
Abstract
N1-Benzylated dihydroquinolin-6-ols and their corresponding esters display exceptional activity against African trypanosomes in vitro, and administration of members of this class of compounds to trypanosome-infected mice results in cures in a first-stage African trypanosomiasis model. Since a quinone imine intermediate has been implicated in the antiparasitic mechanism of action of these compounds, evaluation of the hepatotoxic, mutagenic, and methemoglobin-promoting effects of these agents was performed. 1-Benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol hydrochloride and 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate showed outstanding in vitro selectivity for Trypanosoma brucei compared to the HepG2, Hep3B, Huh7, and PLC5 hepatocyte cell lines. 1-Benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol hydrochloride and 1-(2-methoxybenzyl)-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate were not mutagenic when screened in the Ames assay, with or without metabolic activation. The latter 2 compounds promoted time- and dose-dependent formation of methemoglobin when incubated in whole human blood, but such levels were below those typically required to produce symptoms of methemoglobinemia in humans. Although compounds capable of quinone imine formation require careful evaluation, these in vitro studies indicate that antitrypanosomal dihydroquinolines merit further study as drug candidates against the neglected tropical disease human African trypanosomiasis.
Introduction
Human African trypanosomiasis (HAT) is a parasitic disease spread by the bite of tsetse flies infected with Trypanosoma brucei gambiense or T. brucei rhodesiense. Cycles of HAT epidemics have been reported in sub-Saharan Africa since the time that David Bruce, Aldo Castellani, and others first described the protozoans that cause this disease at the beginning of the 20th century. As recently as 1999, 450 000 cases of HAT were believed to occur annually. 1 The incidence of HAT has shrunk to new lows over the last few years; 7216 cases were reported in 2012 (http://www.who.int/mediacentre/factsheets/fs259/en/). Many more cases are likely to occur than are actually reported, however, 2 and this underreporting of HAT cases and the tendency of HAT to reappear in epidemic fashion are causes for continued vigilance regarding the disease. The World Health Organization recently launched a plan to eliminate HAT. 2 Such efforts to reduce the number of HAT cases to a point where no disease occurs in humans depend on the availability of effective drugs.
During first-stage HAT, parasites are found in the blood and lymph, and infections can be treated effectively using the venerable drugs pentamidine (for T. b. gambiense infection) and suramin (for T. b. rhodesiense infection). Ultimately fatal neurological symptoms of HAT are caused by penetration of the parasites into the central nervous system, however, and the first-stage drugs are ineffective for treating this second stage of the disease. Melarsoprol, an organoarsenical drug introduced over 60 years ago, was the only option for treating second-stage disease for many years. Administration of melarsoprol causes fatal encephalitis in approximately 5% of patients with HAT, 3 and the antiparasitic efficacy of this drug has been diminished due to the development of resistance. 4 More recently, a combination of the injectable drug eflornithine, an ornithine decarboxylase inhibitor, and the oral drug nifurtimox, a nitrofuran, has been implemented as the preferred treatment for second-stage HAT caused by T. b. gambiense. 5 Nonetheless, this regimen requires 14 injections of eflornithine in relatively high doses and is not effective against second-stage disease caused by T. b. rhodesiense. The new oral drug candidates fexinidazole, another nitroheterocycle, and the novel oxaborole SCYX-7158 are currently in clinical trials for treating HAT, 6 but neither has been approved as yet. Continued research is needed to identify drug candidates against HAT to provide therapeutic options in the event of the failure of the compounds currently in trials and/or a resurgence in the number of HAT cases.
