
Abstract
There has been an increased interest in and activity for the use of peptide therapeutics to treat a variety of human diseases. The number of peptide drugs entering clinical development and the market has increased significantly over the past decade despite inherent challenges of peptide therapeutic discovery, development, and patient-friendly delivery. Disparities in interpretation and application of existing regulatory guidances to innovative synthetic and conjugated peptide assets have resulted in challenges for both regulators and sponsors. The Symposium on Development and Regulatory Challenges for Peptide Therapeutics at the 40th Annual Meeting of the American College of Toxicology held in November of 2019 focused on the following specific topics: (1) peptide therapeutic progress and future directions, and approaches to discover, optimize, assess, and deliver combination peptide therapeutics for treatment of diseases; (2) toxicological considerations to advance peptide drug-device combination products for efficient development and optimal patient benefit and adherence; (3) industry and regulatory perspectives on the regulation of synthetic and conjugated peptide products, including exploration of regulatory classifications, interpretations, and application of the existing guidances International Council for Harmonisation (ICH) M3(R2) and ICH S6(R1) in determining nonclinical study recommendations; and (4) presentation of the 2016 Health and Environmental Sciences Institute’s Genetic Toxicology Technical Committee working group assessment of genotoxicity testing requirements. Perspectives were shared from industry and regulatory scientists working in the peptide therapeutics field followed by an open forum panel discussion to discuss questions drafted for the peptide therapeutics scientific community, which will be discussed in more detail.
Introduction
A symposium was held in November 2019 as a part of the 40th Annual Meeting of the American College of Toxicology (ACT). This symposium was intended to bring together industry and regulatory scientists contributing to the peptide therapeutic field. Despite the increased interest in the discovery and development of peptide and peptide-device combination therapeutics, there is currently no guidance for nonclinical safety assessment of synthetic and conjugated peptide products, nor is there alignment on which nonclinical studies need to be considered or conducted for approval. Disparities in interpretation and application of existing International Council for Harmonisation (ICH) and United States Food and Drug Administration (FDA) regulatory guidances have resulted in challenges, inconsistencies, and confusion for both regulators and drug developers. In this symposium, the presenters attempted to showcase the value of peptide therapeutics and identify scientific aspects for potential safety hazard concerns and appropriate nonclinical assessment strategies. The specific topics included 1 : peptide therapeutic progress and future directions and approaches to discover, optimize, assess, and deliver combination peptide therapeutics for treatment of diseases 2 ; toxicological considerations to advance peptide drug-device combination products for efficient development and optimal patient benefit and adherence 3 ; industry and regulatory perspectives on the regulation of synthetic and conjugated peptide products, including exploration of regulatory classifications, interpretations, and application of existing guidances in determining nonclinical study requirements (ICH M3[R2] vs ICH S6); and 4 presentation of the 2016 Health and Environmental Sciences Institute (HESI) working group assessment of genotoxicity testing requirements. By drawing on the experience and expertise of industry and regulatory scientists working in the peptide therapeutic field, the symposium presenters discussed the potential safety concerns of this therapeutic class. They further noted issues that could be addressed and/or resolved by development of an appropriate regulatory guidance for nonclinical safety assessment recommendations specifically focused on synthetic and conjugated peptide products. Such guidance would bring much-needed consistency and clarity to the field and benefit both industry and regulatory scientists. The individual presentations are summarized below.
Addressing the Challenges of Peptide Drug Discovery and Delivery: Combination Peptide Therapeutics to Treat Metabolic Diseases (Paul L. Feldman)
There has been surgent interest and activity in the use of peptide therapeutics to treat a variety of human diseases. 1 Historically, the use of peptide-based drugs was a challenge due to the need to administer via injections, requirement for refrigerated supply chains, short half-lives leading to multiple injections per day, and the cost of manufacturing. Additionally, the discovery and development of peptide therapeutics requires specialized techniques and equipment that further dissuaded their pursuit within the pharmaceutical industry as medications for chronic diseases. Despite this, peptide-based drugs have some significant advantages over small molecules because they can productively engage biologic targets that are intractable for small-molecule drug discovery, and there is a lower attrition rate through development for peptides versus small molecules. 2 More recently, advances in biology, peptide chemistry, and delivery have made discovery and development of peptide therapeutics more accessible and advances in delivery technologies have provided more and better options for patients leading to greater uptake of peptide therapeutics to treat chronic diseases.
The treatment of metabolic diseases, including obesity and types 1 and 2 diabetes, is an area that has benefited from advances in peptide therapeutics, but these disorders remain a significant and growing world health problem. 3 In the United States, the prevalence rate of obesity has approximately doubled over the past 30 years, such that more than 30% of adults are presently classified as obese. Due in part to the rise in obesity rates, greater than 100 million adults in the United States have diabetes or prediabetes. These metabolic diseases lead to complications that include multiple micro- and macrovascular disorders. Drugs with greater efficacy coupled with ease of administration and adherence benefits are needed.
In order to achieve the high level of differentiated efficacy needed to treat patients with type 2 diabetes (T2D) and obesity, scientists at Intarcia Therapeutics have taken inspiration from the patients who have achieved phenomenal metabolic benefits from the Roux-en-Y gastric bypass surgery (RYGB). After undergoing RYGB patients generally experience more than 25% weight loss and greater than 80% of obese patients with T2D who undergo this metabolic surgery experience complete remission of their diabetes within weeks after surgery. 4 It is known that some gut-derived peptide hormones are significantly increased postprandially in RYGB patients who manifest metabolic benefits. Additionally, in a recent clinical report, administration of combinations of some gut peptide hormones, specifically glucagon-like peptide-1 (GLP-1), oxyntomodulin, and peptide tyrosine tyrosine, resulted in significant antidiabetic and weight loss effects. 5 In order to leverage these beneficial metabolic findings from RYGB patients, the company’s strategy was to rigorously assess the metabolic efficacy and safety of these and other peptide hormones alone and in combination in our preclinical models to determine the best combinations of peptide targets to pursue.
This strategy addresses the first significant challenge of a peptide discovery program: choosing the biological target(s). In the end, we profiled more than 150 peptides, including gut peptides and peptides derived from other tissues, in preclinical models to assess food intake inhibition, antiobesity activity, and antidiabetic efficacy and identified 3 combinations of peptides that worked synergistically to deliver RYGB-like efficacy.
The second significant challenge of a peptide discovery program is optimization of peptide leads to generate clinical candidates. This requires simultaneous optimization of peptide physicochemical properties, receptor potency and selectivity, and pharmacokinetics (PK). Our approach to optimizing the PK of peptides includes increasing their resistance to metabolism by peptidases and proteases. 6 This can be achieved by substituting amino acids at specific positions within a peptide that are susceptible to enzymatic hydrolysis. Identification of positions within a test peptide that are vulnerable to enzymatic-mediated cleavage is achieved by in vitro incubation of test peptides with tissues rich with peptidases and proteases and identifying the peptide fragments formed by mass spectrometry. Using synthetic peptide chemistry, judicious amino acid substitutions at these vulnerable positions furnishes peptides resistant to cleavage. Ultimately, an optimized peptide is infused in vivo in a rodent model to determine its clearance and the goal is to achieve a peptide clearance that is equal to the glomerular filtration rate at which point peptidase resistance has been attained and the predominate mechanism for clearance is via filtration by the kidneys. Optimization of the PK must also be performed in concert with refinements in the potency and selectivity for the biologic target of interest and physicochemical properties (solid state and solution stability, solubility) of the peptide. When a peptide candidate is identified it is then assessed in preclinical models of T2D and obesity to determine its efficacy and in vivo potency. The process of peptide optimization described is being completed for all 3 of the peptide combinations identified via the target identification campaign described above. Assessments of the optimized peptide combinations have provided preclinical combination candidates that are able to replicate the efficacy of RYBG in rodent models of T2D and obesity.
Along with the selection of biological targets and peptide candidates, the third key challenge to address is selection of a delivery technology. Some important considerations include drug loading capacity, cost effectiveness, convenience for patients, and robustness of performance. Additionally, for the treatment of chronic diseases, the frequency of administration of the drug via the technology is important to ensure patient adherence to the therapy. It is well known that adherence to therapies for chronic diseases, such as T2D, is poor. Despite the approval of many new medicines to treat T2D most patients do not achieve the desired treatment goals; and it has been shown that the failure to achieve these goals is predominately due to lack of adherence to therapies. 7 Thus, peptide delivery technologies that are convenient and support adherence via infrequent dosing should enable more patients to realize the therapeutic potential of these medicines. Several technologies are available for administration of peptide therapeutics that vary in their frequency of administration. Oral and inhaled therapies are generally delivered daily or more often, injectable therapies are daily to monthly, and implantable technologies can enable delivery over periods of time ranging from months to year(s). Thus, matching the needs of a patient with desired clinical outcomes requires careful consideration of the delivery technology to ensure efficacy, safety, ease of use, cost effectiveness, and adherence.
