
Abstract
The present studies were conducted to compare the dermal absorption, plasma pharmacokinetics, and excretion of phenylethyl alcohol (PEA) by pregnant and nonpregnant rats, rabbits, and humans. The PEA is a natural fragrance material that is widely used in perfumes, soaps, and lotions and is a major ingredient of natural rose oil. Following dermal (430, 700, or 1400 mg/kg body weight [bw]), gavage (430 mg/kg bw), or dietary (430 mg/kg bw) administration of PEA to rats, plasma concentrations of PEA were found to be low regardless of the route of administration. The plasma concentrations of phenylacetic acid (PAA, the major metabolite of PEA) greatly exceeded the concentrations of PEA and were highest after gavage, followed by dermal then dietary administration. Absorption, distribution, metabolism, and excretion were compared following topical application of 14C-labeled PEA to rats, rabbits, and humans (specific activities of dosing solutions: 58-580, 164, and 50 µCi/mL, respectively). In rabbits, the plasma concentration–time profile for PAA was markedly prolonged compared to rats or humans. In humans, only 7.6% of the applied dose of PEA was absorbed, versus 77% in rats and 50% in rabbits. Based on a human dermal systemic exposure of 0.3 mg/kg per day from the use of multiple consumer personal care products containing PEA, a rat dermal no observed adverse effect level of 70 mg/kg per day, and the percentage of dose absorbed in humans, the margin of safety exceeds 2600 concluding that, under normal fragrance use conditions, PEA is not a developmental toxicity hazard for humans.
Introduction
Phenylethyl alcohol (PEA) is a natural fragrance material that is widely used in perfumes, soaps, and lotions and is a major ingredient of natural rose oil. It is also found in other natural products, such as foods, spices, tobacco, and wine. 1 PEA was given Generally Recognized As Safe (GRAS) status by the Flavoring Extract Manufacturers Association and is also approved by the Food and Drug Administration for food use. 2 A comprehensive summary of PEA safety studies has been published by the Cosmetic Ingredient Review Expert Panel. 1 However, Mankes et al 3 reported that single gavage doses of 4.3, 43, and 432 mg/kg of PEA administered to pregnant rats during organogenesis (days 6-15 of gestation) produced developmental effects (fetal death, malformation, and growth retardation) in a dose-related manner. At the lowest dose, in addition to intrauterine growth retardation, there were malformations in 50% and embryolethality in 10% of the pups; in the mid-dose group malformations were seen in 93% and embryolethality in 18% of the pups; in the highest dose group 100% of implants yielded dead and malformed pups. 3
Subsequent rat developmental toxicity studies in which PEA was administered in the diet or by the topical route did not produce any adverse systemic effects in either the dams or their offspring at dosages below 140 mg/kg, which is well above any human exposure levels. A review and safety assessment of these studies concluded that PEA is not considered to be a developmental toxicity hazard for humans. 4 Summaries of these results were shared with the Cosmetic Ingredient Review Expert Panel for their review 1 and have been presented in poster format at the Society of Toxicology5–7 and as proceedings from scientific meetings.8,9 They were also included in a submission to the Environmental Protection Agency under the High Production Volume (HPV) Challenge Program. Nevertheless, species differences in pharmacokinetics, as well as dermal absorption and disposition, play important roles in safety assessment. Therefore, the present studies were conducted to compare the dermal absorption, plasma pharmacokinetics, and excretion of PEA by pregnant and nonpregnant rats, rabbits, and humans.
Methods
Pharmacokinetics in Rats With Unlabeled PEA
Unlabeled PEA (CAS # 60-12-8; Gold Label Grade, Aldrich Chemical Co, Gillingham, UK; >99% pure) was used for an assessment of plasma levels of PEA and its major metabolite, phenylacetic acid (PAA), in female Sprague-Dawley (CD) nonpregnant rats. The PAA was obtained from Fisons Scientific Apparatus (Loughborough, UK). Two groups of 32 rats supplied by Charles River UK, Ltd (Margate, Kent) were administered, 1 topical dosage of either 700 or 1400 mg/kg (0.7 or 1.4 mL/kg) PEA. The liquid was applied to an intact, closely clipped, 3 × 3 cm area of skin on the back of each animal. The area was then covered by an aluminum foil patch held in place by an adhesive bandage until sacrifice, to minimize oral ingestion or evaporation of test agent. An additional 2 groups of 4 rats were untreated and used as predose negative controls. At 0.5, 1, 2, 4, 6, 8, 12, and 24 hours post treatment, 4 rats from each group were euthanatized and exsanguinated. Plasma was immediately frozen at −20°C. The PEA and PAA plasma levels were determined using an HP5792A gas chromatograph coupled directly to a VG16F single-focusing mass spectrometer. The methodology for PEA detection was developed in the Department of Chemical Metabolism and Radiosynthesis at Huntingdon Research Centre Ltd. (Cambridgeshire, UK) and involved separation by capillary gas chromatography (GC) and detection by mass spectrometry (MS), using α-methylbenzyl alcohol as internal standard. Retention time of PEA and α-methylbenzyl alcohol were 3.4 and 3.1 minutes, respectively. The limit of detection (limit of quantification [LOQ]) for PEA was set at 0.03 μg/mL. The methodology for PAA detection was based on the method of Karoum et al, 10 and involved separation by capillary GC and detection by MS of the pentafluoropropyl ester of PAA, using the deuterium-labeled analog, phenylacetic-d7acid, as internal standard. Retention times of PAA and phenylacetic-d7acid were 4.34 and 4.32 minutes, respectively. The limit of detection (LOQ) for PAA was set at 0.1 μg/mL.