Dihydroquinolines have displayed exceptional in vitro potency and selectivity and promising in vivo efficacy in an animal model of first-stage African trypanosomiasis. 7,8 Among these compounds, administration of 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol hydrochloride (OSU-36.HCl, Figure 1) and 1-(2-methoxybenzyl)-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate (OSU-75) at intraperitoneal (ip) doses of 50 mg/kg/d for 5 days resulted in cures in a murine first-stage trypanosomiasis model. 8 The structural feature that is essential for antitrypanosomal activity in this series of molecules is a hydroxyl group at C6 of the 2,2,4-trimethyldihydroquinoline ring system. Such compounds are thus capable of forming a quinone imine intermediate via 2 electron oxidation. This intermediate could then generate reactive oxygen species through single electron reduction and subsequent redox cycling mediated by molecular oxygen, resulting in the production of superoxide. Support for this mechanistic hypothesis was provided by electron spin resonance experiments that demonstrated the formation of radicals when T. b. brucei was treated with dihydroquinoline 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate (OSU-40). RNA interference (RNAi) studies also showed that parasites displaying RNAi-induced decreased expression of the antioxidant enzymes superoxide dismutase and trypanothione synthetase were more susceptible to OSU-40 than wild-type parasites. 9 Trypanosomes may be more susceptible to such compounds due to their antioxidant defense system. This system lacks catalase and thioredoxin reductase and contains trypanothione rather than glutathione, differing from host cells. 10,11
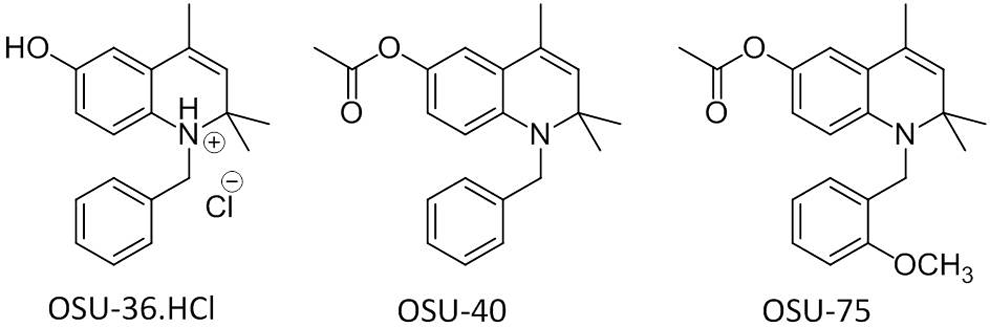
Structures of antitrypanosomal dihydroquinolines.
Given the need for new drug candidates against HAT, dihydroquinolines merit further investigation due to their promising in vitro and in vivo activities. Undesirable side effects could occur in the host due to the formation of a quinone imine species, however. The hepatotoxic side effects caused by acetaminophen and diclofenac are likely to be caused by their conversion to quinone imine metabolites. 12 Quinones derived from the metabolism of estrogens can form adducts with DNA, resulting in genotoxicity due to depurination, and such quinones may also cause genotoxicity through the production of reactive oxygen species formed via redox cycling. 13 Primaquine is an antimalarial drug that is known to cause hemolytic anemia in persons deficient in glucose-6-phosphate dehydrogenase. 5-Hydroxyprimaquine, a primaquine metabolite capable of forming a quinone imine intermediate, has been shown to induce methemoglobin formation in rat erythrocytes and hemolysis in glutathione-depleted red blood cells. 14 Careful assessment of the safety profile of lead dihydroquinolines is therefore necessary. In this study, the evaluation of key antitrypanosomal dihydroquinolines has been assessed for toxicity to human hepatic cell lines, for mutagenic potential in the 2-strain Ames screen, and for their ability to promote methemoglobin formation in human whole blood.
