
Abstract
The 5 known RecQ helicases in humans (RECQ1, BLM, WRN, RECQL4, and RECQ5) have demonstrated roles in diverse genome maintenance mechanisms but their functions in safeguarding the genome from environmental toxicants are poorly understood. Here, we have evaluated a potential role of WRN (mutated in Werner syndrome) and RECQ1 (the most abundant homolog of WRN) in hydroquinone (HQ)- and benzo[a]pyrene (BaP)-induced genotoxicity. Silencing of WRN or RECQ1 expression in HeLa cells increased their sensitivity to HQ and BaP but elicited distinct DNA damage response. The RECQ1-depleted cells exhibited increased replication protein A phosphorylation, Chk1 activation, and DNA double-strand breaks (DSBs) as compared to control or WRN-depleted cells following exposure to BaP treatment. The BaP-induced DSBs in RECQ1-depleted cells were dependent on DNA-dependent protein kinase activity. Notably, loss of WRN in RECQ1-depleted cells ameliorated BaP toxicity. Collectively, our results provide first indication of nonredundant participation of WRN and RECQ1 in protection from the potentially carcinogenic effects of BaP and HQ.
Keywords
Introduction
Exposure to environmental and chemical toxicants pose serious threats to genomic integrity. Polycyclic aromatic hydrocarbons (PAHs), including hydroquinone (HQ) and benzo[a]pyrene (BaP), comprise a large group of environmental organic pollutants and potent carcinogens. 1 Polycyclic aromatic hydrocarbons are present ubiquitously in air, drinking water, tobacco smoke, and grilled or broiled food; and PAHs are metabolized upon their ingestion to more toxic or carcinogenic metabolites that can react with cellular macromolecules. 2 Benzo[a]pyrene is a major carcinogen in cigarette smoke, a leading risk factor for a variety of cancers. 3,4 Indeed, exposure to PAHs has been shown to induce oxidative damage, formation of adducts, and strand breaks in DNA. 2 Despite the fact that PAHs are highly genotoxic, the molecular details of cellular response that combat DNA damage following PAHs exposure are yet elusive.
The RecQ helicase family is a group of highly conserved DNA unwinding enzymes critical in guarding genome stability in all kingdoms of life. 5,6 Human RecQ homologs include RECQ1 (also known as RECQL or RECQL1), BLM, WRN, RECQL4, and RECQ5, 3 of which have been linked to diseases with elevated risk of cancer and growth defects (Bloom syndrome and Rothmund-Thomson syndrome) or premature aging (Werner syndrome). Human RecQ proteins contribute diverse biochemical activities to metabolize toxic DNA structure intermediates that are formed in the genome spontaneously or by the action of extrinsic mutagens. 5,7 A previous study suggested that the functions of RECQ1, the most abundant RecQ protein in humans, and WRN, the protein deficient in Werner syndrome, may be similarly affected by BaP-induced DNA damage that is known to promote mutations by causing replication errors and is associated with cancer development. 8 Specifically, in vitro helicase activity of WRN and RECQ1 proteins was profoundly inhibited by a BaP diolepoxide adduct with the N2 position of guanosine, which is formed upon metabolic activation of BaP in vivo. 8 The BaP-DNA adducts are partially resistant to cellular repair processes, 9 block replication fork progression, and activate the checkpoint kinases ATR and Chk1 in intact cells. 10 A very recent study shows that BaP increases double-strand break (DSB) repair. 11 WRN and RECQ1 are each involved in the repair of stalled and broken replication forks, 12,13 although the loss of RECQ1 or WRN contributes differently to the repair of DSB by homologous recombination (HR). 14
Increased HR is also reported in response to oxidative stress induced by benzene metabolites including HQ. 15 Hydroquinone-induced DNA damage is mediated by the formation of reactive oxygen species. 16 Furthermore, HQ can be metabolized to potentially genotoxic and carcinogenic benzoquinones that can also induce the formation of free radical, predisposing cells to oxidative damage. 17 Genetic association studies have previously suggested a link between WRN and susceptibility to benzene-induced toxicity. 18 Depletion of WRN is reported to enhance DNA damage in HeLa cells exposed to HQ, suggesting that WRN plays a key role in benzene toxicity. 19,20 RECQ1 has also been associated with benzene poisoning, 21 but its impact on cell survival following HQ exposure is yet unknown.
Deficiency of WRN or RECQ1 results in increased sensitivity to a variety of genotoxic agents, accumulation of DNA damage, and chromosomal instability associated with cancer predisposition. 5,22,23 Despite the genetic associations of WRN and RECQ1 with cancers, 23 –25 their roles in governing cellular response to environmental carcinogens have remained poorly characterized. Given the diverse roles of RecQ helicases in nucleic acid metabolism, 23 they are likely to be critical in counteracting adverse effects of DNA lesions caused by BaP and HQ. To assess the importance of RECQ1 and WRN in genome surveillance mechanisms activated upon exposure to BaP or HQ, we determined survival and DNA damage response to HQ and BaP treatment in RECQ1- and WRN-depleted cells. HeLa cells have been previously used to investigate functions of RecQ helicases 14,26 –28 and mechanisms of HQ 20 or BaP toxicity 4,11 ; therefore, we used HeLa cells as the experimental model.
