
Abstract
The role of autophagy in pyocyanin (PCN)-induced toxicity in the central nervous system (CNS) remains unclear, with only evidence from our group identifying it as a mechanism underlying toxicity in 1321N1 astrocytoma cells. Therefore, the aim of this study was to further examine the role of autophagy in PCN-induced toxicity in the CNS. To achieve this, we exposed 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells to PCN (0-100 μmol/L) and tested the contribution of autophagy by measuring the impact of the autophagy inhibitor 3-methyladenine (3-MA) using a series of biochemical and molecular markers. Pretreatment of 1321N1 astrocytoma cells with 3-MA (5 mmol/L) decreased the PCN-induced acidic vesicular organelle and autophagosome formation as measured using acridine orange and green fluorescent protein-LC3 -LC3 fluorescence, respectively. Furthermore, 3-MA (5 mmol/L) significantly protected 1321N1 astrocytoma cells against PCN-induced toxicity. In contrast pretreatment with 3-MA (5 mmol/L) increased PCN-induced toxicity in SH-SY5Y neuroblastoma cells. Given the influence of autophagy in inflammatory responses, we investigated whether the observed effects in this study involved inflammatory mediators. The PCN (100 μmol/L) significantly increased the production of interleukin-8 (IL-8), prostaglandin E2 (PGE2), and leukotriene B4 (LTB4) in both cell lines. Consistent with its paradoxical role in modulating PCN-induced toxicity, 3-MA (5 mmol/L) significantly reduced the PCN-induced production of IL-8, PGE2, and LTB4 in 1321N1 astrocytoma cells but augmented their production in SH-SY5Y neuroblastoma cells. In conclusion, we show here for the first time the paradoxical role of autophagy in mediating PCN-induced toxicity in 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells and provide novel evidence that these actions may be mediated by effects on IL-8, PGE2, and LTB4 production.
Introduction
Pseudomonas aeruginosa is a gram-negative bacterial pathogen, capable of causing infection in a wide range of species, including humans, and is a leading cause of nosocomial infections. 1 Within the human central nervous system (CNS), it is implicated as a causal factor in a number of pathologies, including gram-negative bacillary meningitis, meningoencephalitis, ventriculitis, and brain abscesses. 2 –4 Primary CNS infections by P aeruginosa are rare with the majority occurring secondary to surgical procedures, chronic otitis media, or head trauma. 5 –7 However once established, these infections are difficult to treat with rapid progression and a high mortality rate. 1 Much of the pathogenicity of P aeruginosa has been attributed to the secreted virulence factor pyocyanin (PCN). 8,9
The PCN is a redox-active, blue-green phenazine pigment secreted by P aeruginosa and is considered to be an important virulence and proinflammatory factor for the organism. Sputum from patients with cystic fibrosis colonized by P aeruginosa has been shown to contain PCN at concentrations up to 27.3 µg/mL (∼130 µmol/L), 10 and its presence has also been demonstrated to be crucial for successful P aeruginosa respiratory infection in mice. 11 The PCN has been shown to have numerous damaging effects on eukaryotic cells including reversible ciliary dysfunction in human respiratory 12 and sheep tracheal epithelial cells, 13 bronchoconstriction, 14 and neutrophilia 15 in sheep. In addition, PCN modulates several cellular functions and acts as an immune and inflammatory modulator by reducing mucociliary clearance 16 and increasing interleukin 8 (IL-8) 17 and nuclear factor κB production. 18 The PCN has also been shown to inhibit lymphocyte proliferation by decreasing IL-2 levels and expression of its receptor. 19 The majority of PCN’s toxicity has been attributed to the generation of reactive oxygen species (ROS) and depletion of intracellular antioxidant defences. Our group has causally linked oxidative stress to PCN-induced toxicity and production of proinflammatory proteins in human respiratory epithelial cells. 18 Despite extensive knowledge of PCN’s effects in respiratory models, very little is known about the toxicity of PCN in other models such as the CNS. Due to the low molecular weight and zwitterionic properties of PCN, it has been proposed that it can readily cross biological membranes and enter the systemic circulation. However to our knowledge, the ability of PCN to cross the blood–brain barrier is yet to be demonstrated.
Recently, our group provided the first in vitro evidence on the toxicity of PCN in the CNS. 20 The PCN produced dose-dependent toxicity in 1321N1 astrocytoma cells, but in contrast to respiratory models, oxidative stress was not centrally involved. Instead, autophagy appeared to promote cell death in response to PCN treatment. In general, autophagy refers to an evolutionary conserved process in which intracellular organelles are sequestered in autophagosomes (AVs) and then processed to recycle cellular components and prevent the accumulation of damaged proteins and organelles. 21 This process has been associated with either cell death or survival. 22 Furthermore, there is strong evidence to suggest that autophagy has a protective function in neurons, and its dysregulation has been associated with neurodegeneration 23 and increased neuronal death in response to viral infection. 24 However, the role of autophagy in the etiology of PCN-induced CNS toxicity remains largely unknown with the only evidence provided by a single study from our group. Therefore, the aim of this study was to further investigate PCN-induced CNS toxicity in vitro and establish a role for autophagy in this toxicity. Astrocytes have a key role in maintaining CNS integrity and protecting neuronal cells against infectious agents including viruses and bacteria. 25 Furthermore, astrocytes have also been recently shown to regulate neuroinflammation by preventing microglial overactivation. 26 Therefore, we chose the 1321N1 astrocytoma cell line as our in vitro model of astrocytes in this study. As neurons can also be a direct target for several pathogens, we included SH-SY5Y neuroblastoma cells as our model of neuronal cells. Since autophagy is known to influence inflammatory and immune responses, we tested whether this process contributed to PCN-induced toxicity in 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells.