In summary the 3 key considerations to enable a successful peptide therapeutic discovery program include 1 selection of the biological target, 2 optimization of the peptide lead to a clinical candidate, and 3 selection of delivery technology. These 3 components have been carefully considered in the quest to discover next generation, differentiated peptide therapeutics to treat select metabolic diseases.
Toxicological Considerations for Peptide Drug-Device Combination Product (Doris Zane)
Given the inherent physical property limitations of peptides and poor stability, delivery of peptide therapeutics to their intended patient populations represents a challenging obstacle. Various toxicological considerations to advance a peptide drug-device combination asset for efficient development and optimal patient benefit and adherence was presented. In addition, a brief overview of toxicological expectations, including regulatory requirements for combinations products, along with case studies and experience with peptide drug-device combination therapeutics, including multipurpose study designs to minimize further animal testing, was discussed.
Type 2 diabetes has become a global epidemic with an estimated worldwide prevalence of ∼9%, affecting 463 million people. 8 At current trends, 700 million people are expected to have diabetes in 2045, which is a 52% growth. Approximately 50% of treated diabetic patients are not at goal, with low adherence rates across all classes of therapy and poor persistence on treatment, particularly with injectable drugs. 9 Glucagon-like peptide-1 receptor agonists are effective, well-tolerated therapeutic options but limited by the need to administer by injection which frequently results in early treatment discontinuation. In order to improve therapeutic control, new therapies must address the issue of poor adherence. ITCA 650 is a unique, injection-free combination drug-device preapproval product. ITCA 650 contains the GLP-1 receptor agonist, exenatide, as the active pharmaceutical ingredient (API), in an osmotic mini-pump drug delivery technology. ITCA 650 delivers a continuous exposure of exenatide with a 6-month treatment from a single subdermal placement. The primary mechanism of action of this drug-device combination is attributed to the synthetic peptide exenatide. In clinical trials, ITCA 650 has shown significant improvement in glycemic control, weight loss, and HbA1c goal attainment. 10 -12 Meaningful reductions in HbA1c of 1.4% to 17% with an associated weight loss of 3 to 4 kg was achieved. Over 18,000 placement and removal procedures have been conducted in clinical trials.
Nonclinical regulatory requirements for a New Drug Application for a drug-led combination product, such as ITCA 650, include toxicology assessments (per ICH M3(R2)) 13 and biocompability assessments (per ISO 10993-1 14 ). Using a risk management framework (per ISO 14971 15 ) to reduce unnecessary in vitro and in vivo biocompatibility testing requirements, existing toxicology data were leveraged, where possible, to reduce certain biocompatibility testings. After determining what toxicity information was available (based on prior testing and literature), only biocompatiblity studies that addressed the gaps were conducted.
A case study example of how existing toxicology data were leveraged to reduce certain biocompatibility testings included the use of the cynomologus monkey chronic toxicity study. Per ISO 10993-11, 16 a chronic systemic toxicity biological end point can also include sensitization, irritation/intracutaneous reactivity (per ISO 10993-6 17 ), and implantation (per ISO 10993-6) using the final finished form of the drug-device combination (manufactured, processed, cleaned of contaminants, and sterilized by the method intended for the final product), and implanted into a site most relevant to the intended clinical use of the material and animal species appropriate for the evaluation of the biological safety of the material. Per ICH M3(R2), a 9-month cynomologus monkey chronic toxicity study using the final finished form of the drug-device combination (ITCA 650) was administered by subcutaneous implantation. In this toxicology study, biological end points such as sensitization, irritation/intracutaneous reactivity, and implanation were also included. Therefore, by leveraging the existing toxicology data from the one cynomologus monkey chronic toxicity study, separate biocompatbility testings required for chronic systemic toxicity, senisitization, irritation/intracutaneous reactivity, and implantation end points were reduced.
As demonstrated in the following case study example, the existing genotoxicity studies were not able satisfy the biocompatibility requirement of the peptide drug-device combination. Per ICH M3(R2), 3 genotoxicity studies (2 in vitro studies [bacterial reverse mutation assay and mammalian chromosomal aberration assay in Chinese hamster ovary (CHO) cells] and 1 in vivo study [mammalian erythrocyte micronucleus assay in mice]) were performed using the peptide drug product only as the test article; device biocompatibility was not assessed in any of these genotoxicity studies. In preliminary feasibility/pilot dose range-finding studies, using the final finished form of the drug-device combination (ITCA 650) proved to be problematic. Following extraction of ITCA 650, the test article extract remained cloudy. This extract was used for the dilutions and dosed to the cell culture wells. Following incubation, the cultures were examined under a phase-contrast microscope to identify any systemic cell seeding errors, growth characteristics, and changes in cell morphology. A film was observed over the cell layer and therefore, the cells were not visible. Unfortunately, determination of cytotoxicity or percent viability was not obtained for the study. Therefore, per ISO-10993-3, 3 3 separate stand-alone genotoxicity studies using device components only extracts were conducted in order to satisfy the biocompatibility end point of the peptide drug-device combination product. The definitive genotoxicity tests performed with extracts from device components only also included 2 in vitro studies (bacterial reverse mutation assay and mammalian chromosomal aberration assay in CHO cells) and 1 in vivo study (mammalian erythrocyte micronucleus assay in mice).
Per FDA guidance, Use of International Standard ISO 10993-1 “Biological evaluation of medical devices- Part 1: Evaluation and testing within a risk management process” (2016), 18 section III.C.3. (Animal study experience) “Data from an in vivo animal study of the medical device in its final finished form may be used in lieu of some biocompatibility tests. Testing performed in a relevant animal model can be used if the study was designed to include assessments for biocompatibility end points.” Using this approach, nonclinical toxicology studies were used to evaluate the biological response to ITCA 650 implanted in a clinically relevant implantation site (subcutaneous; Table 1). A comprehensive nonclinical testing strategy was designed to demonstrate and characterize the safety of ITCA 650 using the proposed subcutaneous route of administration for clinical use. Repeat-dose toxicology studies (including systemic—acute, subacute/subchronic, chronic; carcinogenicity; reproductive and developmental) were conducted per ICH M3(R2) guidelines. Dose selection for the toxicology studies was based on information derived from publicly available studies and dose range-finding studies. Specifically for the rat carcinogenicity study, FDA concurred with the proposed doses via the Special Protocol Assessment process and doses were selected based on the ICH recommended 25-fold area under the curve over human exposure. As a part of these toxicology studies, biocompatibility end points as defined in ISO-10993-1 (eg, sensitization, irritation/intracutaneous reactivity, material-mediated pyrogenicity, and implantation) were also assessed thereby significantly reducing stand-alone biocompatability testings. Based on the existing toxicology data and any remaining biological evaluations conducted to address the gaps, the peptide drug-device combination, including the device components used in ITCA 650, has provided adequate evidence of safety to support treatment of patients with T2D and are absent of harmful effects for patient contact.
ITCA 650 Biological End Points That Were Met Using GLP Animal Studies.

Abbreviation: GLP, glucagon-like peptide.