Materials and Methods
Chemicals
Benzo[a]pyrene, HQ, dimethyl sulfoxide (DMSO), NU7026 (DNA-dependent protein kinase [DNA-PK] inhibitor), and KU55933 (ataxia telangiectasia mutated [ATM] inhibitor) were purchased from Sigma (St. Louis, Missouri). The stock solutions of BaP, NU7026, and KU55933 were made in DMSO. Hydroquinone stock was made in phosphate-buffered saline (PBS).
Cell Culture, RNA Interference, and DNA Damage Treatment
HeLa cells (ATCC) were maintained in Dulbecco modified Eagle medium (DMEM; Invitrogen, Carlsbad, California) supplemented with 10% fetal bovine serum (Hyclone Laboratories, Logan, Utah), 100 IU/mL penicillin, and 100 µg/mL streptomycin (Invitrogen) at 37°C in a humidified incubator with 5% CO2. Depletion of WRN and RECQ1 was achieved by reverse transfection of WRN or RECQ1 small interfering RNA (siRNA; 10 nmol/L, siGenome smartpool; Dharmacon, Logan, Utah) using RNAiMax per the manufacturer’s instructions (Invitrogen); HeLa cells transfected with scrambled control siRNA were used as control. Cells were treated with various concentrations of HQ or BaP as indicated.
Cell Proliferation
Twenty-four hours after siRNA transfection, HeLa cells, seeded at a density of 3 × 103 cells per well in 96-well plates, were treated with HQ or BaP in complete DMEM for indicated time. Cell proliferation was evaluated each day by trypan blue exclusion assay and counting the viable cells using an automated cell counter (Bio-Rad TC10, Bio-Rad, Hercules, California). Results presented here are from at least 3 independent experiments performed in triplicates.
Western Blotting
Forty-eight hours after siRNA transfection, cells were treated with HQ or BaP for 24 hours as indicated. Where applicable, NU7026 (20 µmol/L) and KU55933 (10 µmol/L) were added 2 hours before BaP treatment to inhibit DNA-PK and ATM, respectively; 0.1% DMSO was added to untreated cells. Subsequently, cells were washed with cold PBS and lysed in radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl [pH 8.0], 150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing protease and phosphatase inhibitors (Roche, Indianapolis, Indiana). Equal amounts of total protein for each sample was used for immunoblotting with antibodies against RECQ1 (1:1000) and WRN (1:500; Santa Cruz Biotechnology, Dallas, Texas), replication protein A (RPA) 32, γH2AX, phospho-Chk1 Ser345, phospho-Chk2 Thr68, and glyceraldehyde 3-phosphate dehydrogenase (all 1:1000; Cell Signaling, Danvers, Massachusett). Images were captured using GeneGnome XRQ Chemidoc System (Syngene, Frederick, Maryland) and signal intensities were quantified using ImageJ.
Results
Depletion of WRN or RECQ1 Promotes Cellular Sensitivity to HQ Treatment
Previously, Galvan et al have shown that siRNA depletion of WRN in HeLa cells leads to enhanced susceptibility to HQ (100 µmol/L) cytotoxicity. 20 Thus, to directly compare the contribution of WRN and RECQ1 in cellular response to HQ exposure, we utilized individual siRNA knockdown of these proteins in HeLa cells. Efficient knockdown of WRN or RECQ1 protein, detected 36 hours after siRNA transfection, was maintained at 72 hours (Figure 1A). Thirty-six hours after transfection, cells were exposed to HQ for 3 days, and survival was determined by trypan blue exclusion assay. As reported previously, HQ induced significant cytotoxicity, which was further enhanced by WRN depletion. 20 Notably, RECQ1 depletion also resulted in reduced survival following HQ treatment. As compared to cell survival without any treatment, HQ treatment (50 µmol/L, 72 hours) resulted in ∼25, 50, and 70% survival of WRN, RECQ1, and control siRNA-transfected cells, respectively (Figure 1B). This indicates that both WRN and RECQ1 contribute to the survival after HQ exposure but the role of WRN may be more critical in dealing with HQ-induced toxicity.