In a second study, plasma and urine concentrations, as well as the pharmacokinetics of PEA and PAA, were determined after dermal or oral administrations of 430 mg/kg of PEA. Forty female CD nonpregnant rats supplied by Charles River UK, Ltd were administered PEA by topical application (0.43 mL/kg, equivalent to 430 mg/kg) as described above; 44 female CD nonpregnant rats were gavaged once with 0.43 mL/kg PEA (430 mg/kg PEA) in an aqueous PEG 200 solution, and 40 female CD nonpregnant rats were fed for up to 24 hours an ad libitum diet containing encapsulated PEA in a gum arabic matrix providing a nominal dose of 430 mg/kg. All groups had 4 additional rats that provided predose control plasma. Plasma samples were obtained from 4 rats from each treatment method at 10 or 11 time points ranging from 0.25 to 24 hours after the initiation of the treatment. The rats killed at 24 hours post treatment were housed in metabolism cages, and a 0 to 24 hour urine sample was collected and stored at −20°C, along with all plasma samples. Analyses of plasma and urine were by GC/MS, as described above.
Dermal Absorption and Disposition of 14 C-PEA in Rats, Rabbits, and Humans
The radiolabeled PEA studies in rats, rabbits, and humans were designed to provide data on the extent of the dermal absorption of the 14 C-labeled PEA ( 14 C-PEA), its distribution, excretion and biotransformation, and the influence of dose levels and repeated treatment on these parameters. The 14 C-PEA (Imperial Chemical Industries, Cleveland, UK) had a specific activity of 15 mCi/mmol/L and was diluted for the various dosage levels with unlabeled PEA. Purity (>97%) was determined by thin-layer chromatography. The PAA was synthesized (Huntington Research Centre, Gillingham, UK) and its identity was confirmed by MS analysis. Metabolite identification was performed with high-performance liquid chromatography (HPLC)/MS. The chromatography was performed in a reversed phase mode, using a solvent system of 0.1 mol/L sodium acetate-acetic acid: methanol and a flow rate of 2 mL/min at 4000 psi pressure. Hippuric acid, phenaceturic acid, PAA, and PEA had retention times of 4.2, 5.2, 11.4, and 14.2 minutes, respectively. Radioactivity was measured by using an LKB Liquid Scintillation Counter (Model No. 1219 Rackbeta `Spectral', LKB-Wallac, Turku, Finland) or a Philips Liquid Scintillation Analyzer (Model No. PW 4700, Philips NV. Eindhoven, Holland). Radioactivity in amounts less than 2 times background disintegrations per minute (dpm) was considered to be below the limit of accurate measurement.
Studies in rats
Female CD nonpregnant rats, female CD pregnant rats, and male Long-Evans rats were supplied by Charles River UK, Ltd. The 14 C-PEA was applied to a 3 × 3 cm2 area of the back of each rat, as described previously. Dressings were stored at −20°C prior to analysis.
After completion of a pilot range-finding study, plasma kinetics were investigated in 2 groups of 4 female CD nonpregnant rats administered single topical dosages of 14 C-PEA at levels of 140 and 700 mg/kg. Blood samples were taken prior treatment and at 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96, and 120 hours after the initiation of treatment. Plasma was analyzed for radioactivity without prior freezing.