Opportunities and Challenges of Intracellularly Targeted Peptide Drug Discovery (Tomi Sawyer)
Historically, peptide drug discovery (Figure 1) began with a major focus on receptor targets (eg, G protein–coupled receptors [GPCRs]) during the 1970s and this first wave has continued onward to present times. In the late 1980s and into the 1990s, there were noteworthy achievements in the identification of peptides that exhibited cellular permeability and/or the ability to modulate intracellular target activity. This second wave was exemplified by the macrocyclic natural product peptide cyclosporin, several classes of cell-penetrating peptides (CPPs) such as transactivating transcriptional activator (TAT)-based synthetic CPPs, and a worldwide campaign on HIV drug discovery that include many novel synthetic peptidomimetic inhibitors of HIV protease. Over the past 3 decades, powerful and innovative technologies enabling extraordinary diversity (106-1012) of peptides through phage display, mRNA-display, DNA-encoded and other synthetic libraries are significantly driving a third wave of macrocyclic peptide therapeutics. Importantly, it is well-recognized nowadays that peptides are a compelling modality for both “classic” targets as well as “undruggable” targets versus small molecules that may have inability to bind to large, flat binding surfaces or antibodies that have limited cell permeability to access intracellular targets. 19 -27
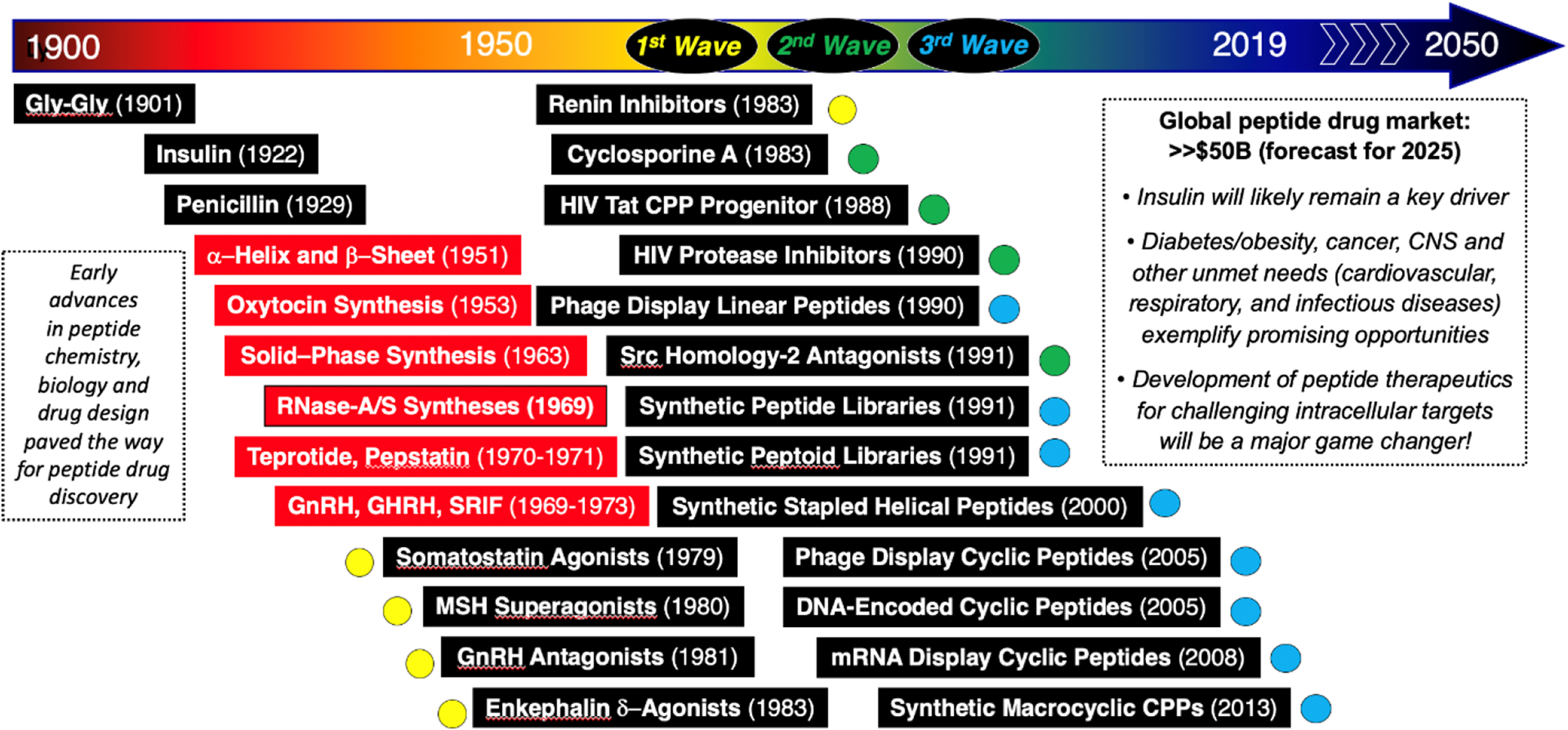
Some historical milestones in peptide drug discovery, including 3 major waves (see text for details).
It is recognized that there has been increased biotech/pharma R&D efforts focused on macrocyclic peptides to challenge varying targets ranging from GPCRs to other classes of receptors, the cell membrane (eg, antibiotics, CPPs) and intracellular proteases, protein–protein interactions, and protein–nucleic acid interactions. In terms of recently FDA-approved peptide drugs (Figure 2), such target space is well exemplified across many therapeutic indications, including endocrine, gastrointestinal, and infectious diseases as well as cancer. Interestingly, there is a significant effort focused on macrocyclic peptides and peptidomimetics 19 -27 that are being advanced, and unquestionably these will further increase relative to the third wave of peptide drug discovery to build upon what has already been accomplished. Yet, a remaining challenge for peptide therapeutics is drug delivery. Albeit, sustained-acting peptides for receptor targets (eg, GLP-1 analogs) have been advanced via methods such as fatty acid conjugation or PEGylation to provide subcutaneous injectable therapeutics, there has been very limited success relative to oral delivery. Nevertheless, it is noteworthy to highlight the success of peptidomimetics with respect to oral delivery as well as recent achievements 28 with the GLP-1 agonist peptide Rybelsus (FDA-approved 2019) which spurring renewed enthusiasm to further investigate the potential of oral delivery technologies for peptides.
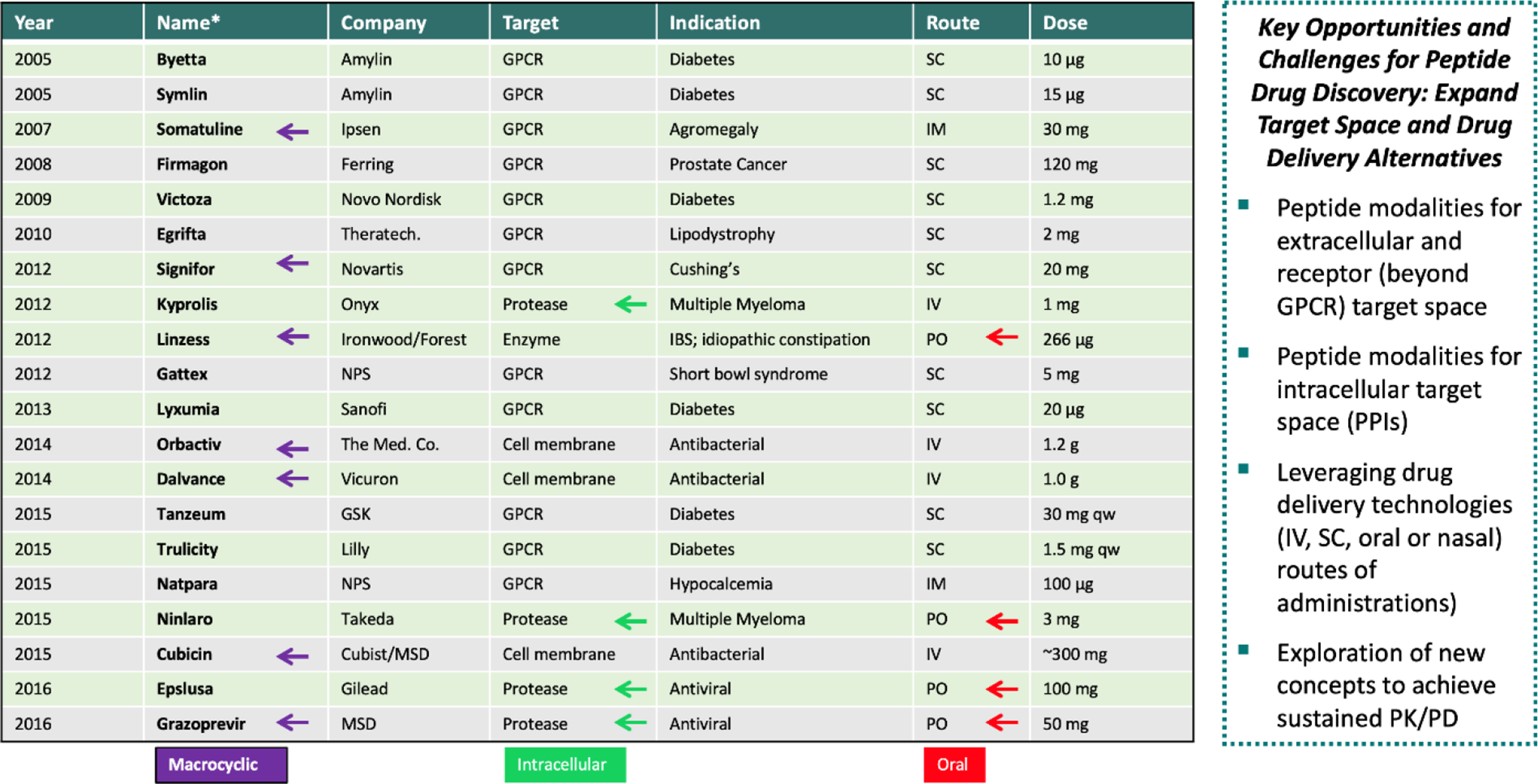
Some Food and Drug Administration (FDA)–approved peptide/peptidomimetic drugs (2005-2015) and future strategies (see text for details).