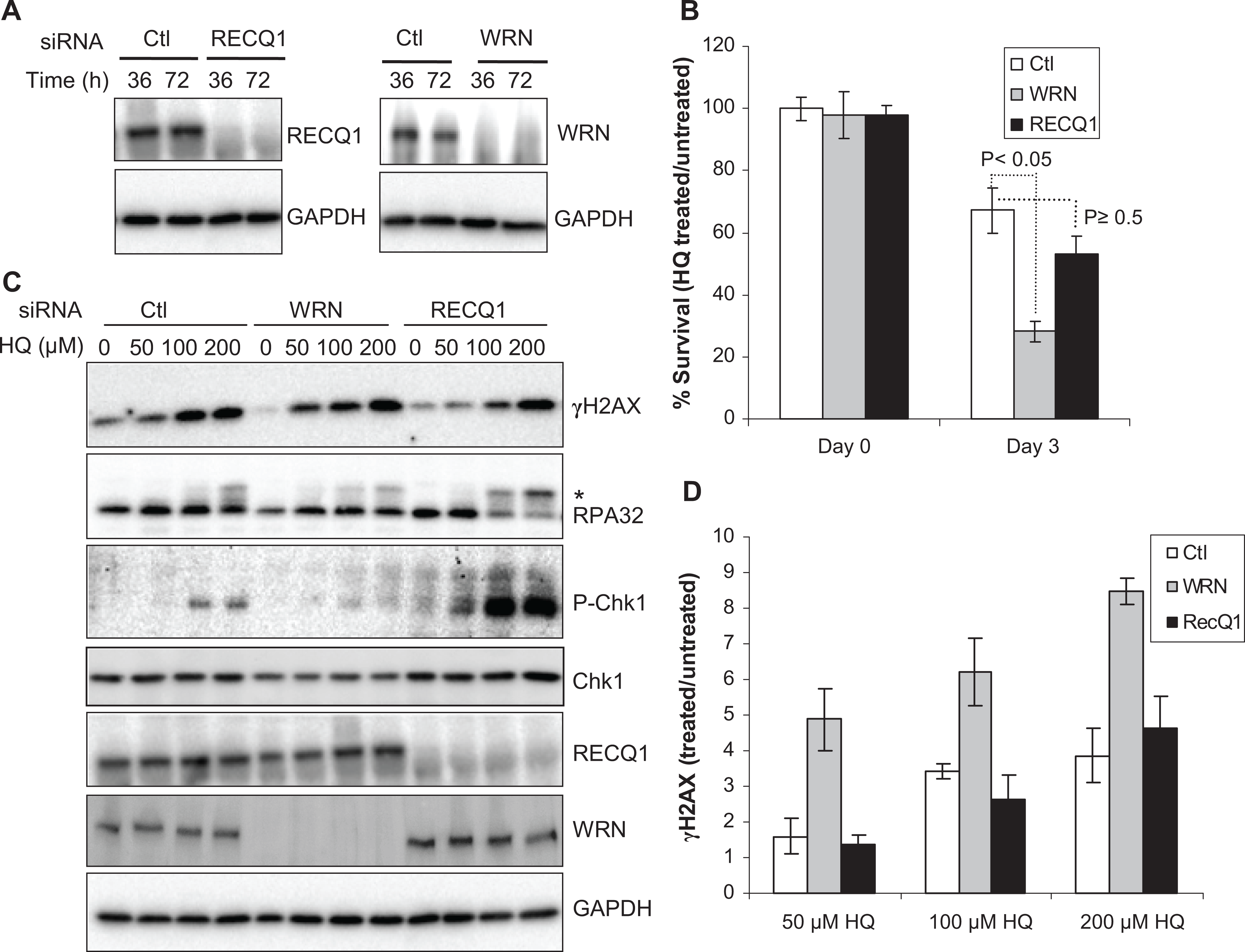
Cell survival and response to HQ treatment. A, Depletion of RECQ1 and WRN in HeLa cells. Western blot shows efficient knockdown of WRN or RECQ1. B, Cell survival following HQ treatment (50 µmol/L, 72 hours). Surviving fraction is presented as the mean ± SD of 3 independent experiments. Statistical significance of differences in survival is indicated by P value. C, DNA damage response. Cells were treated with indicated dose of HQ for 24 hours, and equal amounts of total protein were used for Western blotting with indicated antibodies. Phosphorylated replication protein A (RPA) 32 is indicated by asterisk. D, HQ-induced γH2AX levels. Signals intensities, quantitated using ImageJ, were normalized with GAPDH, and the fold enrichment in γH2AX was calculated. Mean ± SEM from 3 independent experiments is shown. GAPDH serves as loading control. Ctl indicates control; pChk1, phospho-Chk1(Ser345); GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HQ, hydroquinone; SD, standard deviation; SEM, standard error of the mean.
Hydroquinone Induces Greater Phosphorylation of Chk1 and RPA in RECQ1-Depleted Cells
Cytotoxicity of benzene metabolites has been associated with a variety of DNA damage. 29 In fact, treatment with HQ has been shown to induce a dose-dependent phosphorylation of H2AX at Ser139 (γH2AX), which is widely used as surrogate marker of DSBs. 30 To test whether WRN and RECQ1 regulate the formation of HQ-induced DSBs, we determined γH2AX levels in lysates prepared from control, WRN-, or RECQ1-depleted cells that were either untreated or treated with HQ. Consistent with a previous report in HL-60 cells, 30 HQ treatment induced DSBs in control HeLa cells in a dose-dependent manner; HQ treatment resulted in a greater induction of γH2AX in WRN-depleted cells, whereas the level of γH2AX in RECQ1-depleted cells was similar to that in control cells (Figure 1C and D). As compared to their individual untreated condition, exposure to HQ (50 µmol/L) resulted in ∼1.6-, 4.8-, and 1.3-fold increase in γH2AX signal in control, WRN-, or RECQ1-depleted cells, respectively (Figure 1D).
