual enzyme of the cholesterol biosynthetic pathway are controlled by sterol regulatory element binding proteins (SREBPs) while an increase in expression of antioxidant genes in response to oxidative stress is coordinated by nuclear factor erythroid 2-related factor 2 (NRF2).16,53 Weighted gene co-expression network analysis (WGCNA) is a widely accepted unsupervised method with broad application for identifying co-expressed sets of genes (modules of genes) regulated by transcriptional and epigenetic mechanisms in large datasets.31,51 WGCNA teases out networks of highly connected genes that are up or down regulated together (high Pearson correlation) in response to similar treatments and clusters them into modules of co-expressed genes.31,57 Thus, modules of co-expressed genes are assembled in an unsupervised manner based only on the coalescent property of co-expression necessary to alter levels of function in complex biological systems. The question that remained to be answered was whether these networks reflect known and novel biology relevant to understanding pathogenesis of liver injury.
By 2012, two large transcriptomic datasets from rat liver were available in the public domain, DrugMatrix and TG-GATES providing a unique opportunity for additional modeling approaches. 12 Sutherland et al used the DrugMatrix database to construct modules of co-expressed liver genes that respond to hepatotoxicants and demonstrated that the modules were both enriched for specific biological functions and for targets of specific transcription factors.56,57 These authors also demonstrated that some modules showed high statistical correlation with occurrence of liver pathologies. They further demonstrated the value of reducing the dimensionality of gene expression datasets, which leads to high false discovery rates, 27 and showed that difference in time between experiments with the same compound contribute more to variation in gene expression profiles, 56 further highlighting the importance to time dynamics in understanding the contribution of gene expression to pathogenesis.
Callegaro et al took a similar WGCNA approach to extract modules of co-expressed genes from both human hepatocytes and rat liver transcriptome data using the TG-GATES database.9,10 They also created a public analysis tool, the TXG-MAPr, using an R Shiny framework that allows the user to mine modules for biological function and extract mechanistic information using transcriptomic data from new experiments with uncharacterized hepatotoxicants (https://txg-mapr.eu/). Using this analysis approach, they demonstrate the utility of using human primary hepatocytes networks across different analysis platforms, for example, microarray versus RNA-Seq, and by identifying disease associations across individual liver donors when only partial transcriptomic data are available from targeted expression platform, such as the S1500+. 7
Network approaches are not only useful for understanding mechanisms, but they also address one of the most fundamental and difficult questions in risk assessment, whether a hazard characterized in one system translates to equivalent risk in another. This translational risk assessment approach is facilitated by the ability to quantify node-edge relationship within gene networks. The co-expressed genes (nodes) are connected by the correlation value (edges) defining the network. When the node-edge relationships defined in one system, for example, rat liver, are preserved in another system, for example, human primary hepatocytes, it suggests that the biological function represented by the gene network is also preserved. 32 Callegaro et al used this preservation approach to identify a preserved ATF4 gene network that is associated with occurrence of single cell necrosis (apoptosis) of hepatocytes in rat liver, and is preserved in human primary hepatocytes, to identify Tribbles homolog 3 (TRIB3) as a translational biomarker useful for screening compound with drug-induced liver injury liability in human primary hepatocytes. 10
Co-expression approaches have also been applied to derive gene signatures using a seed gene approach. Podtelezhnikov et al used a combination of clustering, correlation analysis, and linear modeling to show that genes identified in the literature as targets for xenobiotic responsive transcription factors are members of co-expressed gene signatures that can be used to characterize new compounds. 47 Interestingly, they found that rat Cyp1A1 and Cyp1A2 alone were sufficient as signature genes to identify AhR activators; this would have been missed using a WGCNA approach in which 5 is generally used as a minimum number of genes required to constitute a network. In subsequent papers, these authors demonstrate that co-expression gene signatures can be used early in drug development to identify potential hazards that can be avoided prior to long-term toxicology studies 36 and identify a generalized signature for risk of drug-induced liver injury. 21
In conclusion, although latecomers to the field of toxicogenomics, network-based approaches offer considerable value in defining early events linked to key transcriptional drivers of toxic compounds with common mechanisms as well as offering insights into more complex pathogenic processes at the level of whole tissues. Utilizing a common analysis platform, such as the TXG-MAPr, information relevant to pathogenesis and risk assessment can be extracted from new gene expression data and hazards characterized. The approach also enables quantitative translational risk assessment based on the preservation of networks that reflect mechanisms of pathogenesis. Additional benefits of the approach include statistical advantages associated with the reduction in dimensionality of gene expression analysis and the ability to quantify the likelihood that events in one model systems may translate to another enabling translational risk assessment.
Footnotes
Acknowledgements
The authors thank Frank Sistare, Giulia Callegaro, Steven Kunnen, and Bob van de Water for valuable discussions and review of manuscript sections. The authors also thank Alex Merrick (DTT/NIEHS) for his critical review of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by the NIEHS Intramural Research Program (ES103376-02).