Perhaps the most challenging, but yet compelling, opportunities for peptide drug discovery is that related to intracellular target space. Many therapeutic targets in the cell have been considered essentially undruggable by conventional small molecules and although quite likely druggable by antibodies there has been very limited success to confer cellular permeability to antibodies or therapeutic proteins. Therefore, peptides, and more precisely, macrocyclic peptides have become highly favored within the molecular armamentarium of biotech/pharma to become a new therapeutic modality to tackle intracellular target space in innovative ways. 19 -27 Some noteworthy examples include cyclosporine-A (CsA), 29 cyclic CPPs, 30 and ATSP-7041 31 (Figure 3).
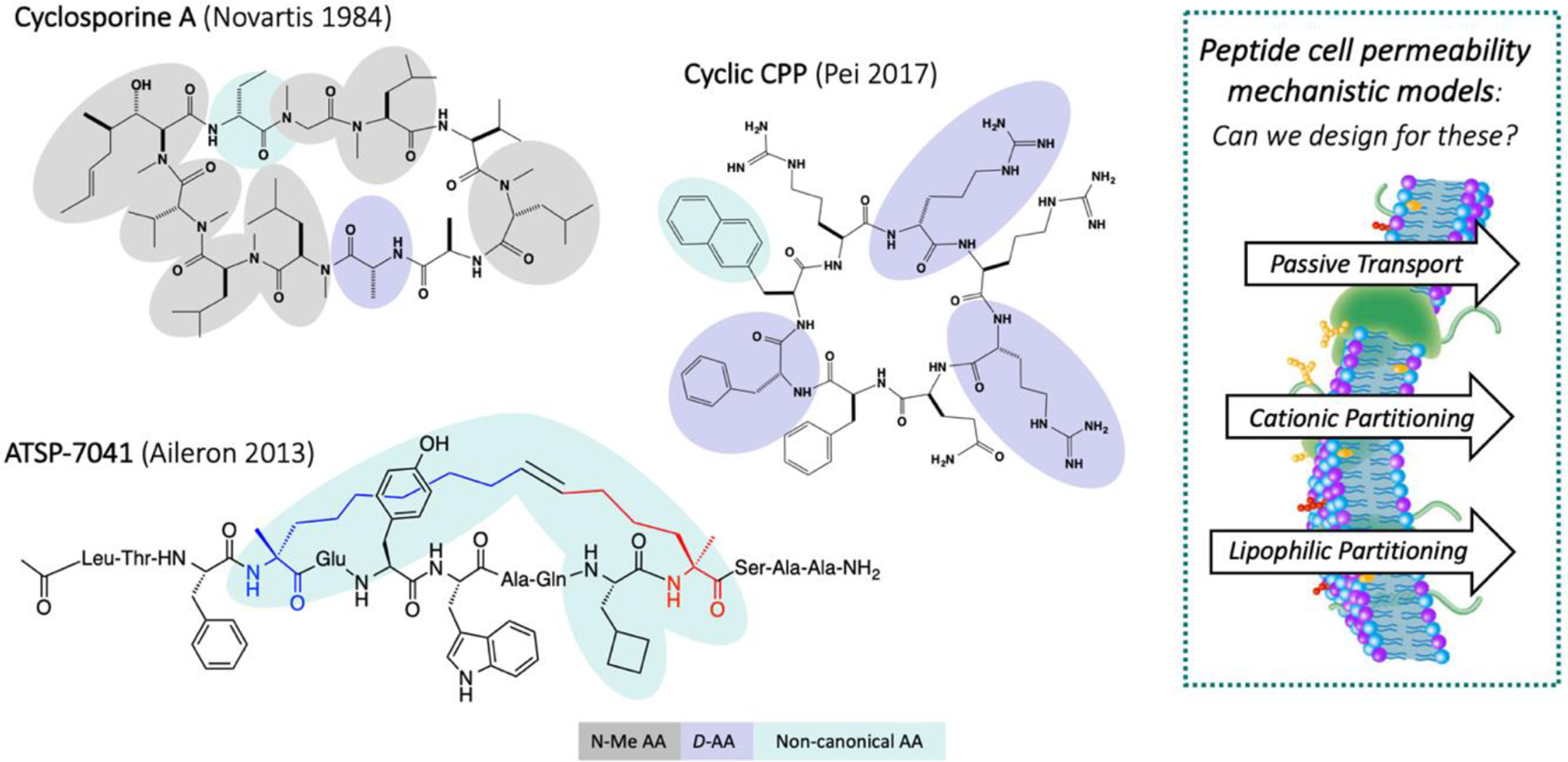
Some macrocyclic peptides exemplifying cell permeability by varying mechanisms (see text for details).
Unquestionably, the natural product peptide CsA was a monumental breakthrough for peptide drug discovery relative to its cell permeability properties and ability to modulate intracellular target function. Albeit its large-size, the specific structural features of CsA, such as N-methylation of several backbone amides as well as a high degree of lipophilicity, have been determined to contribute to its passive permeability. In fact, CsA is regarded as a benchmark for both structure-based design and diversity-driven strategies to advance novel macrocyclic peptides of this general class. 23,25,32 Cyclic CPPs are a relatively new class of macrocyclic peptides which have translated past learnings from linear CPPs (eg, Tat and poly-Arg) in terms of cationic partitioning into cell membranes and endocytosis to achieve cellular uptake and release from endosomes into the cytosol. 22,30,33 Such cyclic CPPs may be used as carriers or be designed to incorporate a peptide pharmacophore for an intracellular therapeutic target. Stapled α−helical peptides have also arisen as a promising new class of macrocyclic peptides 24,26,34 -36 and have recently advanced into the clinic (ie, ALRN-6924). 37 ATSP-7041 31,38 -40 (a progenitor of ALRN-6924) is a benchmark stapled α−helical peptide to explore cellular permeability that seems to fit into a third mechanism which has been dubbed lipophilic partitioning that is different from the passive transport properties of CsA and the cationic partitioning (and endosomal pathway) of cyclic CPPs. These 3 general mechanisms of cellular uptake are recognized to effectively enable peptide entry into the cell to subsequently engage and modulate the functional properties of a therapeutic target (Figure 4). Nevertheless, there are many other factors which may significantly impact the efficacy of such peptides, including intracellular metabolism as well as the extracellular concentration of the peptide and how metabolism, excretion, and serum albumin binding may collectively contribute to exposure levels and PK properties.
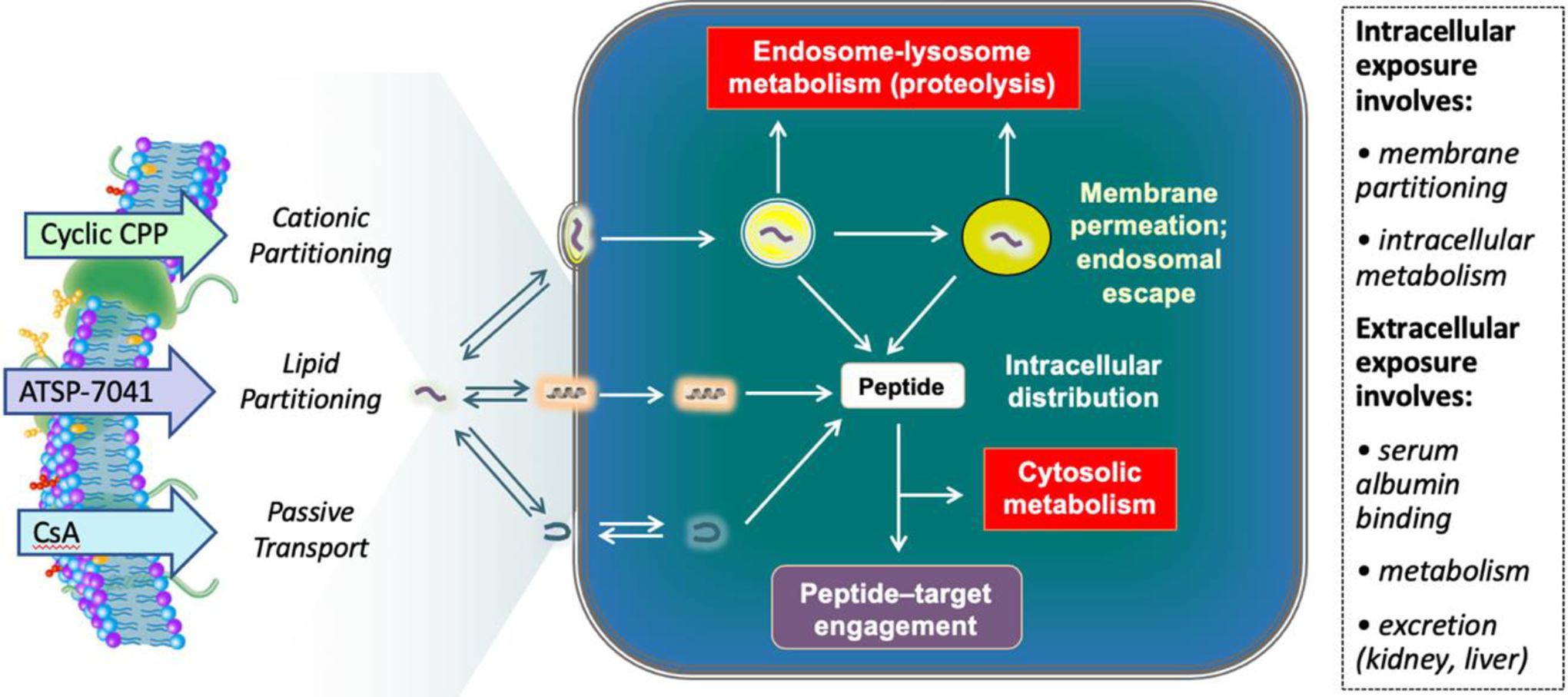
Model for cell uptake and intracellular fate of 3 classes of macrocyclic peptides (see text for details).