Excretion was studied in 3 groups of 4 female CD nonpregnant rats that were housed in glass metabolism cages for the separate collection of urine and feces. The rats were administered single topical dosages of 140, 700, or 1400 mg/kg 14 C-PEA. Urine was collected in solid carbon dioxide-cooled containers during 0 to 6, 6 to 12, 12 to 24, 24 to 48, 48 to 72, 72 to 96, and 96 to 120 hours. Feces were collected at 24-hour intervals up to 120 hours, when the rats were euthanized, and the liver, kidneys, gastrointestinal tract, and treated skin were removed and stored at −20°C. In a supplemental study, 2 female CD nonpregnant rats were administered a single topical dosage of 700 mg/kg C-PEA 14 and housed in closed glass metabolism cages. Expired air was monitored through 2 traps containing 2-ethoxyethanol. Urine and feces were collected over 24-hour intervals and when the rats were euthanatized, and the liver, kidneys, gastrointestinal tract, and treated skin were removed and stored at −20°C.
Two groups of 4 female CD nonpregnant rats were also administered daily topical dosages of 140 or 700 mg/kg 14 C-PEA for 5 days. The doses were applied each morning and a fresh dressing was applied after each dose. Urine and feces were collected for analysis only at 0 to 6 and 6 to 24 hours during the last (5th) day of the treatment period.
Quantitative tissue distribution was determined by applying single topical dosages of 140 mg/kg of 14 C-PEA to 12 male Long-Evans rats and analyzing tissues from major organs for radioactivity. The dosage was applied as described above with the exception that for rats euthanized at 12 hours and later times, the dressings were removed at 6 hours and the treatment area was washed with ethanol-moistened cotton wool to remove residual dose. The tissues were obtained and frozen at −20°C from pairs of rats euthanatized at 3, 6, 12, 24, 48, or 72 hours after treatment. The pigmented Long-Evans rats were used here because this was the strain originally tested by Mankes et al. 3
Qualitative tissue distribution was explored in 2 groups of 5 female CD pregnant rats after topical administration of daily dosages of 140 or 700 mg/kg 14 C-PEA on gestation days (GDs) 6 through 15. Single animals were euthanized at 2 hours (140 mg/kg group) and 4 hours (700 mg/kg group) after the first (GD 6), fifth (GD 10), or tenth (GD 15) dosages and at 24 and 48 hours after the tenth dosage (GD 15). All carcasses were then quick frozen and prepared for whole-body autoradiography. Minimum detectable radioactivity levels corresponded to tissue concentrations of 4 to10 dpm/mg wet tissue.
Studies in rabbits
Eight female New Zealand White (NZW) rabbits were obtained from Buckmasters (Henham, Herts, UK) and divided into 2 groups of 4 rabbits each. The rabbits were supplied with ad libitum feed and water and individually housed in stainless steel metabolism cages that allowed for the collection and separation of urine and feces. Single topical dosages of 140 or 700 mg/kg of 14 C-PEA were applied by a syringe to a 10 × 5 cm2 area of the back that had been shaved previously with an electric clipper. The treatment area remained unoccluded, but the rabbits were fitted with Elizabethan collars to prevent interference with the treated skin. Urine was collected in solid CO2-cooled containers at 0 to 6, 6 to 24, 24 to 48, and 48 to 72 hours after treatment. Feces were collected at the same time periods. Plasma was obtained from blood samples taken from the lateral ear vein before treatment and at 0.5, 1, 2, 4, 6, 9, 12, and 24 hours after treatment. The rabbits were euthanatized 72 hours after treatment, and the treated skin was removed and frozen at −20°C, along with the plasma samples, for analysis of radioactivity.
Studies in humans
At the time these studies were conducted, all applicable laws and regulations regarding human use were followed. After submission of animal metabolic data and calculation of likely radiation dosages to critical organs and whole body by the National Radiological Protection Board in the United Kingdom, the human study was approved by the National Drugs Advisory Board, Ireland, in accordance with the European Economic Community’s directive for the administration of radioactive substances to humans. The study protocol was reviewed and approved by the ethics committee of the Institute of Clinical Pharmacology (St James’s Hospital, Dublin, Ireland). Each participant gave consent, in writing, to participate after the nature and aim of the study were explained and the study was conducted in accordance with the relevant articles of the Declaration of Helsinki (1975).
Two normal healthy male volunteers, aged 36 and 27 years, were selected for the study. The participants were fasted for 12 hours before application of 10 mg 14 C-PEA (in 1 mL of ethanol) to a 100 cm2 area of skin on the upper chest. After 1 hour (to allow for ethanol evaporation), the area was occluded with a gauze dressing, which was removed after 6 hours and retained for analysis of radioactivity. The treated area was wiped with swabs and then washed with swabs moistened with ethanol and a 2 × 5 cm section of treated skin was stripped with 5 successive applications of adhesive tape; the swabs and strips were retained for analysis. Blood samples were taken before treatment and at 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, and 120 hours after treatment. Urine was collected at 0 to 3, 3 to 6, 6 to 12, 12 to 24, 48 to 72, 72 to 96, and 96 to 120 hours after treatment. Feces were collected at 24-hour intervals during 5 days. Plasma, urine, and feces were frozen at −20°C until analysis for radioactivity.