Materials and Methods
Cell Culture
Human glial cells from brain astrocytoma (1321N1) were obtained from Sigma-Aldrich (St Louis, Missouri), and human neuroblastoma cells (SH-SY5Y) were provided by Prof Greg Anderson (QIMR, Herston, QLD). Cells were grown and maintained at 37°C with 5% CO2 in complete Dulbecco’s Modified Eagle Medium (Invitrogen, Victoria, Australia) containing high glucose,
Resazurin (Alamar Blue) Proliferation Assay
Reduction of the redox dye resazurin to resorufin was used to measure the proliferation of cell cultures. 27,28 Cells were seeded at 2 × 105 Trypan blue-excluding cells/mL in 96-well microtiter plates and pretreated with either 5 mmol/L 3-methyladenine (3-MA) or N-acetylcysteine (NAC) for 30 minutes prior to the addition of PCN (0-100 μmol/L). Following incubation for 24 hours, the medium above the cells was removed and replaced with fresh medium containing 44 μmol/L resazurin. After a 3-hour incubation, reduction of resazurin to resorufin was determined by fluorescence (excitation 530 nm; emission 590 nm) using a Fluoroskan Ascent microplate fluorometer (Thermo Scientific, Victoria, Australia). Appropriate cell-free controls were included to test for potential interaction between NAC and PCN with resazurin. Under all conditions tested, extent of resazurin reduction was directly proportional to viable cell counts (data not shown) performed on a Countess automated cell counter (Invitrogen).
Acridine Orange Staining
The presence of acidic lysosomes and development of acidic vesicular organelles (AVOs), a characteristic of autophagy, were visualized by acridine orange staining. 29,30 Following treatment and incubation described earlier, cells were stained with 1 μg/mL acridine orange (Sigma) for 15 minutes at 37°C. Cells were washed twice with phosphate-buffered saline (PBS), and fluorescence was visualized under a Leica DM2000 fluorescence microscope (Leica Microsystems, Victoria, Australia). Contrast was enhanced by 0.4% for all images using ImageJ 1.43u (National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997-2012).
Green Fluorescent Protein-LC3 -LC3 Immunofluorescence
LC-3 expression was detected through the use of the Premo™ Autophagy Sensor LC3B-GFP BacMAM 2.0 kit (Invitrogen). Cells were seeded in 6-well plates at 5 × 104 cells/mL and allowed to attach for 8 hours. Following attachment, the cells were transfected with green fluorescent protein (GFP)-LC3B. After incubation for further 24 hours, the cells were treated with 100 µmol/L PCN in the absence or presence of 5 mmol/L 3-MA. After 24-hour incubation, the cells were washed twice with PBS, then mounted in 1 mL Hanks balanced salt solution. Visualization of green fluorescence was observed using an Olympus Cell-M microscope (Olympus Australia, Mt Waverly, Victoria).
Caspase-3 Measurement
Caspase-3 activation was used as an index for apoptosis. Cells were seeded at a density of 2 × 105 cells/mL, incubated for 24 hours, and then treated as described earlier. Following incubation for a further 24 hours, caspase-3 activity was determined using a caspase-3 fluorescence assay kit (Cayman Chemicals, Michigan). All steps were performed according to the manufacturer’s instructions. Fluorescence was read using a Fluoroskan Ascent microplate fluorometer (excitation 485 nm; emission 535 nm).
Determination of IL-8, Prostaglandin E2 , and Leukotriene B4
Cells were seeded at a density of 2 × 105 cells/mL, incubated for 24 hours, and then treated as described earlier. Following incubation for a further 24 hours, IL-8 was measured in 100 μL of cell culture supernatant by enzyme-linked immunosorbent assay (ELISA; Life Research, Victoria, Australia). Prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) were measured in 50 μL of cell culture supernatant by ELISA (Cayman Chemicals). All steps were performed according to the manufacturer’s instructions.
DCFH-DA Fluorimetry
2′,7′-Dichlorofluorescein diacetate (DCFH-DA) was used to measure the global ROS production as previously described. 31 Cells were seeded at a density of 2 × 105 cells/mL, incubated for 24 hours, and then treated as described earlier. Following incubation for 24 hours, the medium above the cells was replaced with serum-free medium containing DCFH-DA (10 μmol/L) for 30 minutes. The cells were then washed twice with PBS, and fluorescence, due to the oxidized dye (excitation 485 nm; emission 535 nm), was measured using a Fluoroskan Ascent microplate fluorometer (Thermo Scientific).
Statistical Analysis
Data were analyzed with the Tukey-Kramer multiple comparisons test, using Graphpad Instat software v3.10 (San Diego, California). Significance levels were defined as P < .05 (* and #), P < .01 (** and ##), and P < .001 (***and ###). All graphs were drawn using Graphpad Prism v4.01 (San Diego, California).
Results
Autophagy Mediates PCN-Induced Toxicity in 1321N1 Astrocytoma and SH-SY5Y Neuroblastoma Cells
Pyocyanin-induced toxicity in 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells was measured using resazurin reduction to resorufin as an index of proliferation. Exposure of 1321N1 astrocytoma cells to PCN at concentrations greater than 10 µmol/L significantly reduced cell viability (Figure 1A). In contrast, SH-SY5Y neuroblastoma cells were significantly more resistant to PCN-induced toxicity with only 100 µmol/L PCN significantly decreasing cell viability (Figure 1B). This result was confirmed with independent counts of viable cells using Trypan blue (data not shown). Pretreatment of 1321N1 astrocytoma cells with the autophagy inhibitor 3-MA (5 mmol/L) provided complete protection against PCN-induced toxicity (Figure 1A). In contrast, pretreatment with 3-MA (5 mmol/L) increased PCN toxicity in SH-SY5Y neuroblastoma cells at concentrations (0.1-10 µmol/L) previously shown to be nontoxic (Figure 1B). A similar paradoxical effect on PCN toxicity was also observed with 1 mmol/L 3-MA (data not shown). In addition, 3-MA by itself was nontoxic to both cell lines (data not shown).