To further understand both peptide cell permeability and drug delivery, there is great need for robust screening tools (computational and experimental) which can facilitate predictive design rules and iterative testing thereof. As expected, there exists a framework of existing screening tools for passive permeability (eg, transcellular permeability using cellular monolayers and proteolytic stability in varying biological matrices) as well as emerging computational and biophysical methods, 41 -51 including those that address exposed polar surface properties, nuclear resonance/mass spectrometry (NMR/MS) analysis of intramolecular H-bonding, radius of gyration, and aqueous/lipid phase partitioning (Table 2). Cell-based screening methods 52 -56 include MS and/or fluorescence microscopy for unlabeled or fluorescently labeled peptides, respectively, and such may also be used to investigate cell uptake and correlate with cell-based functional properties. More recently, the development of a peptide cell permeability screening using a HaloTag-based assay dubbed CAPA (chloroalkane penetration assay) has provide quantitative and high-throughput analysis to understand the structure-permeability relationship of a diverse set of peptides. Overall, such screening tools will significantly enhance the understanding of peptide cell permeability and the development of intracellularly targeted peptide therapeutics.
Experimental and Computational Screening Tools to Understand Peptide Permeability (See Text for Details).

Genotoxicity Assessment of Peptide/Protein Therapeutics—Perspectives From an HESI Working Group (Zhanna Sobol)
Health and Environmental Sciences Institute’s Genetic Toxicology Technical Committee developed a publication describing the “Genotoxicity assessment of peptide/protein-related biotherapeutics: points to consider before testing.” 57 The goal of this publication was to identify specific challenges in genetic toxicology testing of peptide/protein-related biotherapeutics and provide recommendation for best practice approaches. Earlier initiatives surveyed genotoxicity testing of approved large molecule drugs and showed that many protein/peptide drugs were tested in the standard genotoxicity test battery and almost all were negative. 58,59 The working group sought to understand when standard genotoxicity testing added value to the risk assessment of the molecule and when the testing was unnecessary.
Relevant regulatory guidelines such as ICH S6 and ICH S2 contain gaps related to genotoxicity assessment of protein/peptide therapeutics. International Council for Harmonisation S2 guideline describes the standard genotoxicity test battery (in vitro bacterial reverse mutation assay, in vitro test for chromosome damage, and in vivo tests for chromosome damage) but does not address limitations for testing of proteins/peptides because this guideline was intended for small molecule therapeutics. Although ICH S6 states that genotoxicity testing is not relevant for biotherapeutics/biologics, it stipulates that testing may be needed if there is a cause for concern. The ambiguity around what constitutes a cause for concern often leads to conduct of the standard genotoxicity test battery. Synthetic peptides in particular fall in a gray area between small molecule and biologic and are often tested in the standard genetic toxicology battery even when there is no cause of concern. When synthetic peptides contain only natural amino acids, they are not expected to behave differently toward DNA than recombinant proteins. Likewise, if synthetic peptides contain modified amino acids that have been previously tested, the inclusion of these modified amino acids in a new sequence is not expected to alter the reactivity of the resulting peptide toward DNA. The potential limitations of testing protein/peptide therapeutics according to ICH S2 guideline include lack of uptake in in vitro systems, histidine release confounding the bacterial mutation assay, and various confounding effects on the in vivo assays. Nongenotoxic mechanisms that can lead to induction of micronuclei in vivo include stimulation of erythropoiesis, nucleotide pool imbalance that is secondary to decrease in folate levels and hyper/hypothermia. 60 The potential confounding effects on in vivo genotoxicity tests have been documented for protein/peptide therapeutics such as recombinant human erythropoietin leading to acceleration of erythroblastic maturation or GLP1/GIP dual inhibitor causing a decrease in folate levels as exaggerated pharmacology of the therapeutic. A positive response in the micronucleus assay as a result of such confounding factors would not be indicative of reactivity with DNA.
The cause for concern with protein/peptide therapeutics is often the inclusion of chemical components, such as linkers, that could potentially be released in vivo and lead to genotoxicity. Exposure to the released small molecule component is expected to be low, like impurities in small molecule therapeutics. Since ICH M7 contains a framework for assessment and control of genotoxic impurities, risk assessment concepts described in this guideline are useful when addressing the genotoxic risk of released small molecule components in protein/peptide therapeutics. Although ICH M7 is not applicable to active components or in vivo metabolites, the concept of threshold of toxicological concern (TTC) for mutagenic carcinogens and guidance for in-silico mutagenicity assessment can be useful. For example, if the maximum in vivo exposure to the released small molecule component is below the TTC, then it should not be considered a cause for concern even if its mutagenic potential is unknown. However, if exposure is expected to be above the TTC, then an in-silico assessment of the chemical structure can identify structural alerts for mutagenicity. The presence of structural alerts can warrant testing of the small molecule component; however, selecting the correct chemical species for testing can be challenging as it is important to understand the penultimate form of the small molecule component that will be released upon treatment with the whole therapeutic. The genotoxicity evaluation of the whole therapeutic and products of cleavage could potentially be evaluated as part of the general toxicity studies at doses considered to be compatible with pharmacological activity and therapeutic indication.
Numerous factors weigh into determining the value of genotoxicity testing for a particular protein/peptide therapeutic. The most important consideration is whether the results of the genotoxicity tests provide added information for risk assessment or be uninterpretable.
Nonclinical Studies Supporting Investigational and New Drug Applications for Peptide Products (Jessica Hawes)
Regulatory background, statutes, and guidances related to peptide therapeutics were discussed. Challenges and considerations for peptide therapeutic products were presented from a regulatory perspective. Disparities in interpretations and application of the existing ICH guidances ICH M3(R2) and ICH S6 to synthetic and conjugated peptide therapeutic products were explored. Regulatory perspectives and rationale regarding nonclinical studies evaluating genetic toxicology and metabolic characterization were discussed in further detail.