Results
Pharmacokinetics in Rats With Unlabeled PEA
When plasma concentrations of unlabeled PEA and PAA (major metabolite) were measured by GC/MS after single dermal application of 700 or 1400 mg PEA/kg to female CD nonpregnant rats, PEA was found to reach mean peak levels of 22.7 or 64.4 μg/mL, respectively, at 0.5 hours, after which the levels declined rapidly to 0.91 or 1.93 μg/mL at 6 hours and more slowly to 0.06 or 0.17 μg/mL at 24 hours. On the other hand, PAA levels reached peak values of 520 and 671 μg/mL at 4 hours for both the 700 and 1400 mg/kg doses, respectively, then declined to 188 and 376 μg/mL at 12 hours and to 0.29 and 0.48 μg/mL at 24 hours. Thus, mean plasma concentrations of PAA greatly exceeded those of PEA for most of the 24-hour sampling period, indicating that the rats were exposed for longer time periods to much higher concentrations of the metabolite than to unchanged PEA (Table 1).
Comparison of Mean Plasma Tmax, Cmax, and AUC of PEA and PAA Following a Single Dermal Application of PEA to Female CD Nonpregnant Rats.
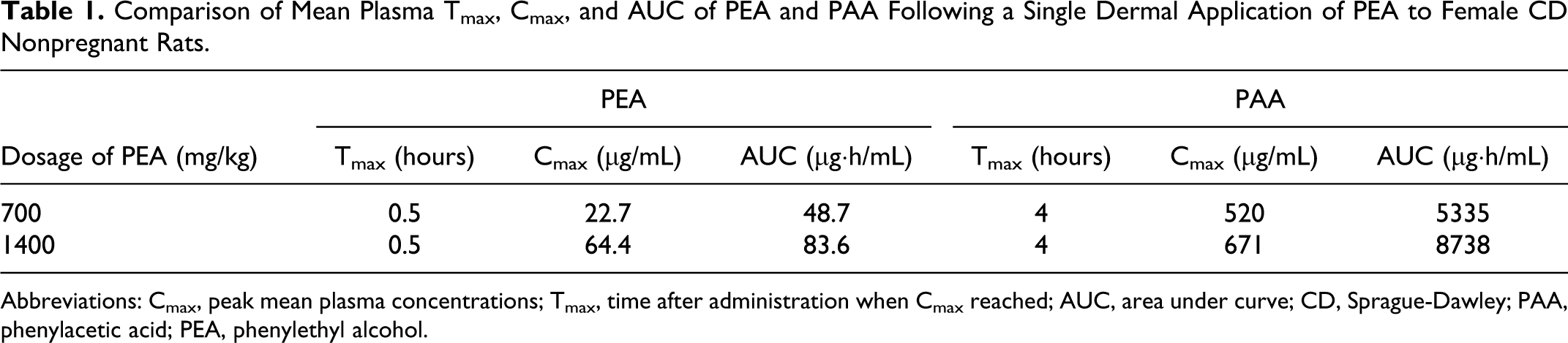
Abbreviations: Cmax, peak mean plasma concentrations; Tmax, time after administration when Cmax reached; AUC, area under curve; CD, Sprague-Dawley; PAA, phenylacetic acid; PEA, phenylethyl alcohol.
When plasma and urine levels of PEA and PAA were compared in female CD nonpregnant rats administered a single gavaged, dietary, or dermal dose of 430 mg/kg (oral projected dose) of unlabeled PEA, the results revealed that for all 3 routes, mean plasma concentrations of PAA greatly exceeded those of PEA (Table 2). It should be noted that the dietary exposure resulted in an actual intake of 261 mg/kg. Plasma Cmax levels of PEA were reached at 0.25 and 0.5 hours after gavage and dermal application, respectively. Plasma concentrations of both PEA and PAA were low and remained relatively constant when PEA was ingested in the diet over 24 hours (Figures 1 and 2). Compared to dermal application, the greatly reduced level of plasma PEA after oral administration was presumed to be due to metabolic conversion of PEA in the liver, resulting in markedly higher plasma PAA levels. Approximately, 70% of the administered PEA was excreted in urine as PAA or its conjugates within 24 hours after oral administration (both gavage and dietary). Only 27% of the dermal dose was recovered in urine (Table 3). The lower excretion of the dermal dose is probably due to evaporation from the dermal application site and absorption to the bandage, as was shown in the supplemental metabolism study described below.