Benzene exposure activates checkpoint proteins such as p21 and p53. 31 p21 cooperates with Chk1 to prevent apoptosis during replication stress. 32 Activation of Chk1 by ATR-dependent phosphorylation is central to the normal DNA damage response to prevent excessive formation of DSBs during replication arrest 33 and the RPA-coated single-stranded DNA is the predominant signal for activation of the ATR-Chk1 signal transduction pathway. 34 Given the critical roles of RecQ helicases in genome maintenance during replication stress, 7 we examined the status of Chk1 phosphorylation at Ser345 and RPA32 phosphorylation in control, WRN-, or RECQ1-depleted cells exposed to HQ (Figure 1C). In control HeLa cells, HQ treatment induced a limited phosphorylation of Chk1, and a detectable signal was observed at >50 µmol/L HQ. This is consistent with a previous observation that treatment with HQ or phenyl-HQ, which shares similar molecular structure, did not cause phosphorylation of Chk1 in Saccharomyces cerevisiae. 35,36 A significantly more robust Chk1 activation and phosphorylated RPA32 were observed in RECQ1-depleted cells as compared to control or WRN-depleted cells (Figure 1C). In contrast, WRN-depleted cells did not induce appreciable Chk1 activation following HQ treatment and exhibited less RPA32 phosphorylation than control cells (Figure 1C). Exposure to HQ (up to 200 µmol/L) did not induce a detectable Chk2 phosphorylation in HeLa cells and this pattern was not altered by silencing of either RECQ1 or WRN (data not shown), indicating that the ATM–Chk2 pathway may not be significantly involved in cellular response to HQ treatment. 35,36
Depletion of WRN or RECQ1 Enhances Cytotoxicity From Exposure to BaP
We next determined relative responses of RECQ1- or WRN-depleted cells to BaP treatment, which induces DNA adducts shown to block replication fork progression. 37 In preliminary experiments, we determined that 48 hours treatment with 1 μmol/L BaP led to minimal (<10%) loss in cell viability in HeLa cells using trypan blue exclusion assay. HeLa cells that were transfected with control siRNA maintained ∼96% survival following treatment with BaP (1 μmol/L, 48 hours; Figure 2A). Depletion of RECQ1 or WRN enhanced cytotoxicity of BaP; notably, RECQ1-depleted cells displayed greater sensitivity to BaP than WRN-depleted cells. As compared to their respective untreated condition, BaP treatment reduced survival to ∼64% in WRN-depleted cells and to ∼42% in RECQ1-depleted cells (Figure 2A).
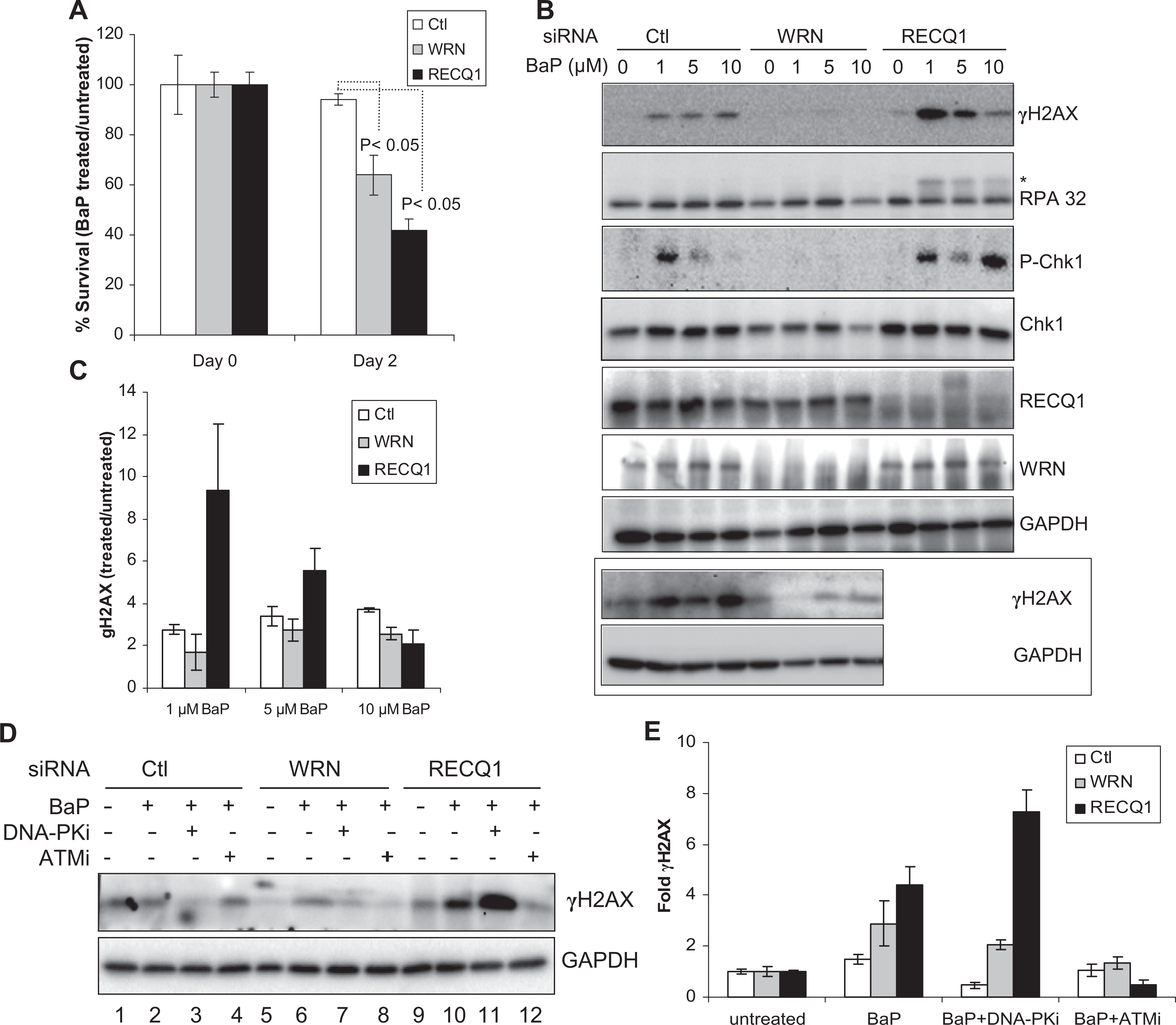