Materials and Methods
Chemicals
OSU-36.HCl, OSU-40, and OSU-75 were prepared as described previously. 7,8 1-Benzyl-6-chloro-2,2,4-trimethyl-1,2-dihydroquinoline (JR-I-78) was synthesized as outlined in Scheme 1. Briefly, a solution of 4-chloroaniline (1.0 g, 7.9 mmol), acetone (1.2 mL), and iodine (202 mg, 0.79 mmol) was heated to reflux in toluene (10 mL) for 36 hours. The reaction mixture was cooled, washed with brine, and the solvent was removed. The residue was purified on silica gel using hexanes/dichloromethane (1:1) as the mobile phase to yield 6-chloro-2,2,4-trimethyl-1,2-dihydroquinoline (742 mg, 45%). Proton nuclear magnetic resonance (1H-NMR; 300 MHz, CDCl3) δ 1.28 (6H, s), 1.98 (3H, s), 3.70 (1H, s), 5.36 (1H, s), 6.37 (1H, d, J = 8.4), 6.94 (1H, d, J 1 = 8.4 Hz, J 2 = 2.3 Hz), 7.02 (1H, d, J = 2.3 Hz). 6-Chloro-2,2,4-trimethyl-1,2-dihydroquinoline (500 mg, 2.4 mmol) was added to benzyl bromide (4.3 mL, 3.6 mmol) in toluene (50 mL) containing triethylamine (503 μL, 3.6 mmol). The reaction mixture was allowed to stir at room temperature for 24 hours, then the solvent was evaporated under reduced pressure. The residue was purified on a silica gel column using hexanes/ethyl acetate (6:1) as the mobile phase to yield JR-I-78 as yellow crystals (256 mg, 36%). 1H-NMR (300 MHz, CDCl3) δ 1.39 (6H, s), 2.02 (3H, s), 4.50 (2H, s), 5.40 (1H, s), 6.16 (1H, d, J = 8.7 Hz), 6.85 (1H, dd, J 1 = 8.7 Hz, J 2 = 2.5 Hz), 7.02 (1H, d, J = 2.5 Hz), 7.23-7.27 (5H, m); carbon nuclear magnetic resonance (13C-NMR; 75 MHz, CDCl3) δ 18.7, 28.5, 48.0, 57.2, 112.9, 121.0, 123.3, 124.4, 126.1, 126.7, 127.1, 127.9, 128.6, 130.5, 139.1, 142.6; electrospray ionization-mass spectrometry: [M + H]+ m/z 298.14 (calcd 297.13) corresponding to C19H20ClN. Anal (C19H20ClN) Calcd C, 76.62; H, 6.77; N, 4.70. Found: C, 76.38; H, 6.76; N, 4.73. Unless otherwise noted, all other chemicals were obtained from Sigma-Aldrich (St Louis, Missouri).
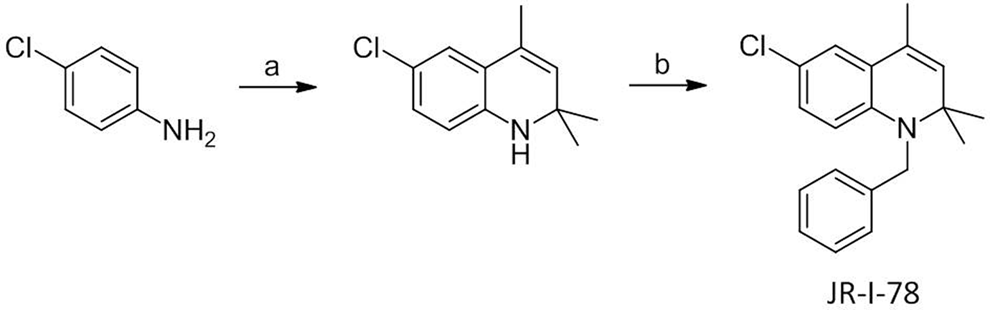
Synthesis of JR-I-78. Reagents and conditions: (A) acetone, I2, toluene, reflux and (B) benzyl bromide, toluene, 24 hours. JR-I-78 indicates 1-benzyl-6-chloro-2,2,4-trimethyl-1,2-dihydroquinoline.