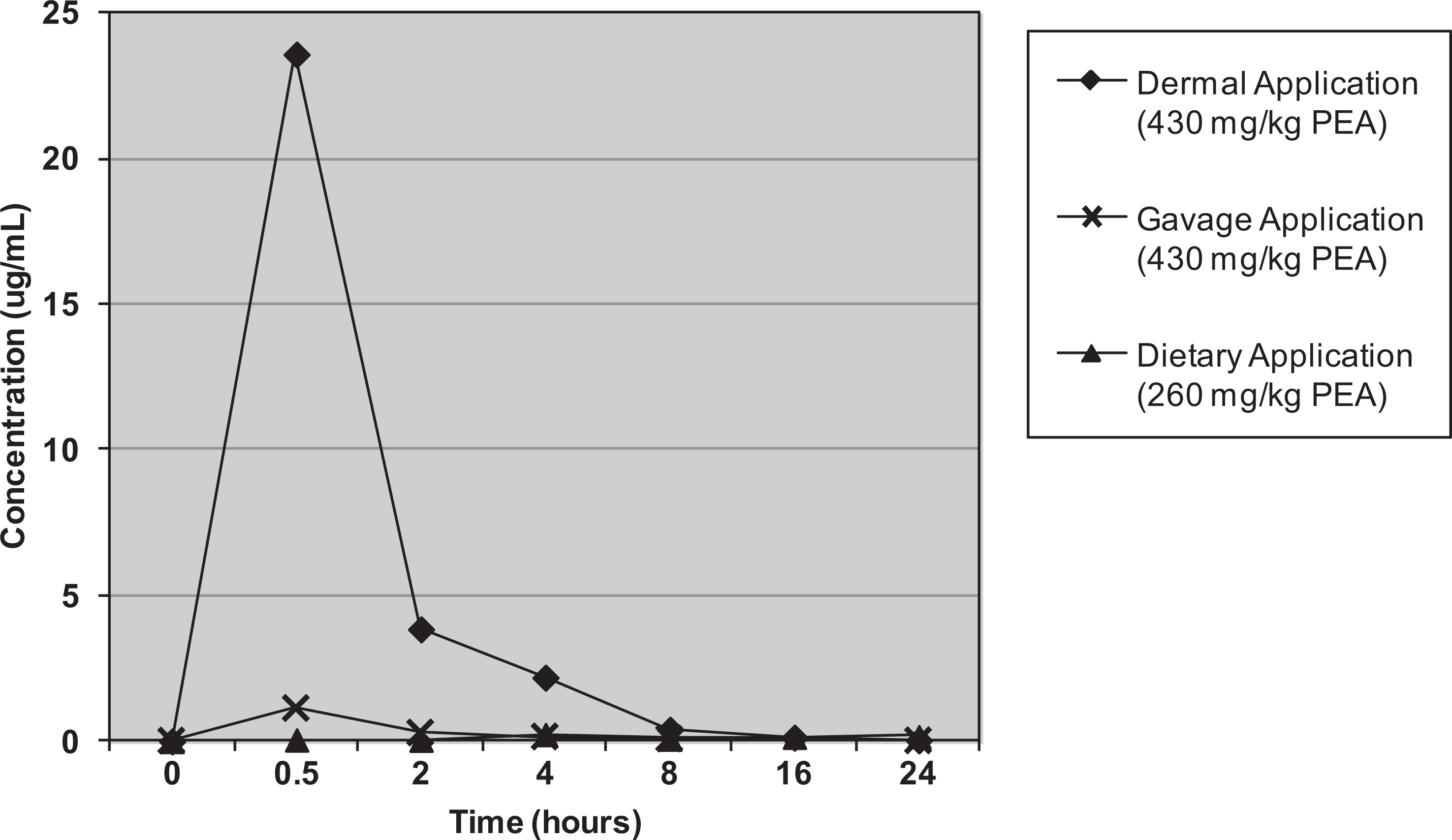
Mean plasma concentrations (Linear Scale) of phenylethanol (PEA) after administration of PEA to rats via 3 different routes.