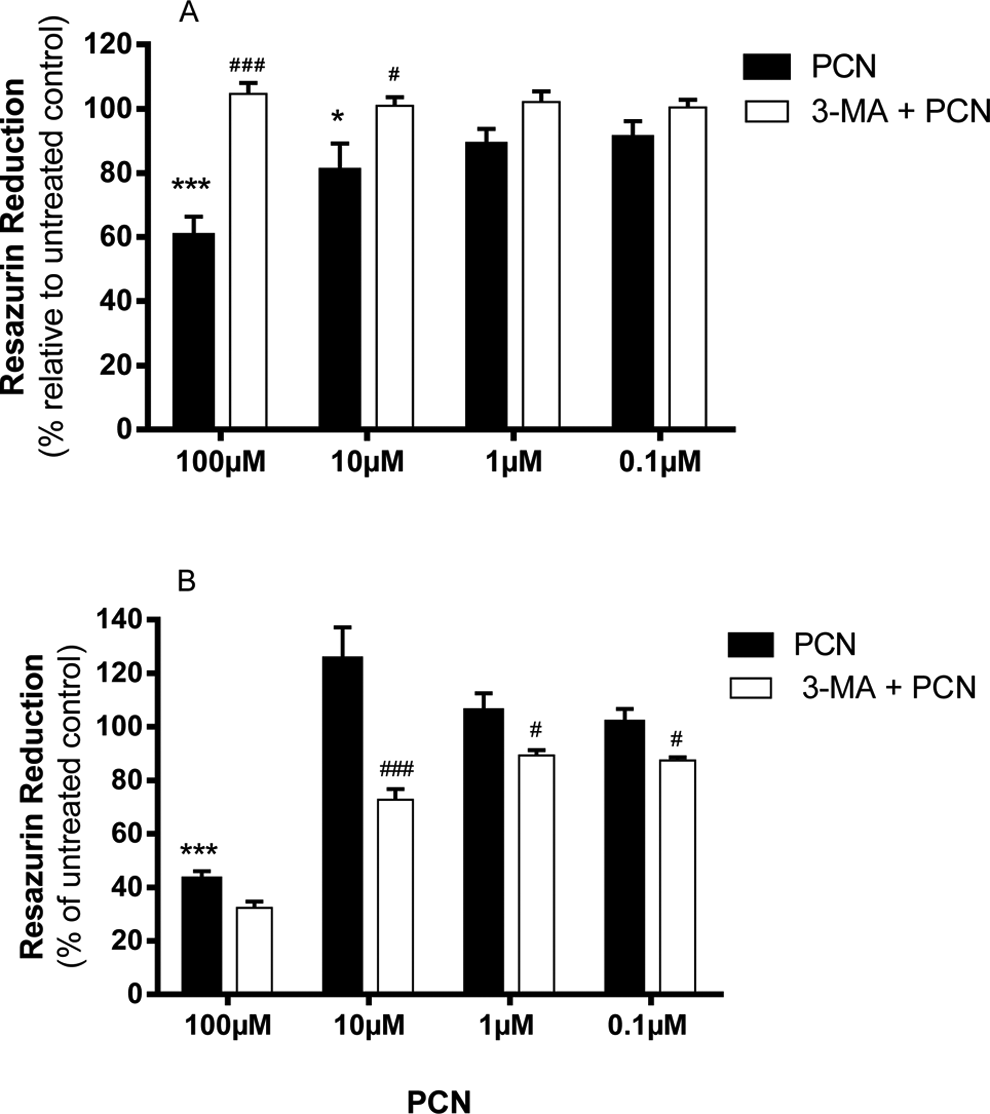
Impact of 3-MA on PCN-induced injury. A, 1321N1 and (B) SH-SY5Y cells were treated with PCN (0-100 μmol/L) in the absence or presence of 3-MA (5 mmol/L) for 24 hours. Cytotoxicity was measured using resazurin reduction to resorufin as an index of proliferation. Data shown are the mean ± standard deviation of 3 independent experiments. Comparisons: * untreated control versus PCN; # PCN versus treatments. PCN indicates pyocyanin; 3-MA, 3-methyladenine.
Parallel staining with acridine orange revealed that untreated 1321N1 astrocytoma control cells demonstrated a strong green diffuse fluorescence from the cytoplasm and nucleus with occasional punctate orange fluorescence, indicating the presence of AVOs(Figure 2). Basal AVO levels were visibly higher in SH-SY5Y neuroblastoma cells compared to 1321N1 astrocytoma cells as indicated by greater orange punctate fluorescence (Figure 2). Exposure of 1321N1 astrocytoma cells to PCN (100 μmol/L) increased basal AVO formation (Figure 2). However, in SH-SY5Y neuroblastoma cells, PCN (100 μmol/L) decreased basal AVO formation (Figure 2). Pretreatment with 3-MA (5 mmol/L) reduced AVO formation to below basal levels in 1321N1 astrocytoma cells, while in SH-SY5Y neuroblastoma cells, AVO formation was comparable to that of PCN treatment (Figure 2).