Regulatory Background
Understanding the regulatory history of peptide products can be useful to understanding the complications and questions that have arisen regarding nonclinical study requirements for peptide therapeutics. The FDA Center for Drug Evaluation and Research (CDER) regulates synthetic, recombinant, and endogenous peptide products as drugs under the Food, Drug, and Cosmetic Act (FD&C). 61 The 2009 Biologics Price Competition and Innovation Act (BPCI Act) created an abbreviated licensure pathway in section 351(k) of the Public Health Service (PHS) Act for biological products shown to be biosimilar to, or interchangeable with, an FDA-licensed biological reference product. 62 This also amended the statutory definition of a “biological product” in the PHS Act to include a protein, except any chemically synthesized polypeptide (csPP). At the time of the November 2019 symposium, the statutory definitions for peptides, csPPs, proteins, and biological products discussed were based on the descriptions provided in the December 2018 Draft Guidance for Industry New and Revised Draft Q&As on Biosimilar Development and the BPCI Act Revision 2. 63 Consistent with the scientific literature, FDA interprets the term “protein” in the statutory definition of a biological product to not include “peptides.” In the simplest context, a peptide is considered to be any polymer composed of less than or equal to 40 amino acids, regardless of how it is made. Thus, “peptides” and “chemically synthesized polypeptides” are not “biological products” and will be regulated as drugs under the FD&C Act in the United States, unless they otherwise meet the statutory definition of a “biological product” (eg, peptide vaccine). Under the 2018 draft Q&A Revision 2, the proposed statutory definition of a csPP was any α amino acid polymer that is both 1 made entirely by chemical synthesis and 2 greater than 40 amino acids, but less than 100 amino acids. Subsequently, a protein was defined as any α amino acid polymer with a specific defined sequence that is greater than 40 amino acids in size “(except any chemically synthesized polypeptide).” In early 2020, section 605 of the Further Consolidated Appropriations Act 64 further amended the definition of a “biological product” under section 351(k) of the PHS Act by removing the parenthetical “(except any chemically synthesized polypeptide)” from the statutory definition of a protein that was added by the BPCI Act. The final ruling on the definition of a protein (Excerpt 1) focuses strictly on the number of amino acids in the polymer in order to “provide regulatory certainty and provide a bright-line interpretation of the term ‘protein,’” and went into effect on March 23, 2020. In other words, amino acid polymers less than or equal to 40 amino acids will be regulated as peptide drugs and any amino acid polymers greater than 40 amino acids will be regulated as protein biologics in the United States as of March 23, 2020. 65
Excerpt 1: Final Statutory Definition of “Protein,” as stated in 21 CFR Part 600 paragraph (h)
6
(Food and Drug Administration, HHS, 2020) 6A protein is any α amino acid polymer with a specific, defined sequence that is greater than 40 amino acids in size. When 2 or more amino acid chains in an amino acid polymer are associated with each other in a manner that occurs in nature, the size of the amino acid polymer for purposes of this paragraph (h)6 will be based on the total number of amino acids in those chains, and will not be limited to the number of amino acids in a contiguous sequence.
International Council for Harmonisation M3(R2) was written for the development of pharmaceuticals and is intended to be a “guide for drug development.” Since peptides are regulated as drugs in the United States, ICH M3(R2) has often been applied in the past to guide peptide therapeutic nonclinical programs even though peptides are not mentioned specifically. Within the scope of ICH M3(R2), consultation of ICH guidances for specific product areas is recommended if they exist, such as biotechnology-derived products which are covered under ICH S6. The scope of ICH M3(R2) under Section 1.3 indicates that only the timing of nonclinical studies described in ICH M3(R2) applies to biotechnology-derived products, and that the most appropriate type of nonclinical studies for these products are described in the 1997 ICH S6 Guideline for Preclinical Safety Evaluation for Biotechnology-Derived Pharmaceuticals (ICH S6). Revisions to ICH S6 have since been made to include updates on species selection, study design, immunogenicity, reproductive and developmental toxicity, and carcinogenicity, which are described in the current widely used ICH S6(R1), published in June of 2011. 66
The definition of biotechnology-derived products ICH S6(R1) has not always been interpreted the same way by scientists working with peptide therapeutics. According to the scope of ICH S6(R1) under Section 1.3, biotechnology-derived peptide products are those purified from plants, animals, or cell cultures. Paragraph 2 of Section 1.3 goes on to state that the principles outlined in ICH S6(R1) may also apply to chemically synthesized peptides. Thus, ICH S6(R1) has also been applied to peptide therapeutics in the past. However, potential safety concerns for synthesis-related processes are not discussed in ICH S6(R1). Thus, it is important to note that ICH S6(R1) will not always be considered by regulators to apply to all chemically synthesized peptide therapeutics.
It is notable that neither ICH M3(R2) nor ICH S6(R1) specifically apply to synthetic or conjugated peptides, with many peptide-based products essentially falling somewhere between both guidances. The nonclinical studies recommended by each guidance have several notable differences. Thus, disagreements as to the most appropriate ICH guidance to apply to synthesized and conjugated peptide products have arisen, particularly between industry and regulatory scientists. When considering the culmination of imperfectly fitted guidances, revisions, terminology refinement, and interpretation differences compounded by transitions between Review Centers and changes in regulatory processes, it becomes understandable that lack of clarity for nonclinical expectations for peptide therapeutics has led to confusion and frustration for both investigators and regulators. Historically, differences in interpretation and application of guidances ICH M3(R2) and ICH S6(R1) have resulted in additional information and study requests from Review Divisions, which have been associated with development delays and clinical holds for investigators.
Clarifying the Peptide Therapeutic Drug Space
Both synthetic and recombinant peptide products are regulated as drugs in the United States by CDER. Thus, strictly speaking, all peptide products may be subject to ICH M3(R2) principles. Peptides purified from tissues or cells, including recombinant peptides produced in cell culture, clearly meet the definition for biotechnology-derived drug products and are subject to recommendations outlined by ICH S6(R1). In contrast, synthetic peptides are not biotechnology-derived products and may be subject to ICH M3(R2) recommendations. Nevertheless, there are some scenarios where application of ICH S6(R1) principles is acceptable for synthetic peptides. For example, safety assessment recommendations outlined by ICH S6(R1) may be sufficient for synthetic peptides that contain only natural proteinogenic amino acids, particularly if there are no significant safety concerns related to the synthesis process, such as synthesis-related impurities. Proteins are generally regulated as biotechnology-derived biologics by the FDA Center for Biologics Evaluation and Research.
Proteins, biotechnology-derived peptides and synthetic peptide products can be conjugated; however, conjugation generally does not affect determination as to whether a product will be regulated as a drug or biologic. Conjugated moieties can include nonpeptide components, such as fatty acids, lipids, nucleic acids, or chemical entities, including small molecules, toxins, or metal nanoparticles, such as bionanomaterials. Conjugation to nonpeptide components is made possible using linkers, which can be small molecules that have inorganic and/or organic chemistries that are static, hydrolysable, or cleavable. Antibody-drug conjugates are an example of protein biologics conjugated to nonpeptide components. Safety concerns related to the conjugation process, the linker, or other non-amino acid components may be subject to additional studies. Since potential safety concerns for conjugated products or conjugation-related processes are not discussed in ICH S6(R1), application of ICH M3(R2) to conjugated products has been implemented in the past. For example, if a nonnatural or small molecule new molecular entity (NME) is part of a conjugated product, applications of ICH M3(R2) principles may be deemed appropriate.
International Council for Harmonisation M3(R2) Versus ICH S6(R1) Nonclinical Study Recommendations
The primary differences between ICH M3(R2) and ICH S6(R1) in nonclinical study recommendations pertain to the duration and type of general toxicology, metabolism, and genetic toxicity studies. Although both guidances recommend general toxicology studies in 2 species (1 rodent and 1 nonrodent), the maximum duration is 6 months for both species under ICH S6(R1), but 9 months for nonrodents under ICH M3(R2). Furthermore, studies longer than 1 month may only be necessary in one species under ICH S6(R1), depending on the 1-month toxicology findings and the species pharmacodynamic activity. Historically, both approaches have been applied to peptide products with acceptability of abbreviated nonclinical programs determined on a case-by-case basis. However, the factors contributing to acceptance of abbreviated studies in place of longer term nonrodent toxicity studies (eg, 6 months vs 9 months) for some peptide products remain unclear. Given the large costs of animal studies, greater understanding of regulatory expectations regarding the duration of nonrodent toxicity studies could be very beneficial to many drug developers in the field of peptide therapeutics. Furthermore, since drug development costs are often transferred to prescription costs for patients, which can affect patient accessibility to needed medications, further discussions regarding the scientific rationale and clarity concerning regulatory expectations are warranted.
Metabolism of amino acid polymers involves catabolism into individual amino acid components. For peptides composed of only naturally occurring amino acids, there are no safety concerns after catabolism. Thus, ICH S6(R1) does not recommend metabolism studies for biotechnology-derived products. However, for peptide products containing nonnatural amino acids (NNAAs), of which there are nearly 600 to date, or other nonnatural components, such as linkers or NMEs, the potential for safety concerns of catabolic products may not always be clear and ICH M3(R2) principles may be applied. In most cases, catabolism would likely result in nonnatural components bound to an adjacent amino acid. Nevertheless, there may be instances where it may be appropriate to assess free forms of nonnatural components or reactive ends, such as when hydrolysable or cleavable linkers are used in conjugated products. A risk-based rationale may also warrant evaluation of catabolic characterization and/or PK of nonpeptide components on a case-by-case basis. Examples include a positive in vivo genetic toxicity result, unusual or unexpected adverse toxicity compared to other members of a drug class, and scenarios when linkers or nonpeptide moieties are part of a prodrug leaving group. Although hazard analysis of some nonnatural components commonly used in peptide products may have been previously evaluated by regulators, without a right of reference to the GLP study results, new studies may be expected.