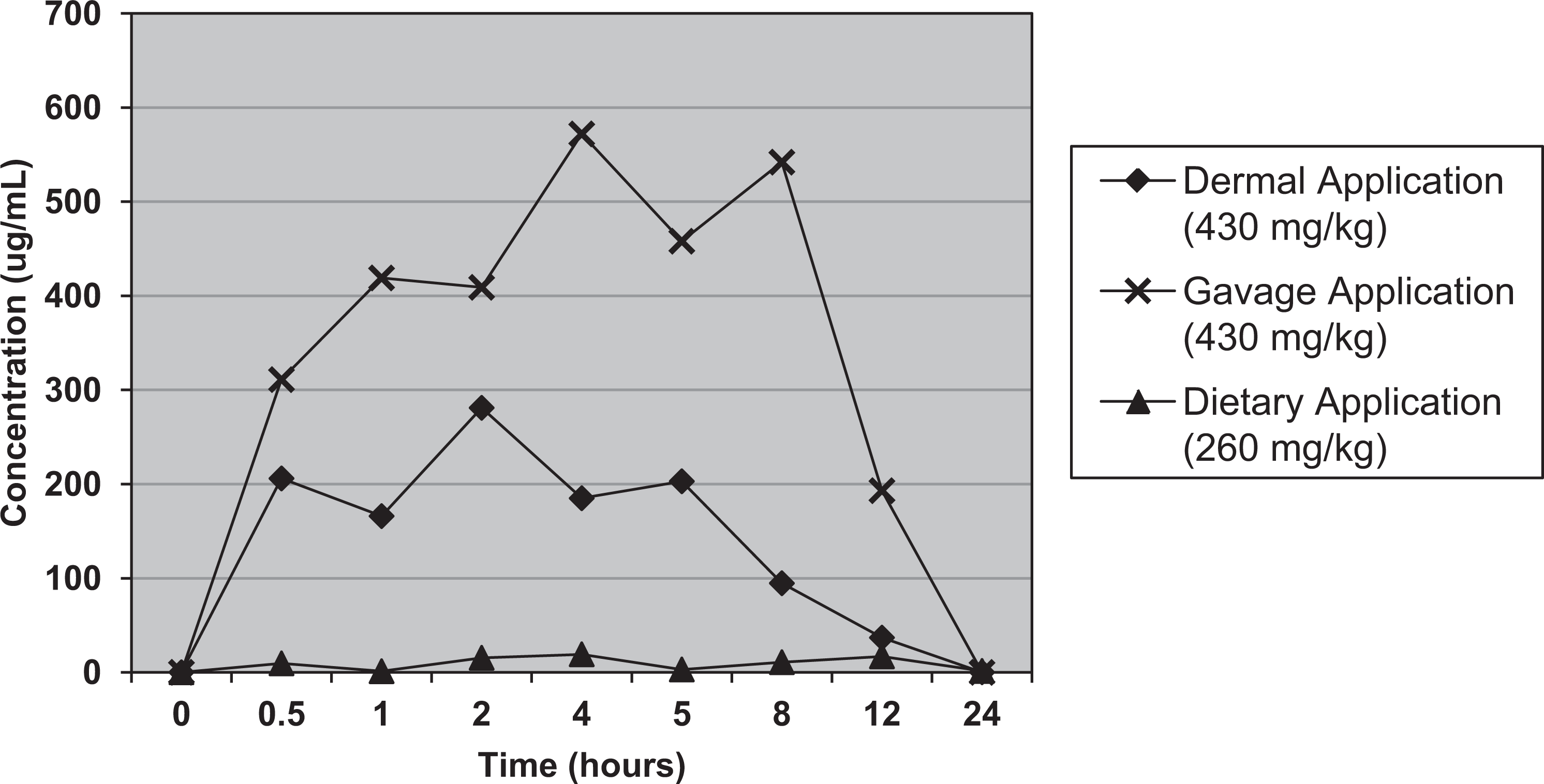
Mean plasma concentration (Linear Scale) of phenylacetic acid (PAA) after administration of phenylethanol (PEA) to rats via 3 different routes.
Comparison of Mean Plasma Tmax, Cmax, and AUC of PEA and PAA following 3 modes of PEA Administration to Female CD Nonpregnant Rats.