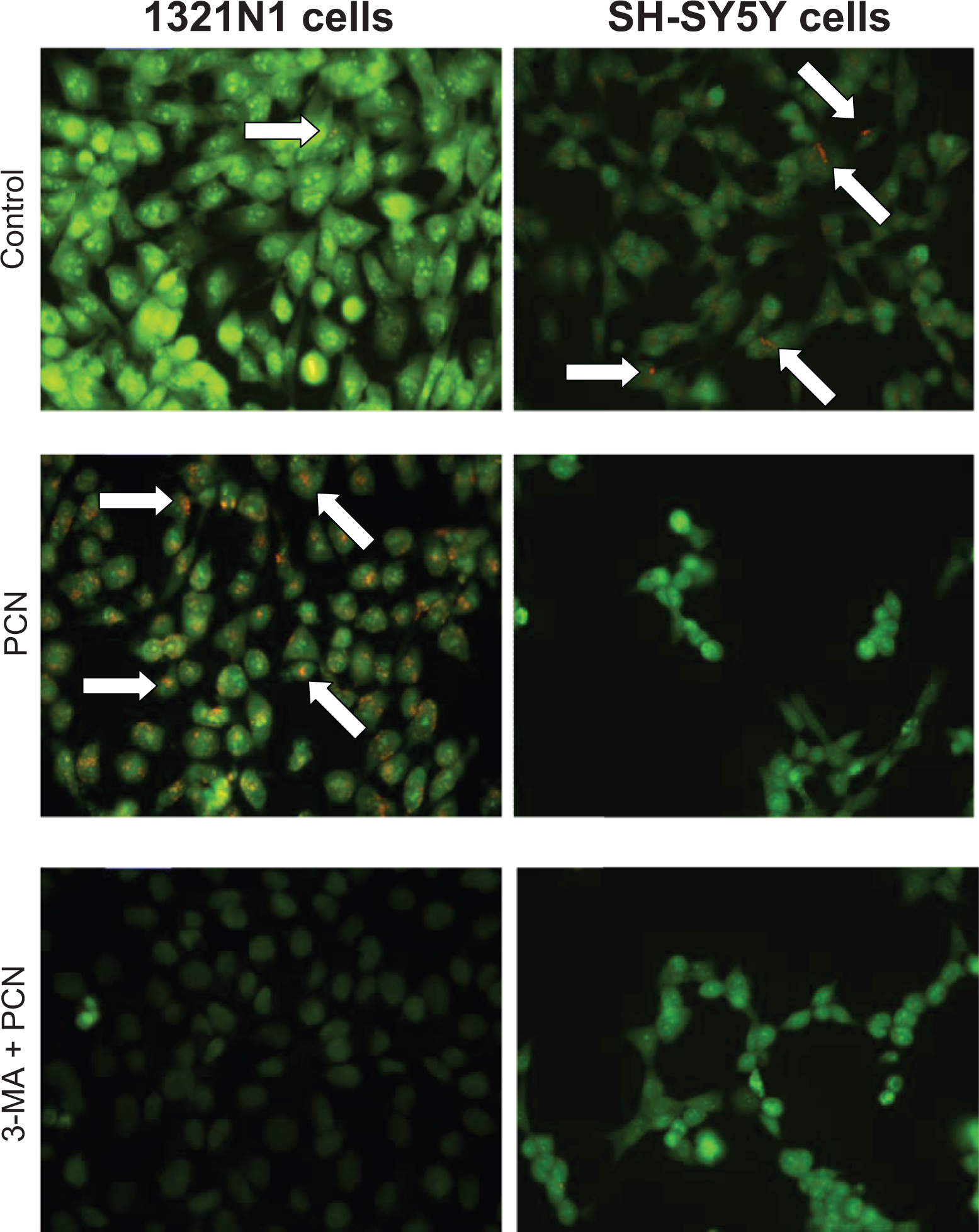
Pyocyanin effects on accumulation of AVOs. A, 1321N1 and (B) SH-SY5Y cells were treated with pyocyanin (0-100 μmol/L) in the absence or presence of 3-methyladenine (5 mmol/L) for 24 hours. The presence of AVOs (white arrows) was visualized by acridine orange staining. Contrast was enhanced by 0.4% for all images using ImageJ 1.43u. Images are at ×400 magnification and are representative of 3 independent experiments. AVO indicates acidic vesicular organelle.
Cells were transfected with GFP-LC3 to allow AV visualization. Similar to the observations of AVOs from acridine orange staining, both 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells displayed constitutive AV as indicated by green punctate fluorescence (Figure 3). Consistent with staining for AVOs, SH-SY5Y neuroblastoma cells displayed higher basal AV levels compared to 1321N1 astrocytoma cells (Figure 3). Exposure of 1321N1 astrocytoma cells to PCN (100 μmol/L) significantly increased AV levels compared to untreated control cells (Figure 3). Pretreatment with 3-MA (5 mmol/L) decreased PCN-induced AV formation to levels comparable to those observed in control cells (Figure 3). In SH-SY5Y neuroblastoma cells, PCN (100 μmol/L) decreased basal AV levels. Prior treatment with 3-MA (5 mmol/L) completely abolished green punctate fluorescence in these cells indicating decreased AV formation.