In Vitro Cytotoxicity Assays
HepG2, Hep3B, Huh7, and PLC5 cells were maintained in Dulbecco’s modification of Eagle’s medium (Invitrogen, Carlsbad, California) containing 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin in 5% CO2 at 37°C. The cells were trypsinized and passed every 2 to 4 days. For assays with all 4 liver cell lines, 5 × 103 cells were seeded and incubated overnight in 96-well plates. Medium containing compounds in serial dilutions was added in a volume of 100 μL/well and incubated at 37°C in a humidified 5% CO2 atmosphere for 72 hours. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide at 20 μL/well (2.5 mg/mL in autoclaved water) was added, and the plates were incubated for 1 hour. Liquid was removed from each well, and dimethyl sulfoxide (DMSO) was added (100 μL/well). Optical densities for each well were measured at 570 nm with the aid of a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA). Half maximal inhibitory concentration (IC50) values were determined with the aid of the software program SoftMax Pro (Version 5.4, Molecular Devices, Sunnyvale, CA).
Ames Assay
Salmonella typhimurium indicator strains TA98 and TA100 were kept frozen at −80°C in Oxoid Nutrient Broth No. 2 (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% sterile glycerol. Cultures were inoculated into 50 mL nutrient broth, were allowed to sit unshaken for 2 to 4 hours, and were then gently shaken (100 rpm) for 11 to 14 hours at 37°C. To a sterile 13 × 100 mm2 test tube placed in a 43°C heating block was added 2 mL of molten top agar, 0.1 mL of indicator organisms (about 108 bacteria), the desired amount of the test article dissolved in 100 µL DMSO, and 0.5 mL of metabolic activation mixture or buffer (the metabolic activation mixture consisted of 10% Aroclor 1254-induced rat liver S9 fraction; Molecular Toxicology, Inc, Boone, North Carolina, containing 8 mM MgCl2, 33 mM KCl, 5 mM glucose-6-phosphate, 4 mM nicotinamide adenine dinucleotide phosphate, and 100 mM sodium phosphate, pH 7.4). This mixture was stirred gently and was then poured onto plates containing about 25 mL of minimal glucose agar. After the top agar was set, the plates were incubated at about 37°C for approximately 48 hours. Revertant colonies were counted using an automated colony counter, Sorcerer Image Analysis System (version 2.2), and data were managed through the Ames Study Manager (version 1.21), both manufactured by Perceptive Instruments (Suffolk, England). When accurate counts could not be obtained due to test compound precipitation on the plates, colonies were counted manually using an electric probe colony counter. Experiments were considered valid when solvent controls were within 10% of historical limits for spontaneous revertants and when positive control mutagens elicited a positive response (≥5-fold increase over the mean value for the solvent for the respective strain). In this assay, test articles are considered mutagens when a dose-related increase in revertants is observed at 1 or more dose levels.
In Vitro Methemoglobin Assay
Test or control articles were spiked into human whole blood (purchased from Bioreclamation LLC, Westbury, New York; collected using sodium heparin as the anticoagulant) and incubated in a 37°C shaking water bath. Samples contained a final organic solvent concentration of 1% DMSO. Aliquots were removed at selected time points (10, 30, and 60 minutes) and stored on wet ice prior to analysis by co-oximetry using an IL-682 CO-Oximeter (Instrumentation Laboratory, Lexington, Massachusetts). Zero time point samples were prepared by spiking vehicle (DMSO or phosphate-buffered saline) into blood at a final concentration of 1% (v/v) and storing the samples on wet ice until analysis. All conditions were assayed in duplicate.
Results
Cytotoxicity of Dihydroquinolines to Human Liver Cell Lines
In earlier work, the dihydroquinolines (DHQs) OSU-36.HCl and OSU-40 displayed promising selectivities for T. b. rhodesiense over L6 rat myoblasts (antitrypanosomal IC50 values of 0.013 and 0.012 μM, respectively, and IC50 values of 11 and 24 μM against the L6 cell line in vitro, respectively). 7,8 The cytotoxicity of these lead compounds against other mammalian cell lines was not examined, however. Due to the potential hepatotoxicity of compounds that generate a quinone imine, we measured the cytotoxicity of OSU-36.HCl and OSU-40 against 4 human liver cancer cell lines. The results from the panel of human liver cells (Table 1) agreed with the previous data obtained using L6 cells, with OSU-36.HCl exhibiting IC50 values ranging from 5.2 to 20 μM and OSU-40 displaying IC50 values varying from 8.0 to 36 μM. As a trend, OSU-40 showed lower toxicity than OSU-36.HCl to all 4 human hepatic cell lines.