Cell survival and response to BaP treatment. A, Cell survival following BaP treatment (1 µmol/L, 48 hours). Surviving fraction is presented as the mean ± SD of 3 independent experiments. Statistical significance of differences in survival is indicated by P value. B, DNA damage response. Lysates prepared from cells treated with indicated dose of BaP for 24 hours were used for Western blotting with indicated antibodies. Western blot of γH2AX in control and WRN-depleted cells from an independent experiment is included in boxed panel. Phosphorylated replication protein A (RPA) 32 is indicated by asterisk. C, BaP-induced γH2AX levels. Relative fold enrichment in γH2AX was quantitated as in Figure 1D, and mean ± SEM from 3 independent experiments is shown. D, Pharmacological inhibition of ATM or DNA-PK prior to BaP exposure. Lysates prepared from cells treated with BaP (1 µmol/L, 24 hours) with or without pretreatment with indicated kinase inhibitor were used for Western blotting with indicated antibodies. E, Fold change in γH2AX. Relative fold enrichment in γH2AX was quantitated from experiments represented in panel (D) after normalizing with GAPDH. Data represent mean of 3 independent experiments with SEM shown by error bars. GAPDH serves as loading control. Ctl indicates control; DNA-PKi, DNA-dependent protein kinase inhibitor; ATMi, ataxia telangiectasia mutated inhibitor; BaP, benzo[a]pyrene; SD, standard deviation; SEM, standard error of the mean.
Consistent with its known genotoxic potential, BaP treatment induced DSBs as indicated by γH2AX (Figure 2B and C). Treatment with BaP (1 μmol/L) induced a greater γH2AX signal (∼9-fold) in RECQ1-depleted cells than in control cells (∼2.5-fold). Unexpectedly, however, increasing BaP dose (5 and 10 μmol/L) resulted in a progressive decline in γH2AX signal in RECQ1-depleted cells, whereas γH2AX formation in control cells did not change appreciably (Figure 2B and C). In contrast, WRN-depleted cells displayed relatively similar γH2AX signal as compared to control cells at BaP dose of 1 and 5 μmol/L (Figure 2B and C). Benzo[a]pyrene treatment did not induce appreciable Chk1 activation in WRN-depleted cells as determined by phospho-Chk1 (Figure 2B). Comparable Chk1 activation was observed in control or RECQ1-depleted cells treated with 1 μmol/L BaP but increasing the BaP dose to 5 μmol/L resulted in reduced phospho-Chk1 signal in control or RECQ1-depleted cells (Figure 2B); treatment with BaP (10 μmol/L) lead to phospho-Chk1 signal exclusively in RECQ1-depleted cells, indicating a biphasic response (Figure 2B). Benzo[a]pyrene treatment induced only a modest phosphorylation of RPA32 in control cells, whereas increased RPA phosphorylation was observed in RECQ1-depleted cells at all concentrations of BaP tested (Figure 2B). Overall, these analyses revealed distinct DNA damage response to BaP treatment in control, WRN-, or RECQ1-depleted cells. The dose–response relationships between BaP exposure and checkpoint activation appear to be more complex and could not be established within these experiments.