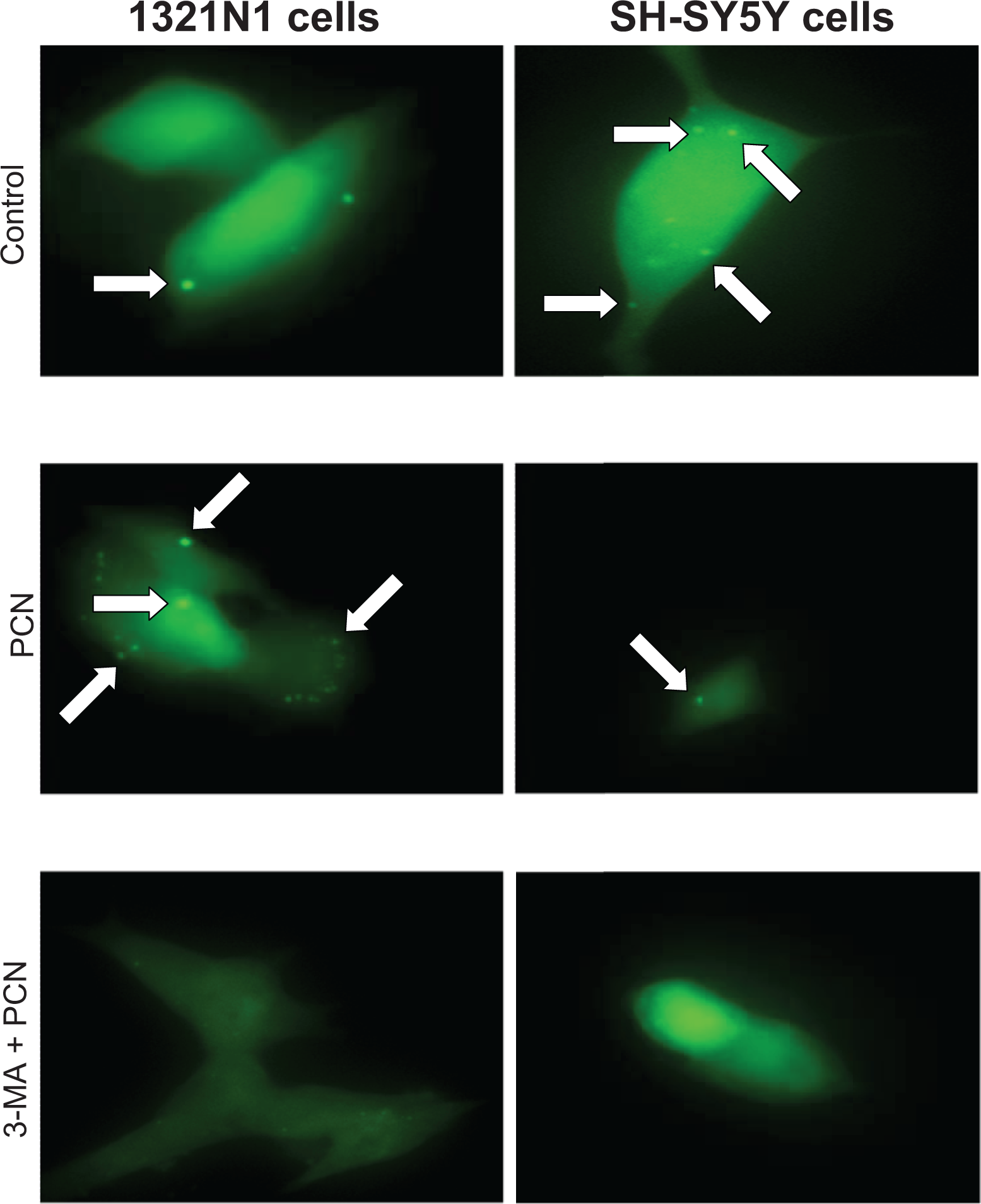
Pyocyanin effects on autophagosome formation. A, 1321N1 and (B) SH-SY5Y cells were treated with pyocyanin (0-100 μmol/L) in the absence or presence of 3-methyladenine (5 mmol/L) for 24 hours. The presence of AVs (white arrows) were visualized by transfecting cells with GFP-LC3. Contrast was enhanced by 0.4% for all images using ImageJ 1.43u. Images are at ×600 magnification and are representative of 3 independent experiments.
Caspase-3 activity was used an index of apoptosis in this study. As shown in Figure 4, PCN (100 μmol/L) significantly increased caspase-3 activity in both cell lines compared to untreated control cells suggesting increased apoptosis. However, pretreatment with 3-MA (5 mmol/L) significantly attenuated PCN-induced caspase-3 activity in both cell lines.
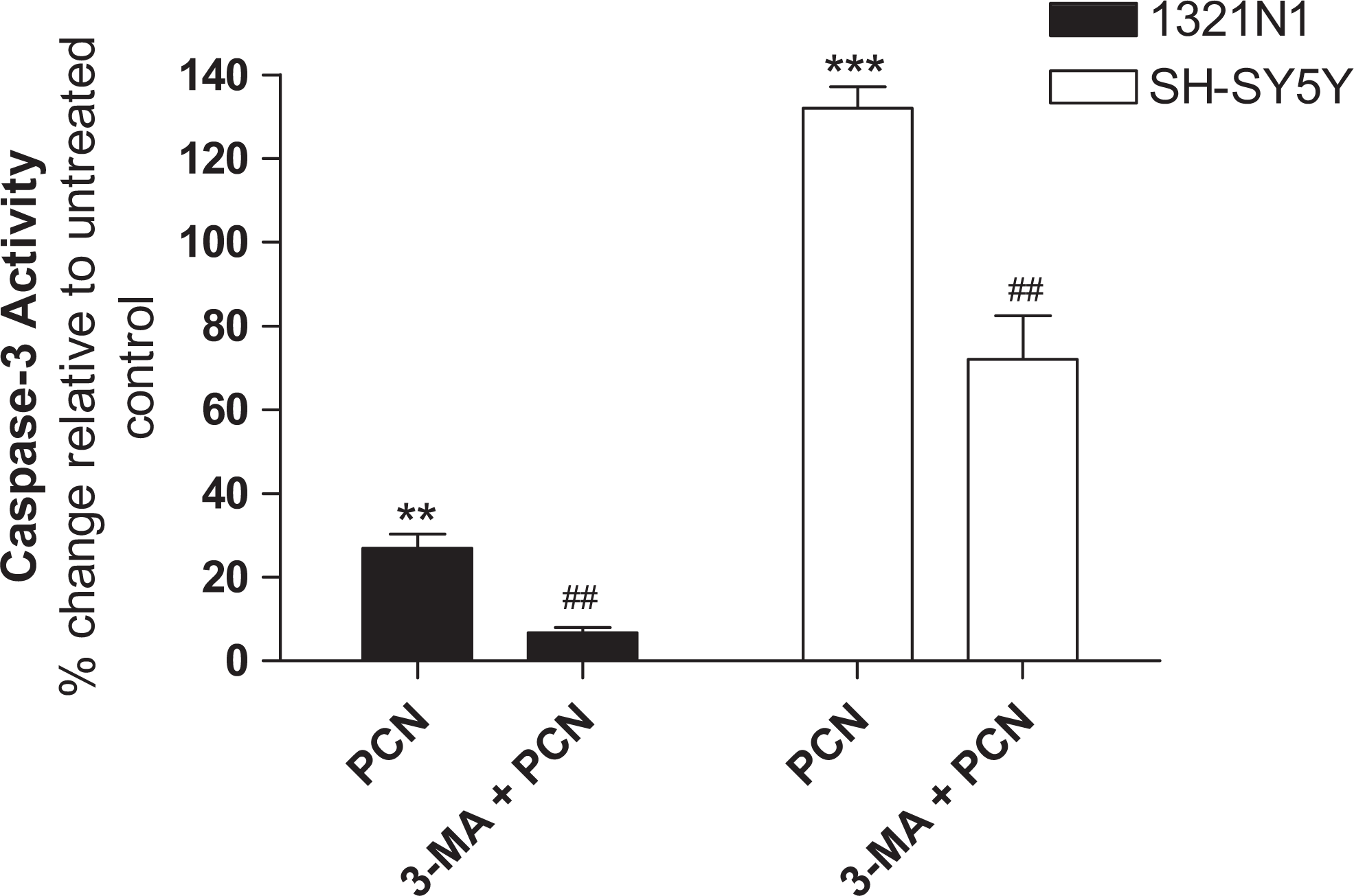
Pyocyanin-induced caspase-3 activation. Cells were treated with either PCN (100 μmol/L) in the absence or presence of 3-methyladenine (5 mmol/L) for 24 hours. Caspase-3 activation was then measured using a Cayman Chemicals assay kit. Data are shown as the percentage increase relative to untreated control and are the mean ± standard deviation of 3 independent experiments. Comparisons: * untreated control versus PCN; # PCN versus treatments. PCN indicates pyocyanin.