A significant difference between ICH M3(R2) and ICH S6(R1) nonclinical study recommendations is genetic toxicity safety assessments. According to ICH S6(R1), genetic toxicity studies are not warranted for biotechnology-derived products. However, ICH M3(R2) recommends that a full battery of genetic toxicity studies is completed according to ICH S2(R1) guidelines prior to phase 2 clinical trials. This battery includes an in vitro mutation assay in bacteria and an in vivo rodent assay with chromosomal damage assessment in either 2 tissues (option 2) or in 1 tissue along with an in vitro cytogenetic test for chromosomal damage in mammalian cells (option 1).
Discussion of Genetic Toxicity Assessments
Some of the genetic toxicity recommendations may not be relevant to peptides. Since the presence of histidine or tryptophan resulting from degradation of amino acid chains can result in false positives and confound bacterial mutation Ames assays, positive Ames assay results for peptide products are questionable and do not contribute meaningful data to the overall hazard assessment. In vitro cytogenetic assays in mammalian cells with peptide products that are not cell penetrable are also unlikely to provide meaningful genetic toxicity data. Traditionally, it was assumed that most intact peptide products do not penetrate cells, which implies that in vitro genetic toxicity assessments would only provide relevant assessment of process-related impurities.
Although assessment of genotoxic potential of process contaminants associated with purification of biotechnology-derived products from tissues or cell culture is not warranted as described by ICH S6, it is reasonable to expect evaluation of nonpeptide impurity profiles of synthesis-related and conjugation-related processes by quantitative structure activity relationship (QSAR) analysis. Thus, it follows that it would also be reasonable to expect positive alerts to be followed-up with assessments for mutagenic potential according to ICH M7(R1), and that impurities would be identified and/or qualified if above ICH Q3A and ICH Q3B thresholds. Furthermore, it is notable that ICH S6 indicates that cases with cause for concern for genetic toxicity testing in relevant systems include examples of conjugated amino acid polymer products with organic linkers.
Assuming that there are no valid in vitro approaches to assess genetic toxicity of peptide products, the in vivo assay becomes the most relevant approach for evaluating genotoxicity concerns of nonbiotechnology-derived peptide products, conjugated amino acid polymers, and potential catabolic byproducts. However, as cell-penetrable peptides become more abundant, and as assays reveal that more peptides may penetrate cells than previously thought, the potential usefulness of in vitro clastogenicity assays to rule out clastogenic potential of cell-penetrable peptide products was discussed. Given the history of overall lower genotoxicity concern for peptide products compared to other drug products, the potential for genetic toxicity to be sufficiently evaluated for cell-penetrable peptides using in vitro clastogenicity assays was discussed.
The Discourse on the Method published by René Descartes, 67,68 a renowned 16th Century French mathematician, metaphysician, and natural philosopher, used the analogy of architectural destruction and rebuilding of a town and buildings to describe a quasi-mathematical procedure for achieving knowledge that is based on scientific fact and unbiased from previous potential misconceptions that have been accepted to be true in the past. Descartes’s philosophical method of throwing out previously accepted scientific philosophies and rebuilding scientific principles based only on indubitable scientific facts continues to be applicable in describing phenomena in the natural sciences today, particularly when scientific observations have been described that don’t completely align with current scientific philosophies. Thus, this process can be useful in reevaluating scientific areas of disagreement. At the ACT symposium on Developmental and Regulatory Challenges for Peptide Products, attendees explored application of this process to genetic toxicology causes for concern with peptide products and associated nonclinical studies. It was discussed that it may be possible to construct ideal nonclinical program expectations by considering the most scientifically justifiable concepts and rationales coming from both ICH M3(R2) and S6(R1) guidances, while keeping in mind emerging technologies, future directions of the peptide product industry, and developmental capabilities and challenges. Continued discussions regarding safety assessments for peptide therapeutics, including rationale for 6-month versus 9-month nonrodent chronic toxicology studies, and genetic toxicity potential will be necessary to reach a consensus among scientists in the field.
Discussion of Genotoxic Potential for Peptide Therapeutics
In an effort to explore the genotoxicity potential of peptide products from a conservative perspective, the following question was asked: is there a scientific basis for reassurance of nongenotoxicity without testing? Subsequently, this leads one to ask: are there peptides that test positive in genotoxicity assays? To address these questions, a broad literature search was conducted and evidence of 9 peptides and amino acid polymers with positive results in genotoxicity assays were identified (Table 3), which were categorized into 2 groups.
Literature Results of Peptides and Proteins With Positive Genotoxicity Assay Results.
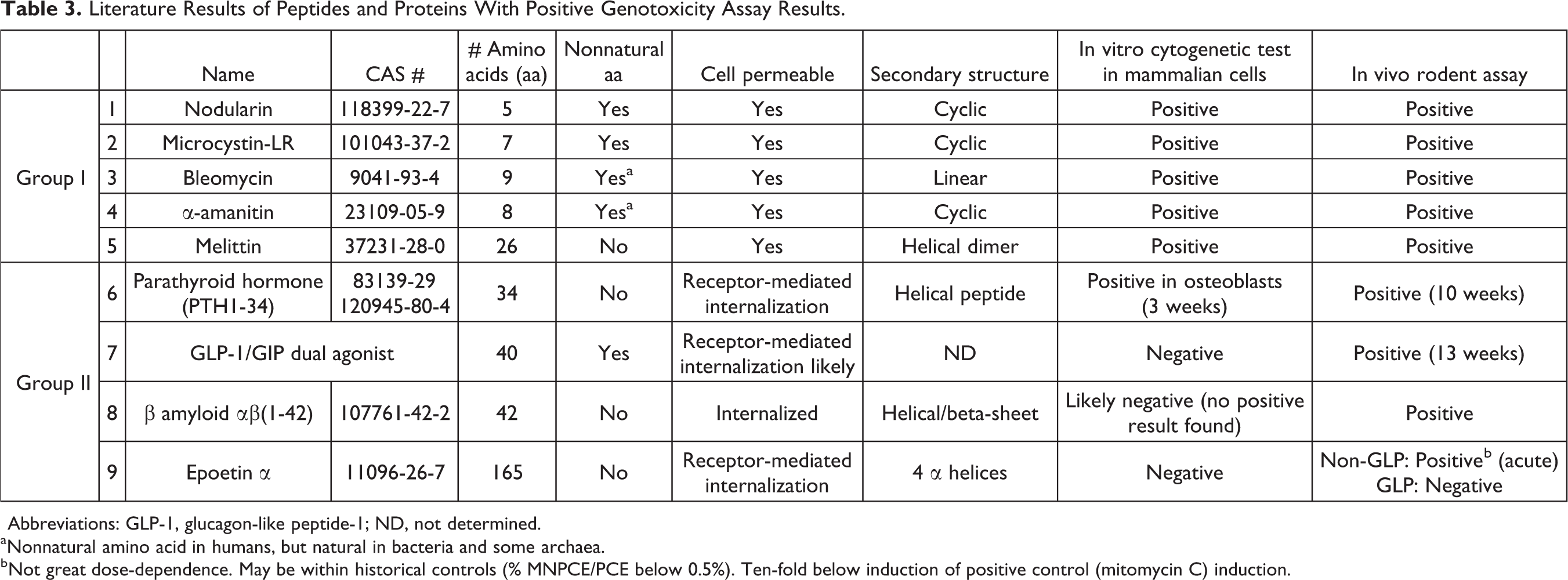
Abbreviations: GLP-1, glucagon-like peptide-1; ND, not determined.
a Nonnatural amino acid in humans, but natural in bacteria and some archaea.
b Not great dose-dependence. May be within historical controls (% MNPCE/PCE below 0.5%). Ten-fold below induction of positive control (mitomycin C) induction.
Group I includes the 5 peptides nodularin, 69 microcystin, 69 bleomycin, 70 α-amanitin, 71 and melittin, 72 which have various tertiary structures and all tested positive for both clastogenicity in mammalian cell in vitro cytogenetic tests and genotoxicity in rodent in vivo assays. In fact, it may be argued that the genotoxic potential was predicted by the in vitro clastogenicity assay, which was then confirmed by the in vivo rodent assay. It is notable that all 5 group I peptides are cell permeable, 73,74 but only 4 of the 5 contain NNAAs. Interestingly, not only are each of the group I peptides clastogenic genotoxins, but they are also known natural toxins.