Ataxia telangiectasia mutated and DNA-PK Modulate BaP-Induced DSBs
Members of the phosphatidylinositol 3-kinase family, including ATM, ATR, and DNA-PK, differentially contribute to the BaP-induced H2AX phosphorylation. 38 Thus, we utilized small molecule inhibitors of DNA-PK and ATM and examined the level of BaP-induced γH2AX in control and RECQ1- or WRN-depleted cells. Preincubation with a specific chemical inhibitor of DNA-PK (NU7026), but not ATM (KU55933), reduced BaP-induced γH2AX in control HeLa cells (Figure 2D and E). Inhibition of either DNA-PK or ATM reduced overall γH2AX level in WRN-depleted cells, but ATM inhibitor was more efficient (Figure 2D and E). While ATM inhibition also reduced BaP-induced γH2AX in RECQ1-depleted cells, inhibition of DNA-PK activity prior to BaP exposure led to increased γH2AX signal in RECQ1-depleted cells (Figure 2D and E). Our data indicating that DNA-PK activity is critical to the repair of BaP-induced damage are supported by a recent report that DNA-PK plays a major role in Chk1 activation in response to bulky DNA damage. 39 Our observations also indicate a possible switch, which is largely dependent on RECQ1 level, between the DNA-PK and ATM signaling when cells are exposed to BaP.
Concurrent Knockdown of WRN in RECQ1-Depleted Cells Reduces BaP Sensitivity
To determine the effect of simultaneous depletion of WRN and RECQ1 proteins on cellular sensitivity and response to BaP treatment, we cotransfected cells with siRNA against RECQ1 and WRN and compared them with those transfected either with RECQ1 siRNA or control siRNA. In these experiments, HeLa cells that were transfected with control siRNA maintained ∼89% survival following treatment with BaP (1 μmol/L, 48 hours; Figure 3A). As compared to their respective untreated condition, BaP treatment reduced survival to ∼51% in RECQ1-depleted cells and to ∼78% in cells codepleted of RECQ1 and WRN (Figure 3A). Consistent with their improved survival, codepletion of RECQ1 and WRN led to reduced γH2AX and reduced RPA phosphorylation as compared to RECQ1 depletion (Figure 3B). These results suggest that knockdown of WRN in RECQ1-depleted HeLa cells ameliorates BaP-induced DNA damage and improves survival.
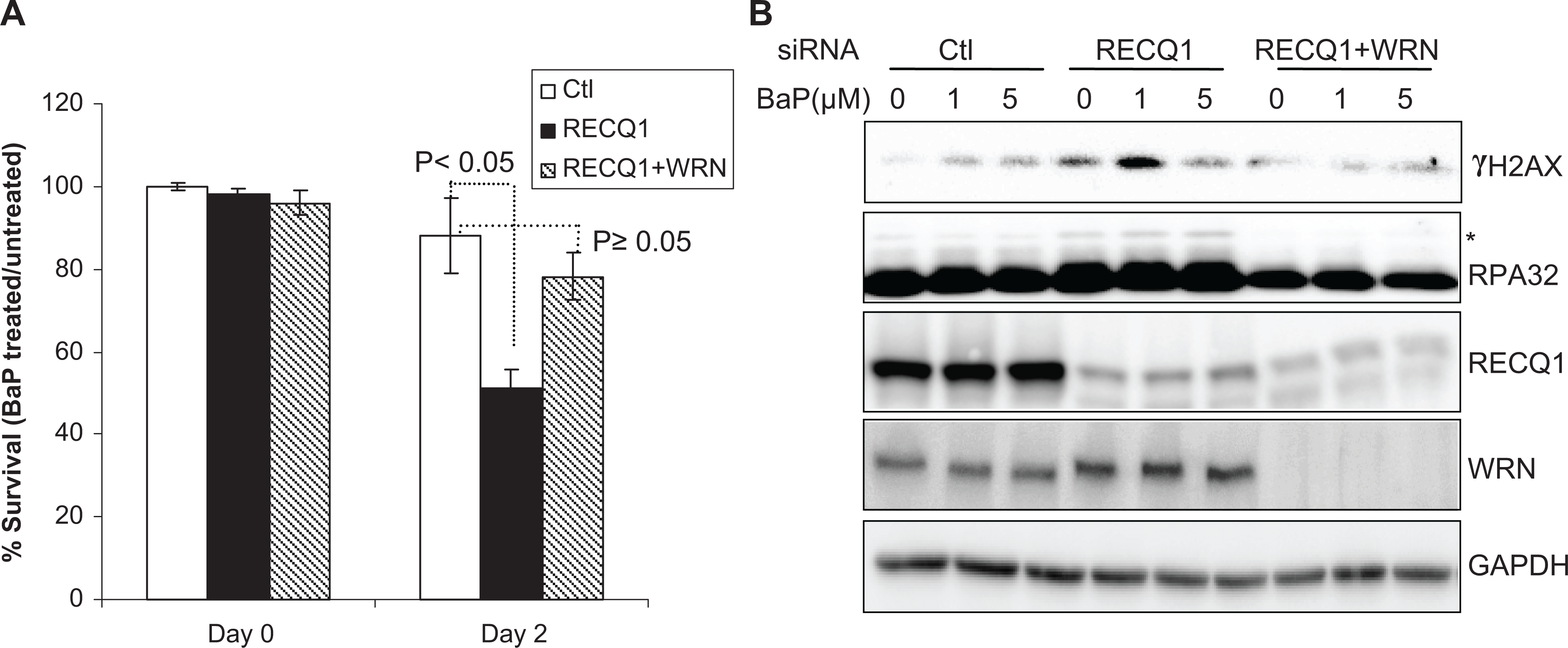
Concurrent depletion of WRN reduces genotoxicity of BaP in RECQ1-deficient cells. A, Cell survival following BaP treatment (1 µmol/L, 48 hours). Surviving fraction in each case is presented as the mean ± SD of 3 independent experiments. B, DNA damage response. Lysates prepared from cells treated with indicated dose of BaP for 24 hours were used for Western blotting with indicated antibodies. GAPDH serves as loading control. BaP indicates benzo[a]pyrene; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; SD, standard deviation.