Modulation of PCN Toxicity in 1321N1 Astrocytoma and SH-SY5Y Neuroblastoma Cells may Involve Production of Inflammatory Mediators
To determine the effect of PCN on inflammatory markers in CNS models, we exposed 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells to PCN (100 μmol/L) and measured the release of IL-8, PGE2, and LTB4 after 24 hours. As shown in Figure 5, PCN significantly increased the production of IL-8, PGE2, and LTB4 in both cell lines.
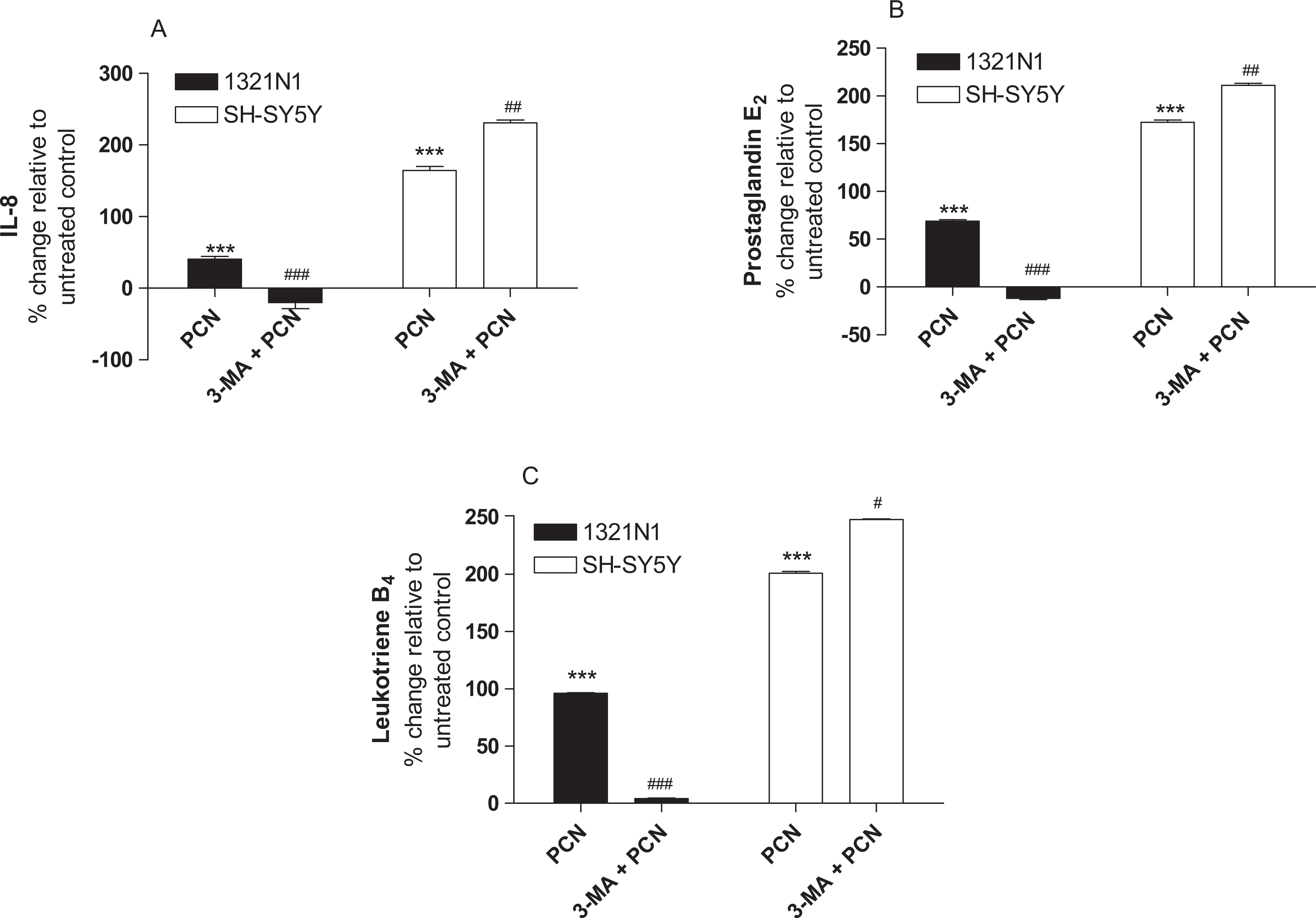
Pyocyanin-induced production of IL-8, PGE2, and LTB4. Cells were treated with PCN (100 μmol/L) in the absence or presence of 3-methyladenine (5 mmol/L) for 24 hours. IL-8, PGE2, and LTB4 levels were then measured by ELISA. Data are shown as the percentage increase relative to untreated control and are the mean ± standard deviation of 3 independent experiments. Comparisons: * untreated control versus PCN; # PCN versus treatments. PCN indicates pyocyanin; IL-8, interleukin-8; ELISA, enzyme-linked immunosorbent assay. PGE2, Prostaglandin E2; LTB4, leukotriene B4.