Cytotoxicity of OSU-36.HCl and OSU-40 Against Human Liver Cancer Cell Lines In Vitro.

Abbreviations: IC50, half maximal inhibitory concentration.
a Mean ± standard error of 3 independent measurements.
Synthesis of Control Compound JR-I-78
A control compound from the dihydroquinoline class was desired for Ames and methemoglobin assays that could not be metabolized to a quinone imine intermediate. JR-I-78 was envisioned as such a compound since it has clear structural resemblance to the trypanocidal dihydroquinolines shown in Figure 1, yet cannot form a quinone imine due to the presence of a chlorine atom at the C6 position of the dihydroquinoline core rather than a hydroxyl group. Previous work showed that dihydroquinolines lacking the ability to form a quinone imine were 2 to 3 orders of magnitude less potent against trypanosomes than 6-O-acetyl and 6-hydroxy dihydroquinolines. 7 JR-I-78 was synthesized in 2 steps from 4-chloroaniline (Scheme 1) by the same general route used to prepare other dihydroquinolines. 7,8
Assessment of DHQs in the Ames Test
OSU-36.HCl and OSU-75 were screened for potential mutagenicity using the S. typhimurium tester strains TA98 and TA100. Assays were performed in the presence and absence of a metabolic activation mixture containing 10% Aroclor 1254-induced rat-liver microsomes (S9) with JR-I-78 included as a control compound belonging to this structural class. Mean plate counts are presented in Table 2. OSU-36.HCl was cytotoxic to the TA98 strain at doses ≥100 μg/plate in the absence of metabolic activation and at doses ≥500 μg/plate in the presence of metabolic activation, while OSU-75 and JR-I-78 were not cytotoxic to TA98 under any test condition. Although OSU-36.HCl exhibited cytotoxicity at high doses, there were noncytotoxic doses to evaluate possible mutagenicity. Precipitate was seen on the plates at doses ≥500 µg/plate for OSU-36.HCl and OSU-75. Since