Discussion
Polycyclic aromatic hydrocarbons are ranked among the top 10 in the national priority list of hazardous substances which are determined to pose the most significant potential threat to human health due to their known or suspected toxicity and potential for human exposure (http://www.atsdr.cdc.gov/spl/). We provide first evidence that the cellular levels of individual RecQ helicase proteins contribute to the genotoxicity induced by these agents. Our results have potentially significant implications for understanding genome surveillance and protection mechanisms provided by WRN and RECQ1 in response to HQ and BaP exposure. Furthermore, our results support the notion that functional compensation by redundant and alternative mechanisms involving multiple RecQ proteins ensures genomic stability in humans.
Increased intracellular production of reactive oxygen species by HQ exposure has been implicated in the formation of DSBs, subsequent repair of which is likely to involve HR. 15 Defects in HR are known to be exacerbated in the presence of oxidative stress. 14 WRN has an established role in the repair of oxidative DNA lesion; consequently, WRN-deficient cells accumulate oxidative damage and are HR defective. 12 Earlier work had shown that WRN protects cells from DNA damage induced by HQ. 19,20 Our results suggest that in the absence of WRN, DSBs accumulate leading to enhanced cytotoxicity of HQ. In contrast, RECQ1-depleted cells that exhibit relatively normal DSB-induced HR 14 displayed reduced γH2AX induction upon HQ treatment. Phospho-Chk1 was negligible in WRN-depleted cells, while it was hugely induced in RECQ1-depleted cells. This indicates that these 2 members of the RecQ family are involved in different pathways of HQ-induced oxidative DNA damage repair. Since phospho-Chk1 senses stalled replication forks, an interpretation of our results may be that in the absence of WRN either stalled forks are not formed or they are preferentially resolved into DSBs; alternatively, WRN may be required for activation of ATR-Chk1 pathway, while RECQ1 appears to suppress it. Increased HQ-induced γH2AX in WRN-depleted cells is consistent with increased apopotosis 19 since initiation of DNA fragmentation during apoptosis also leads to phosphorylation of H2AX at Ser 139. 40 Although the mechanisms of HQ-induced DNA damage are poorly understood, our results are consistent with previously documented differences in WRN- or RECQ1-deficient cells in oxidative stress response 14 and warrant future investigation of the potential roles of RECQ1 and WRN in ATR-Chk1-dependent DNA damage checkpoint activation following HQ exposure as the ATR-Chk1 checkpoint activation has been recently implicated in oxidative stress-induced DNA damage repair. 41
The roles of RecQ proteins in checkpoint activation and fork restart are distinct and dependent on the causative source of genotoxic stress. 23,42 Our data do not exclude the possibility that WRN may be differentially involved in replication stress checkpoint signaling induced by HQ or BaP exposure. Benzo[a]pyrene-DNA adducts activate Chk1 signaling, which inhibits association of Cdc45 with origins of replication thereby suppressing initiation of late origins, 43 and stabilize stalled replication forks. 33 Regression of stalled forks allows the leading strand, which could not be synthesized beyond adduct (lesion), to utilize the newly synthesized lagging strand as template and continue the extension of the blocked polymerase. Reversal of regressed fork restores replication fork progression beyond the lesion (Figure 4). 23,43 As this allows time for the lesion to be repaired, fork reversal is implicated in preventing chromosome breakage upon exogenous replication stress. 44 Reverse branch migration of the regressed fork potentially allows replication bypass of the lesion in a nonrecombinogenic mode (Figure 4). 23 Alternatively, or in addition, regressed fork may be cleaved by structure-specific nucleases to generate a DSB, which is subsequently repaired by HR (Figure 4). 23 Both RECQ1 and WRN are recruited at arrested replication forks. 45 Demonstrated preference of RECQ1, but not WRN, for fork reversal 13 supports increased DSBs in RECQ1-depleted cells exposed to BaP. Our observation that loss of WRN suppresses requirement for RECQ1 suggests that WRN helicase may generate DNA structures that are toxic to the cell if RECQ1 isn’t available. Role of WRN in BaP-induced toxicity may involve resolution of recombinational intermediates as well as checkpoint regulation and preventing fork collapse. 46 It is plausible that RECQ1 and WRN, through their preferred biochemical activities, 23 are essential to maintain fork progression in the presence of BaP-induced replication-blocking lesions, and alternate mechanisms of DNA damage tolerance such as translesion DNA synthesis (TLS) and postreplicative homology-directed repair contribute to cellular resistance to BaP in their combined absence. 47 WRN interacts with TLS polymerases 48 and participates in the TLS pathway to prevent genomic instability 49 but the involvement of RECQ1 in TLS and replication-induced HR remains to be determined.