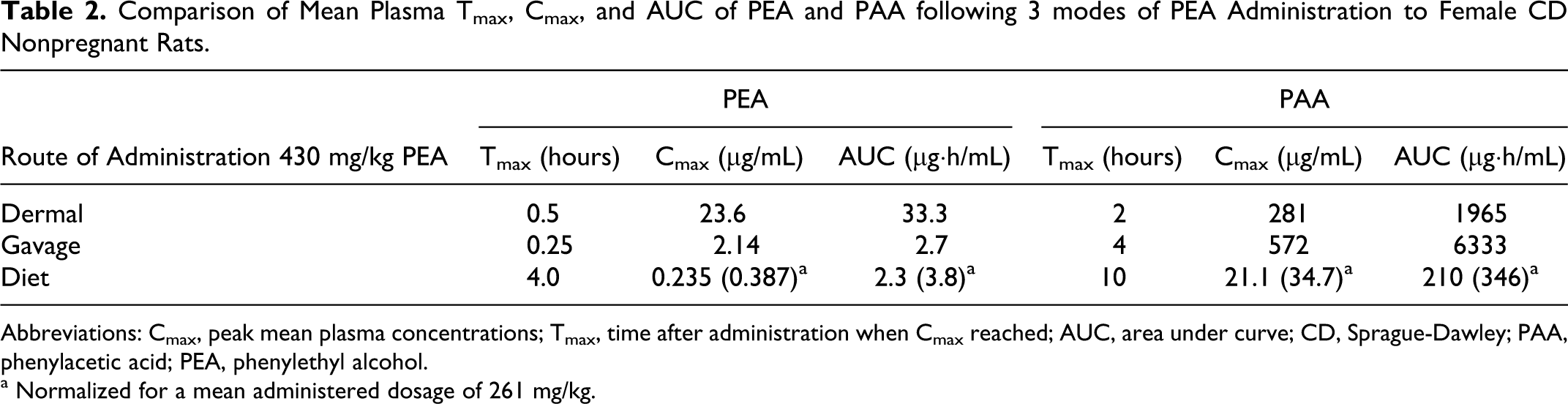
Abbreviations: Cmax, peak mean plasma concentrations; Tmax, time after administration when Cmax reached; AUC, area under curve; CD, Sprague-Dawley; PAA, phenylacetic acid; PEA, phenylethyl alcohol.
a Normalized for a mean administered dosage of 261 mg/kg.
Comparison of Concentrations of PAA Equivalents in 0 to 24-Hour Urine Following 3 Modes of PEA Administration to Female CD Nonpregnant Rats.
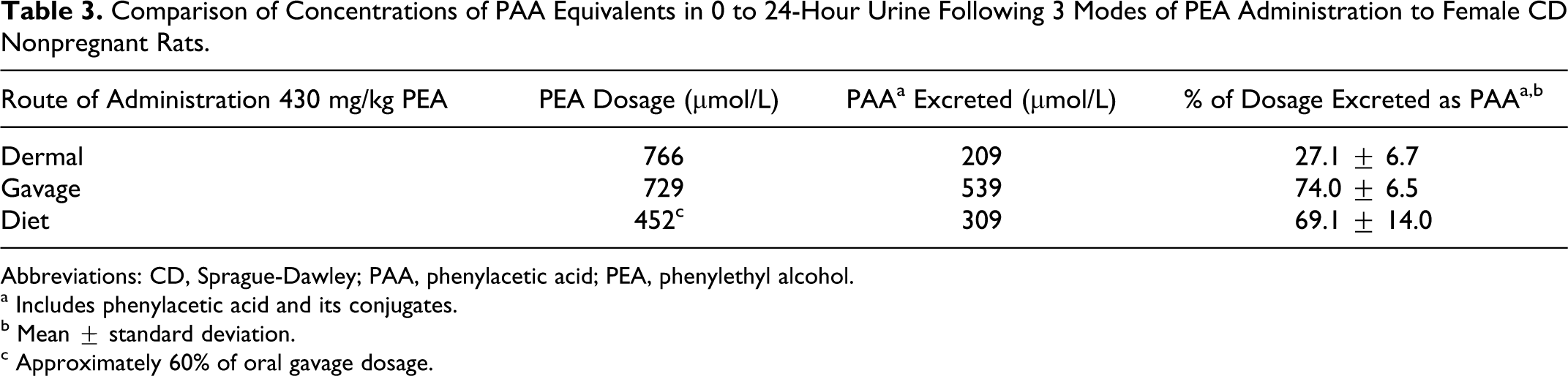
Abbreviations: CD, Sprague-Dawley; PAA, phenylacetic acid; PEA, phenylethyl alcohol.
a Includes phenylacetic acid and its conjugates.
b Mean ± standard deviation.
c Approximately 60% of oral gavage dosage.
Dermal Absorption and Disposition of 14 C-PEA in Rats, Rabbits, and Humans
Studies in rats
In female CD nonpregnant rats, a plasma Cmax concentration of 180 μg/mL of 14 C-PEA equivalents was achieved 2 hours after a single dermal application of 140 mg/kg of 14 C-PEA. Mean concentrations then declined rapidly to 103, 1.48, and 0.47 μg/mL at 4, 12, and 24 hours, respectively. A slower decline to 0.23 and 0.20 μg/mL (limit of accurate measurement) then occurred at 48 and 72 to 120 hours post treatment. A single dermal application of 700 mg/kg produced a Cmax of 543 μg/mL at 4 hours, followed by a slow decline to 522, 427, and 214 μg/mL at 6, 8, and 12 hours post treatment, respectively. After that, concentrations declined rapidly to 1.1 μg/mL at 72 hours and below accurate measurement at 96 to 120 hours (Table 4). These results indicated that dermal dosages of 140 mg/kg of 14 C-PEA were rapidly absorbed and excreted in rats, but that the 700 mg/kg dosage was more slowly absorbed and excreted than the 140 mg/kg dosage.