Pretreatment of 1321N1 astrocytoma cells with 3-MA (5 mmol/L) decreased the production of IL-8, PGE2, and LTB4 in response to PCN (Figure 5). In contrast, prior exposure of SH-SY5Y neuroblastoma cells to 3-MA (5 mmol/L) augmented the PCN-induced production of all 3 mediators (Figure 5). The effects of 3-MA (5 mmol/L) on IL-8, PGE2, and LTB4 are consistent with its paradoxical role in PCN-induced toxicity.
Oxidative Stress Is Not a Primary Mediator of PCN Toxicity in 1321N1 Astrocytoma and SH-SY5Y Neuroblastoma Cells
To test whether or not toxicity observed in 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells was causally linked to oxidative stress, we pretreated cells with the antioxidant NAC (5 mmol/L). The PCN (100 μmol/L) significantly increased ROS production in both 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells as measured by oxidation of DCFH-DA (Figure 6A). Consistent with its antioxidant properties, NAC (5 mmol/L) significantly attenuated this increase in ROS (Figure 6A). However, pretreatment of cells with NAC (5 mmol/L) did not protect either cell line against PCN-induced toxicity (Figure 6B and C). Increased NAC concentrations up to 25 mmol/L also failed to protect cells against PCN-induced cell injury (data not shown) consistent with our earlier findings. 32
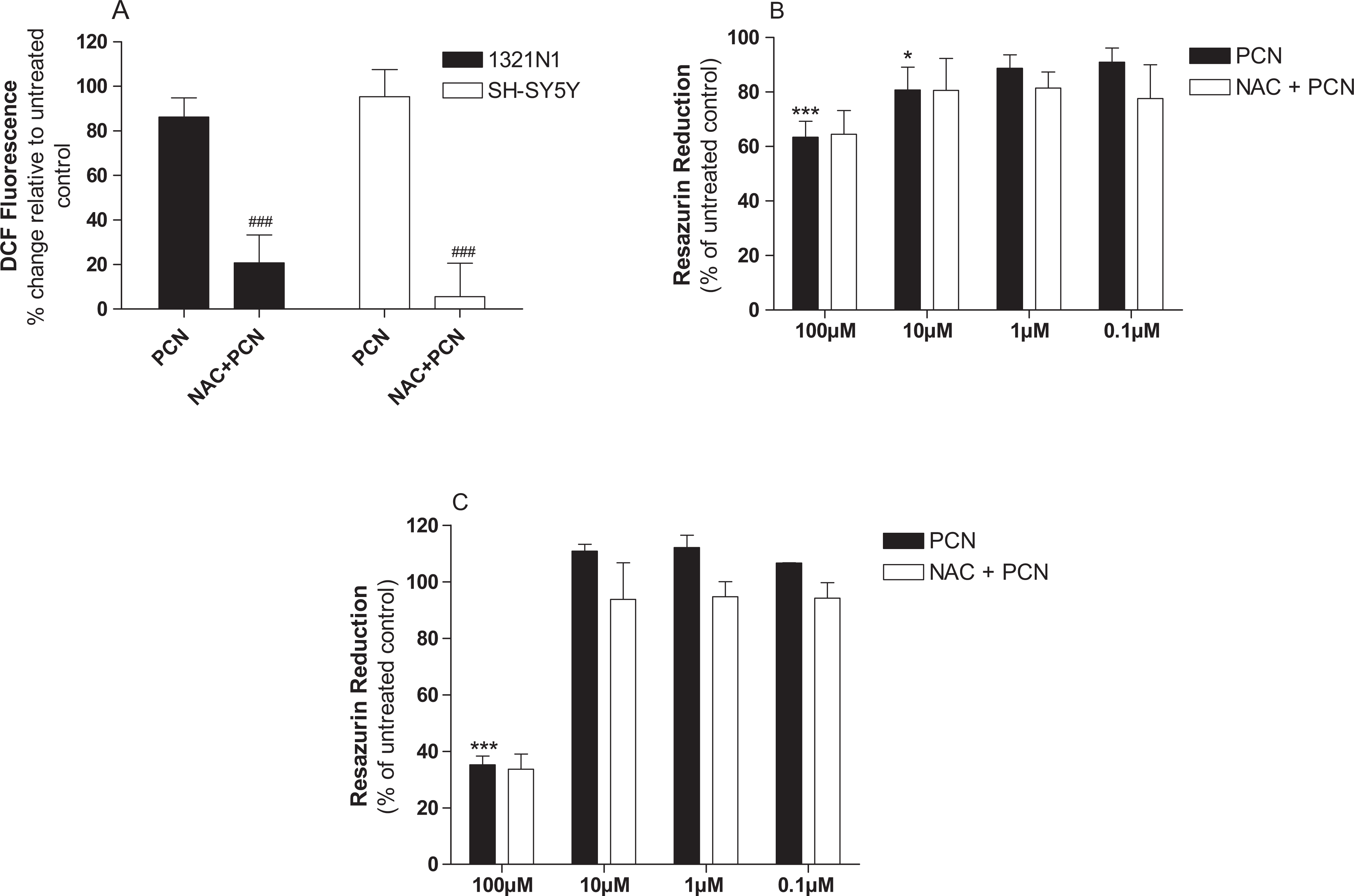
Impact of NAC on pyocyanin-induced ROS production. Cells were treated with PCN (100 μmol/L) in the absence or presence of NAC (5 mmol/L) for 24 hours. Global ROS production was then measured using DCFH-DA (A). Impact of NAC (5 mmol/L) on PCN-induced toxicity was then measured in (B) 1321N1 astrocytoma and (C) SH-SY5Y neuroblastoma cells. Data shown are the mean ± standard deviation of 3 independent experiments. Comparisons: * untreated control versus PCN; # PCN versus treatments. PCN indicates pyocyanin; ROS, reactive oxygen species; NAC, N-acetylcysteine; DCFH-DA, 2′,7′-dichlorofluorescein diacetate.