Group II includes 2 peptides, parathyroid hormone (PTH1-34) 75 and a GLP-1/GIP dual agonist, 76 and 2 proteins, β amyloid αβ 1 -41,77,78 and epoetin α, 79 that were not associated with positivity in standard GLP clastogenicity in vitro assays but were associated with positive results in the in vivo rodent assay. However, for each group II amino acid polymer, the positive in vivo micronucleus assay results were secondary to downstream pharmacodynamic activity and were likely false positives. Thus, all 4 are decidedly not genotoxins. Nevertheless, it is important to acknowledge that they may still be nongenotoxic carcinogens, which is assessed in chronic carcinogenicity assays addressed in both ICH M3(R2) and S6(R1) guidances.
Comparison of Perspectives
When considering the potential for whether or not new peptide products could be identified as group I clastogenic genotoxins, philosophies coming from liberal and conservative perspectives were explored. From the liberal perspective, it may be argued that there is no concern for inappropriate clinical exposure because toxins are likely to be identified early in development and either eliminated early on or used according to the toxic characteristics, such as with antibody-drug combination products for cancer treatment. However, from the conservative perspective, it may be argued that there remains a need for some sort of reassurance of nongenotoxicity for new peptide products in absence of conducting a battery of genotoxicity studies.
To determine whether structural characteristics could be associated with predicting genotoxicity risk, types of structural modifications present in the group I clastogenic peptides were explored. Although cell penetration alone would not be expected to drive genotoxicity, it is noticeable that all of the group I peptides are cell penetrable. Another noticeable structural characteristic is that 4 of 5 contain NNAAs. At least 593 NNAAs have been reported in the literature 80 and the lack of genotoxic potential of each one has not been verified. Other nonnatural modifications, such as nonnatural linkers or other nonnatural components, may also raise concerns of genotoxic risk for peptide products. It is also notable that naturally occurring chemical modifications, such as ferrous chelation 81 and advanced glycation end products, have been reported to be genotoxic in the literature. 82 Advanced glycation end products are produced by reaction of glucose with primary amino groups, which are likely to be increased under hyperglycemic physiological conditions. 83
In order to provide reassurance of the lack of genotoxicity without additional testing for each NNAA, nonnatural component, and potential catabolic products containing the NNAA or nonnatural components, a public database of experimental GLP data could be created. Furthermore, such a database could potentially be used to construct an in silico assessment tool using a 3-D spectroscopic data-activity relationship (SDAR) modeling approach 84 in order to predict the genotoxicity potential of NNAAs and peptide components that are otherwise too large for QSAR assessment. Such an in silico assessment could then be used alongside QSAR analyses of synthesis-related and conjugation-related impurities to determine a more complete prediction of genotoxic risk for peptide products. It is intriguing to imagine that perhaps the combination of in silico approaches, such as impurity QSAR analyses and a hypothetical NNAA SDAR analysis, could represent the first step in genotoxic assessment, such that positive alerts could be followed up with additional nonclinical studies, but the absence of alerts may not. However, taking on such an endeavor would likely be costly and the potential for success in reducing potential genotoxicity concerns and number of additional genotoxicity studies requested by regulators remains uncertain.
Summary of Current Regulatory Expectations for Peptide Products
When considering which regulatory guidance to apply to peptide products, it is important to remember that ICH S6 does not specifically apply to synthetic peptide products in that they are not considered to be biotechnology-derived products and regulators may apply ICH M3R2 principles. On the other hand, not all studies outlined by ICH M3R2 may be relevant for all synthetic peptide products. It is important to keep in mind that CDER Review Divisions may make nonclinical study requirement determinations on a case-by-case basis, particularly for inclusion of genotoxicity studies and acceptance of 6-month nonrodent general toxicology studies in place of a 9-month study. Furthermore, in silico evaluation of synthesis-related and conjugation-related impurities can be expected, including follow-up of positive alerts according to ICH M7 guidelines and identification and/or qualification of impurities according to ICH Q3A and/or Q3B thresholds. Additionally, in vitro and/or in vivo genetic toxicology studies may be expected. For conjugated products, additional studies that may be expected by CDER Review Divisions include assessment of reactive end potential, assessment of linker-related or nonnatural catabolites, and PK assessments of nonpeptide components. The need for submission of catabolic characterization may be based on risk-based rationale, which could include a positive in vivo genetic toxicity result, unusual or unexpected adverse toxicity compared to other members of a drug class, and/or the presence of linker of nonpeptide moieties that are part of a prodrug leaving group. Given that it is not always clear which regulatory guidance or which parts of each guidance should be applied in nonclinical development programs for all peptide products, and given that nonclinical requirements may be determined on a case-by-case basis, it is wise to contact the applicable CDER Review Division with questions regarding design of the nonclinical program design. Submission of rational supporting the proposed nonclinical studies may also be useful in determining the most appropriate path forward with the least degree of potential for future delays in regard to the nonclinical package.
Questions for the Peptide Therapeutic Community
The speakers and organizers of the 2019 ACT Symposium on “Development and Regulatory Challenges for Peptide Therapeutics” shared a list of peptide questions aimed at promoting discussion and gathering feedback from the peptide therapeutics community. Although it was ultimately not possible to present the peptide questions as an anonymous survey, the authors would like to invite readers to submit their responses and opinions as directed in the Supplemental Peptide Therapeutics Survey so that their experiences and perspectives can be captured. All sources of survey responses will be kept confidential.
The intent of the peptide questions is to identify scientific aspects for potential safety hazard concerns and appropriate nonclinical assessment strategies by drawing on the experience and expertise of industry, regulatory, and academic scientists working in the Peptide Therapeutics field, in order to inform, continue discussions, and ultimately benefit the eventual development of an appropriate regulatory guidance for nonclinical safety assessments of synthetic and conjugated peptide products. Although the questions were drafted by the organizers and speakers Symposium, the FDA CDER Pharmacology Toxicology Immediate Office and Pharmacology Toxicology Coordinating Committee Biologics and Genetic Toxicology Subcommittees were invited to comment and provide additional questions.
Concluding Comments
The Symposium on Development and Regulatory Challenges for Peptide Therapeutics at the 40th Annual Meeting of the ACT was held in November of 2019, as there has been increased interest in the use of peptide therapeutics to treat a variety of human diseases. In this symposium, the specific topics included: 1 peptide therapeutic progress and future directions, and approaches to discover, optimize, assess, and deliver combination peptide therapeutics for treatment of diseases 2 ; toxicological considerations to deliver peptide drug-device combination products for efficient development and optimal patient benefit and adherence 3 ; industry and regulatory perspectives on the regulation of synthetic and conjugated peptide products, including exploration of regulatory classifications, interpretations, and application of existing guidances in determining nonclinical study requirements (ICH M3[R2] vs ICH S6); and 4 discussion of the 2016 HESI working group assessment of genotoxicity testing requirements. With the discussion sparked from this symposium, it is our hope that we have identified some scientific aspects for potential safety hazard concerns and appropriate nonclinical assessment strategies from our shared experience and expertise which will lead to the development of an appropriate regulatory guidance for nonclinical safety assessment of synthetic and conjugated peptide products. Similarly, a recent publication from Mitra et al, also concluded that there is considerable ambiguating around the development of peptide therapeutics. 85 Their analysis of a data set consisting of 47 peptides approved in the United States between 1998 and 2019 provides a general framework for nonclinical development of peptide therapeutics and also highlights the need for specific regulatory guidance in terms of genotoxicity, impurity, metabolite, and nonproteogenic amino acid assessment for novel peptide development.
Supplemental Material
Supplemental Material, Supplemental_Peptide_Therapeutics_Survey - Development and Regulatory Challenges for Peptide Therapeutics
Supplemental Material, Supplemental_Peptide_Therapeutics_Survey for Development and Regulatory Challenges for Peptide Therapeutics by Doris Zane, Paul L. Feldman, Tomi Sawyer, Zhanna Sobol and Jessica Hawes in International Journal of Toxicology
Footnotes
Authors’ Contribution
D.Z., P.L.F., T.S., Z.S., and J.H. contributed to conception and design; acquisition, analysis, and interpretation; drafted manuscript; and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Acknowledgments
The authors would like to thank Dr Timothy McGovern for participating in the Symposium panel discussion and for helpful comments on preparation of this manuscript and the Peptide Therapeutics Survey. The authors would also like to thank the CDER Pharmacology Toxicology Coordinating Committee (PTCC) Biologics and Genetic Toxicology Subcommittees for their feedback, comments and contributions to the Peptide Therapeutics Survey.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.