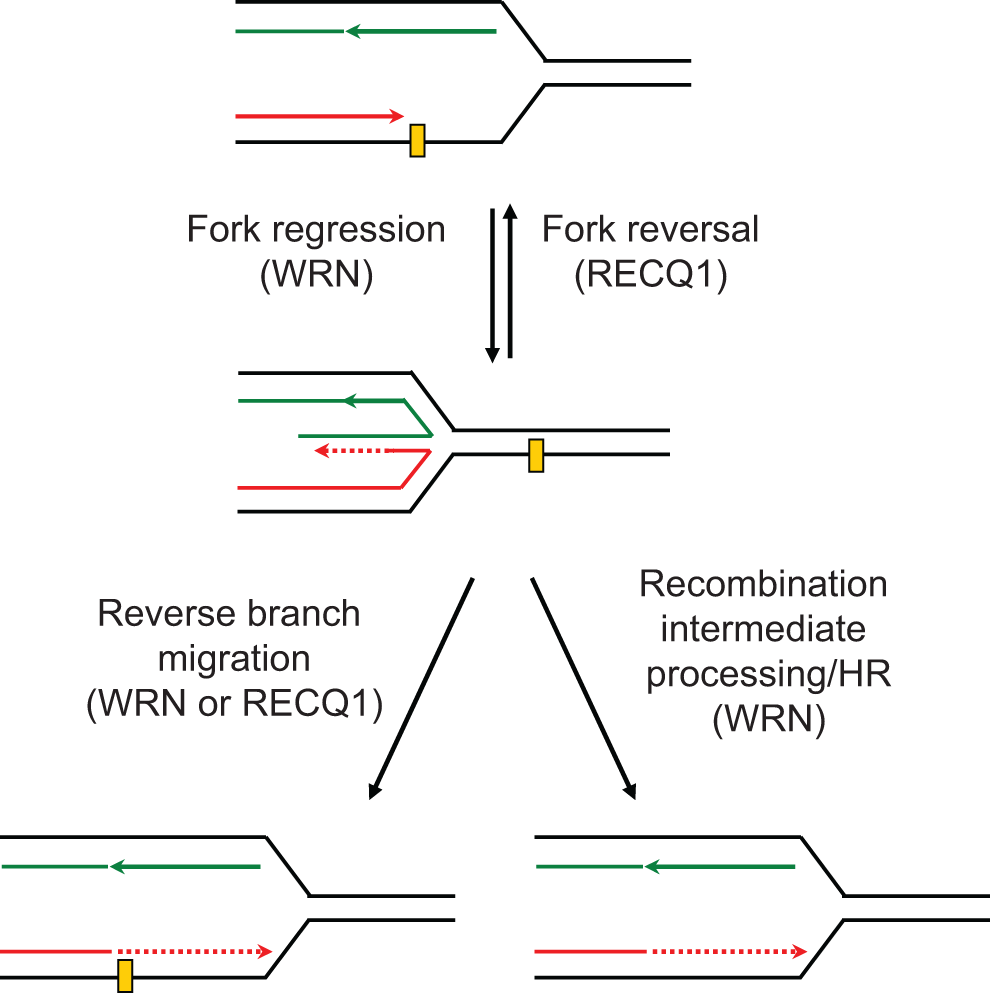
Proposed roles of RECQ1 and WRN in replication fork restart. Known biochemical activities are implicated in this proposed model described in the “Discussion” section.
In conclusion, results presented here provide first evidence of differential contribution of RecQ homologs WRN and RECQ1 in dealing with DNA lesions induced by cellular exposure to HQ and BaP, representative of an important class of environmental carcinogens. These RecQ helicase homologs may sense particular types of DNA damage and facilitate DNA repair mechanisms by specific protein–protein interactions. 5,23 Therefore, the presence of multiple RecQ homologs in humans may be an important safeguarding mechanism to protect from PAH-induced genomic instability.
Footnotes
Authors’ Note
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contribution
M. Garige contributed to acquisition, analysis, or interpretation; gave final approval; and agrees to be accountable for all aspects of work ensuring integrity and accuracy. S. Sharma contributed to conception or design, acquisition, analysis, or interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MG is supported by the Edward Bouchet Scholars Fellowship (Howard University Graduate School). This research was supported by the NIGMS/NIH under award number SC1GM093999 (to SS) and the NIMHD/NIH under award number G12MD007597.