no dose-related increase in the number of revertant colonies was observed in any of the samples, OSU-36.HCl, OSU-75, and JR-I-78 did not elicit a mutagenic response under the conditions employed in this screening study. Similar results were obtained with these compounds using the S. typhimurium tester strain TA100.
Ames Screen of Dihydroquinolines Using Salmonella typhimurium.
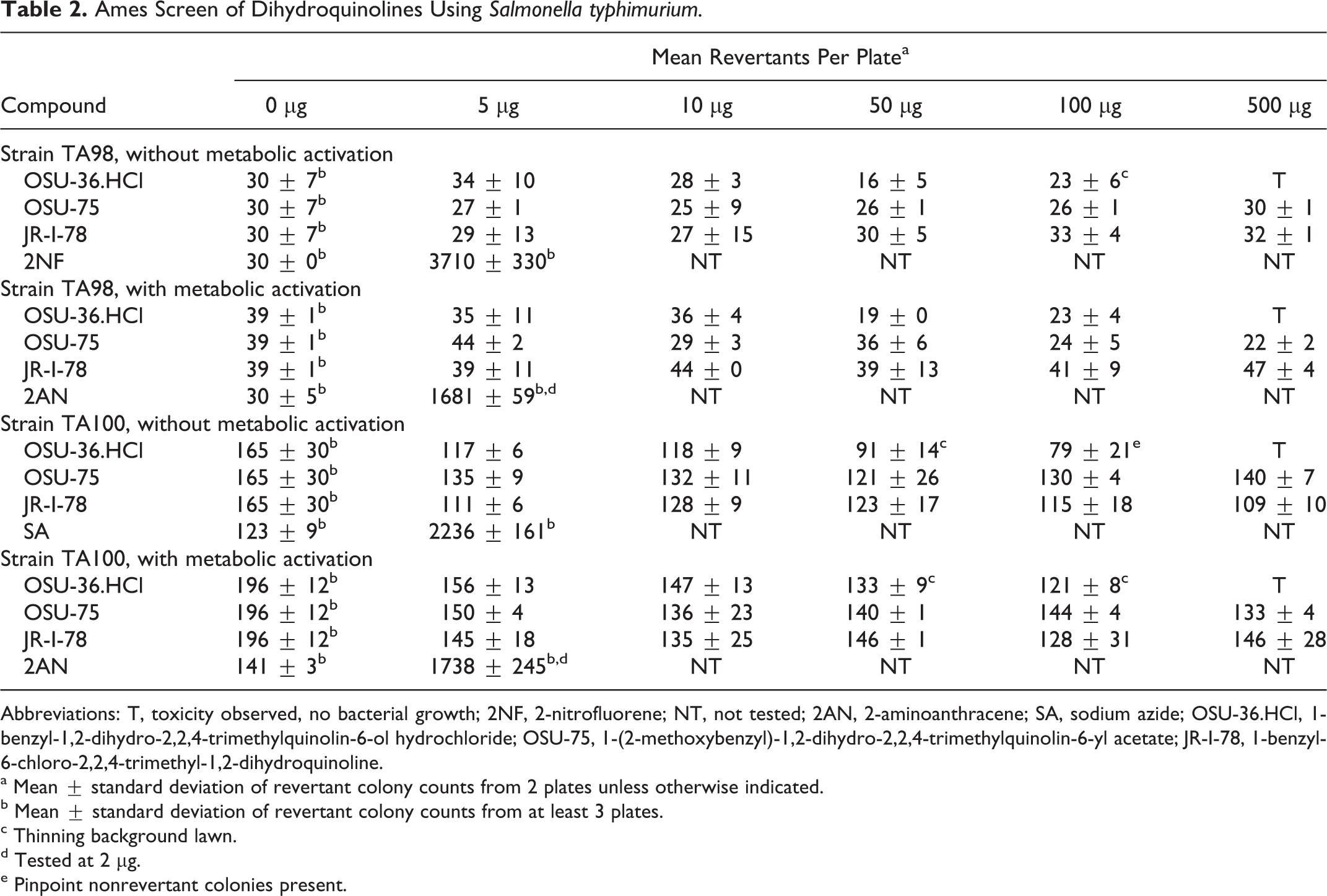
Abbreviations: T, toxicity observed, no bacterial growth; 2NF, 2-nitrofluorene; NT, not tested; 2AN, 2-aminoanthracene; SA, sodium azide; OSU-36.HCl, 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol hydrochloride; OSU-75, 1-(2-methoxybenzyl)-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate; JR-I-78, 1-benzyl-6-chloro-2,2,4-trimethyl-1,2-dihydroquinoline.
a Mean ± standard deviation of revertant colony counts from 2 plates unless otherwise indicated.
b Mean ± standard deviation of revertant colony counts from at least 3 plates.
c Thinning background lawn.
d Tested at 2 µg.
e Pinpoint nonrevertant colonies present.
Effect of DHQs on In Vitro Methemoglobin Levels
The in vitro effect of OSU-36.HCl and OSU-75 on methemoglobin levels in human whole blood is shown in Figure 2. Both OSU-36.HCl and OSU-75 increased methemoglobin levels in human whole blood when incubated at 50 μM (9.3% and 3.8% for OSU-36.HCl and OSU-75, respectively, at 60 minutes) compared with the vehicle control (0.8% at 60 minutes). At 10 μM, only OSU-36.HCl showed a very slight increase in methemoglobin levels compared with the vehicle control (2.1% vs 0.8% at 60 minutes). JR-I-78 had no effect on methemoglobin levels in human whole blood at either 10 or 50 μM (0.8% at 60 minutes for both concentrations). Sodium nitrite, the positive control, increased methemoglobin levels to 37.4% at 10 minutes, to 41.2% at 30 minutes, and to 50.6% at 60 minutes when incubated in blood at a concentration of 5 mM.