Discussion
Pyocyanin is a secreted virulence factor that is widely accepted to account for much of the pathogenicity of P aeruginosa. In contrast to the numerous studies identifying oxidative stress as a causative mechanism underlying PCN-induced toxicity in respiratory models, there is little known about the toxicity of this virulence factor in the CNS. Recently, our group provided the first in vitro evidence of PCN toxicity in the CNS. 32 Furthermore, we established that unlike respiratory models, oxidative stress was not primarily involved, but that autophagy promoted PCN-induced toxicity in 1321N1 astrocytoma cells. Since autophagy has a key role in maintaining neural function and protecting neurons from toxic and microbial insult, the aim of this study was to further examine the role of autophagy in PCN-induced CNS toxicity in vitro.
Consistent with our previous work, 32 we show here that autophagy, and not oxidative stress, mediates PCN-induced toxicity in both 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells at physiologically relevant concentrations. Autophagy has been long known to provide a survival advantage to cells undergoing nutrient deprivation and other stresses but has also recently been linked to promoting death itself, a process termed autophagic or type II cell death. 33 Here, we show that pretreatment of 1321N1 astrocytoma cells with the phosphoinositide 3-kinase inhibitor 3-MA protects against PCN-induced toxicity. Furthermore, 3-MA prevents PCN-induced AVO formation and increased accumulation of GFP-LC-3 in AV. The AVO formation in the cytoplasm has been shown to be a characteristic of cells undergoing autophagic or type II cell death in response to toxic insult. 29 Similarly in response to autophagy triggers, LC3 is associated with AV formation and is commonly used to monitor autophagy. Taken together these data would confirm our previous work identifying a role for autophagic or type II cell death in PCN-induced toxicity to 1321N1 astrocytoma cells.
In contrast to 1321N1 astrocytoma cells, pretreatment with 3-MA sensitized SH-SY5Y neuroblastoma cells to PCN-induced toxicity. Increased basal AVO and AV levels in SH-SY5Y neuroblastoma cells suggest that these cells possess higher basal autophagy activity compared to 1321N1 astrocytoma cells. Interestingly, PCN and 3-MA decreased basal AVO and AV levels suggesting protective role for autophagy against PCN-induced toxicity. This is consistent with previously observed protective role for autophagy against the development of neurodegenerative diseases 23 and neuronal damage caused by viral infection of astrocytes. 24 Furthermore, loss of autophagy has been demonstrated to cause neurodegeneration in mice even in the absence of any disease-associated mutant proteins. 23 A complex relationship exists between autophagy and apoptosis 34 with these pathways either acting in synergy or as counters to each other. 33 Eisenberg-Lerner and colleagues state that studies showing true inhibition of autophagy accompanied by changes in cell death must verify that apoptotic cell death is unaffected. 33 Although PCN increased caspase-3 activity in both cells lines, which is indicative of increased apoptosis, pretreatment with 3-MA decreased caspase-3 activity in both cell lines. This would suggest that apoptosis may be a secondary or even a separate event to autophagy in both cell lines. Furthermore, although apoptosis may be a contributing factor to PCN-induced toxicity in 1321N1 astrocytoma cells, it appears that its role in SH-SY5Y neuroblastoma cells is insignificant. However, this requires further investigation.
Pyocyanin is known to be responsible for the proinflammatory effects associated with P aeruginosa. Consistent with this, we show here that PCN increased the production of IL-8, PGE2, and LTB4 in both 1321N1 astrocytoma and SH-SY5Y neuroblastoma cells. Increased IL-8 production has been observed previously and is well established as a proinflammatory response following PCN exposure. 17 However, to our knowledge this is first study showing increased PGE2 and LTB4 levels in response to PCN exposure. The PCN has been previously been shown to inhibit LTB4 production in human neutrophils 35 and alveolar macrophages. 15 Similarly, PCN decreases prostaglandin production in porcine pulmonary endothelial cells. 36 LTB4 is known to promote neural stem cell development at physiological concentrations but promote cell death at higher concentrations. 37 Elevated PGE2 levels are known to exacerbate neurotoxicity by facilitating glutamate release from astrocytes. 38 Furthermore, increased cyclooxygenase 2 activity and subsequent PGE2 production have been causally linked to lipopolysaccharide-induced neuronal death. 39 Therefore, it is possible that increased PGE2 and LTB4 production observed here contributes to PCN-induced toxicity in both cells lines.
It is now widely accepted that autophagy can influence innate immune and inflammatory responses with loss of autophagy associated with inflammatory disease states. 40 Furthermore, recent novel evidence suggests that lipid-derived inflammatory mediators such as leukotrienes positively influence autophagy. 41 To test whether the modulation of PCN-induced toxicity by autophagy and production of proinflammatory mediators were related, we tested the impact of 3-MA inhibition of autophagy on the production of IL-8, PGE2, and LTB4. Consistent with its protective role against PCN-induced toxicity in 1321N1 astrocytoma cells, 3-MA significantly reduced the production of IL-8, PGE2, and LTB4. Conversely, 3-MA significantly augmented PCN-induced production of IL-8, PGE2, and LTB4 in SH-SY5Y neuroblastoma cells consistent with their increased sensitization to PCN-induced toxicity when autophagy is blocked. Therefore, it appears that in both of these cell lines, autophagy regulates the production of key inflammatory mediators upon exposure to PCN.
Conclusions
In conclusion, this study provides novel evidence for the role of autophagy in modulating PCN-induced toxicity in the CNS. Paradoxically, autophagy promotes cell death in 1321N1 astrocytoma cells but serves as a protective mechanism in SH-SY5Y neuroblastoma cells. Furthermore, we provide novel evidence that this paradoxical role of autophagy in PCN-induced toxicity may be related to its ability to control the production of proinflammatory mediators including IL-8, PGE2, and LTB4. These findings have significant implication in the management of CNS infections caused by P aeruginosa and warrant further investigation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Griffith Health Institute, Griffith University.