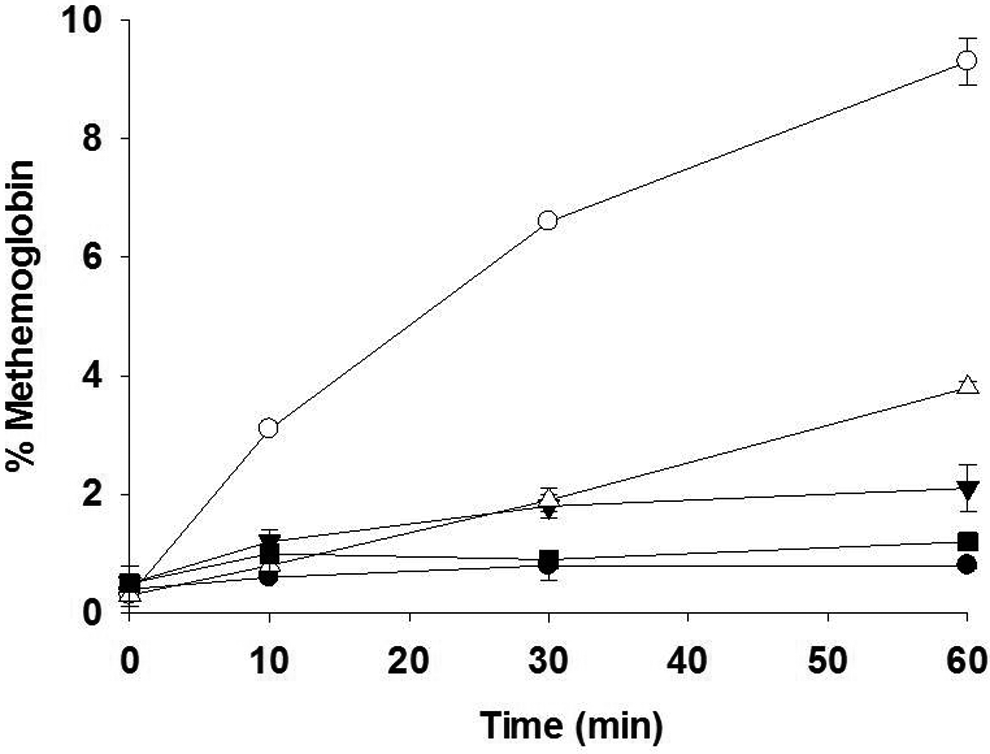
Effect of dihydroquinolines on methemoglobin levels in human whole blood in vitro. Test compounds were incubated in blood for the indicated times and the percentage of methemoglobin was measured by co-oximetry. Open circles—OSU-36.HCl, 50 μM; closed inverted triangles—OSU-36.HCl, 10 μM; open triangles—OSU-75, 50 μM; closed squares—OSU-75, 10 μM; and closed circles—DMSO control. Error bars represent the range of duplicate measurements. Data are not shown for JR-I-78 for clarity in presenting the DMSO control data (see the text). DMSO indicates dimethyl sulfoxide.
Discussion
Evaluation of the lead dihydroquinolines OSU-36.HCl and OSU-40 in 4 human hepatic cell lines (Table 1) demonstrated that the IC50 values of these compounds were in the same range of previously determined values of 11 μM 8 and 24 μM 7 against L6 rat myoblasts, respectively. These observations support the argument that the cytotoxicity caused by oxidation of antitrypanosomal dihydroquinolines to a quinone imine species is neither cell line specific nor cell type specific. Ester hydrolysis of OSU-40 results in the formation of 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol, the same compound that occurs by deprotonation of OSU-36.HCl. The cytotoxicity observed for OSU-36.HCl and OSU-40 in the 4 human hepatic cell lines thus follows the same rank order (Hep3B > PLC5 > Huh7 > HepG2). As observed previously in L6 cells, the requirement for ester hydrolysis may also reduce the cytotoxicity of OSU-40 compared to OSU-36.HCl. 7,8
Although OSU-36.HCl was toxic to S. typhimurium strains TA98 and TA100 at high-dose levels both in the presence and in the absence of S9, neither this compound nor the esterified dihydroquinoline OSU-75 was mutagenic in the Ames screen (Table 2). It is interesting to note that OSU-75 did not have toxic effects on the bacteria even up to a dose level of 1000 µg/plate (data not shown). A possible explanation for the lower toxicity of OSU-75 toward the Salmonella tester strain could be low esterase activity in the S9 fraction and/or the bacteria. Alternatively, the ester hydrolysis product of OSU-75, which contains an additional methoxy group that is not present in OSU-36.HCl (Figure 1), may possess lower intrinsic antibacterial activity.
Data shown in Figure 2 indicate that high (50 μM) concentrations of OSU-36.HCl, and to a lesser extent OSU-75, increased methemoglobin levels in vitro when added directly to human blood (9.3% and 3.8% methemoglobin formation at 60 minutes of incubation, respectively). OSU-75 acetate may produce less methemoglobin than OSU-36.HCl due to (1) lower levels of quinone imine formation resulting from incomplete ester hydrolysis of OSU-75 or (2) lower intrinsic ability of the hydrolysis product of OSU-75 to generate a quinone imine. The second explanation seems less likely considering that OSU-36.HCl and OSU-75 display indistinguishable in vitro antitrypanosomal activity and both compounds provide cures in a murine African trypanosomiasis model. 8 Although preliminary pharmacokinetic studies in rats indicated that OSU-40 was rapidly deacetylated, 15 species-specific differences in esterase activity have been noted in human versus rat plasma. 16 The specific esterase responsible for the hydrolysis of these dihydroquinolines has not been identified. At lower (10 μM) concentrations of OSU-36.HCl and OSU-75, only 2.1% and 1.2% methemoglobin were observed in human blood after 60 minutes of incubation, respectively. Methemoglobin makes up approximately 1% of total hemoglobin in the blood of healthy individuals; symptoms of methemoglobinemia in humans are typically observed when levels exceed 20%. 17 Intravenous administration of OSU-36.HCl at a dose of 2 mg/kg resulted in an OSU-36 plasma concentration of approximately 150 ng/mL (˜0.53 μM) measured at 60 minutes, 15 while a 50-mg/kg ip dose of OSU-36.HCl given each day for 5 days was required to cure a first-stage trypanosome infection in mice. 8 Although dihydroquinolines have the potential to increase methemoglobin levels in vivo if they are present in blood at sufficient concentrations, it is not clear whether the blood levels of a dihydroquinoline would be high enough to cause such side effects during the treatment of a trypanosome infection.
In conclusion, human hepatic cell lines appear to be no more sensitive to dihydroquinolines than the L6 rat myoblast cell line, and antitrypanosomal dihydroquinolines are not mutagenic with or without metabolic activation. Methemoglobin formation was observed in human blood incubated with the dihydroquinolines OSU-36.HCl and OSU-75, but the plasma levels of these compounds may not be high enough when used therapeutically against trypanosomes to cause symptoms of methemoglobinemia in the human host. The esterified dihydroquinoline OSU-75 produced lower levels of methemoglobin than the dihydroquinolin-6-ol OSU-36.HCl. Dihydroquinoline structure–activity relationship demonstrates that ester substitution has little impact on in vitro activity, 7 raising the possibility that a DHQ prodrug could be selectively activated by a trypanosome esterase to facilitate delivery of the active compound to the parasite. The synthesis of a range of DHQ prodrugs is thus a promising strategy to minimize methemoglobin formation in the host if necessary. Although monitoring of host toxicity will be important during their development, the results of the current studies justify the further investigation of dihydroquinolines for their antitrypanosomal potential.
Footnotes
Acknowledgments
We thank Dr. Ching-Shih Chen for supplying the human hepatic cell lines and Dr. Michael Wang and Ahmed Abdelhameed for helpful discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work conducted by SRI International was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases, and Division of Microbiology and Infectious Diseases, under Contract No. HHSN272201100022I. The work done at The Ohio State University (OSU) was supported by the OSU College of Pharmacy.