Concentrations of Radioactivity in the Plasma of Rats Following a Single Dermal Application of 14 C-PEA.a
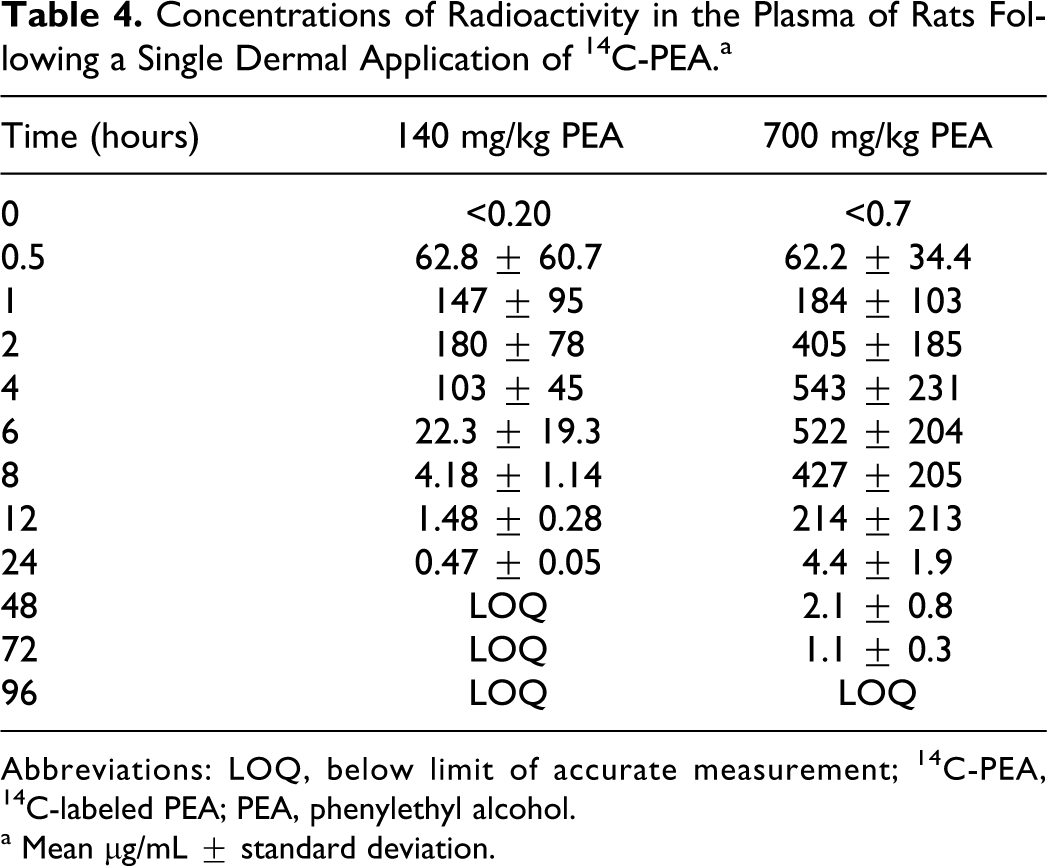
Abbreviations: LOQ, below limit of accurate measurement; 14 C-PEA, 14 C-labeled PEA; PEA, phenylethyl alcohol.
a Mean μg/mL ± standard deviation.
In female CD nonpregnant rats administered a single dermal application of 14 C-PEA, excretion of 14 C-PEA, and its metabolites occurred predominantly via the kidneys; 77%, 36%, and 29% of the dosage was excreted in the urine by 24 hours and 81%, 39%, and 35% was excreted within 120 hours after treatment with 140, 700, and 1400 mg/kg 14 C-PEA, respectively (Table 5). The low recovery of radioactivity at the higher dosages prompted a supplemental study in which chamber air was collected and monitored over 0 to 120 hours and bandaging was adjusted. These modifications resulted in a recovery of 37% in the urine by 24 hours and 76% at 120 hours after treatment with 700 mg/kg 14 C-PEA. These results suggest that higher percentages of the 700 and 1400 mg/kg dosages were lost due to evaporation at the application site. Only 1.3%, 1.6% (1.8% in the supplemental study), and 0.76% of the respective dosages were excreted via the feces.
Mean Excretion and Retention of Radioactivity by Rats, Rabbits, and Humans Following a Single Dermal Application of 14 C-PEA.a
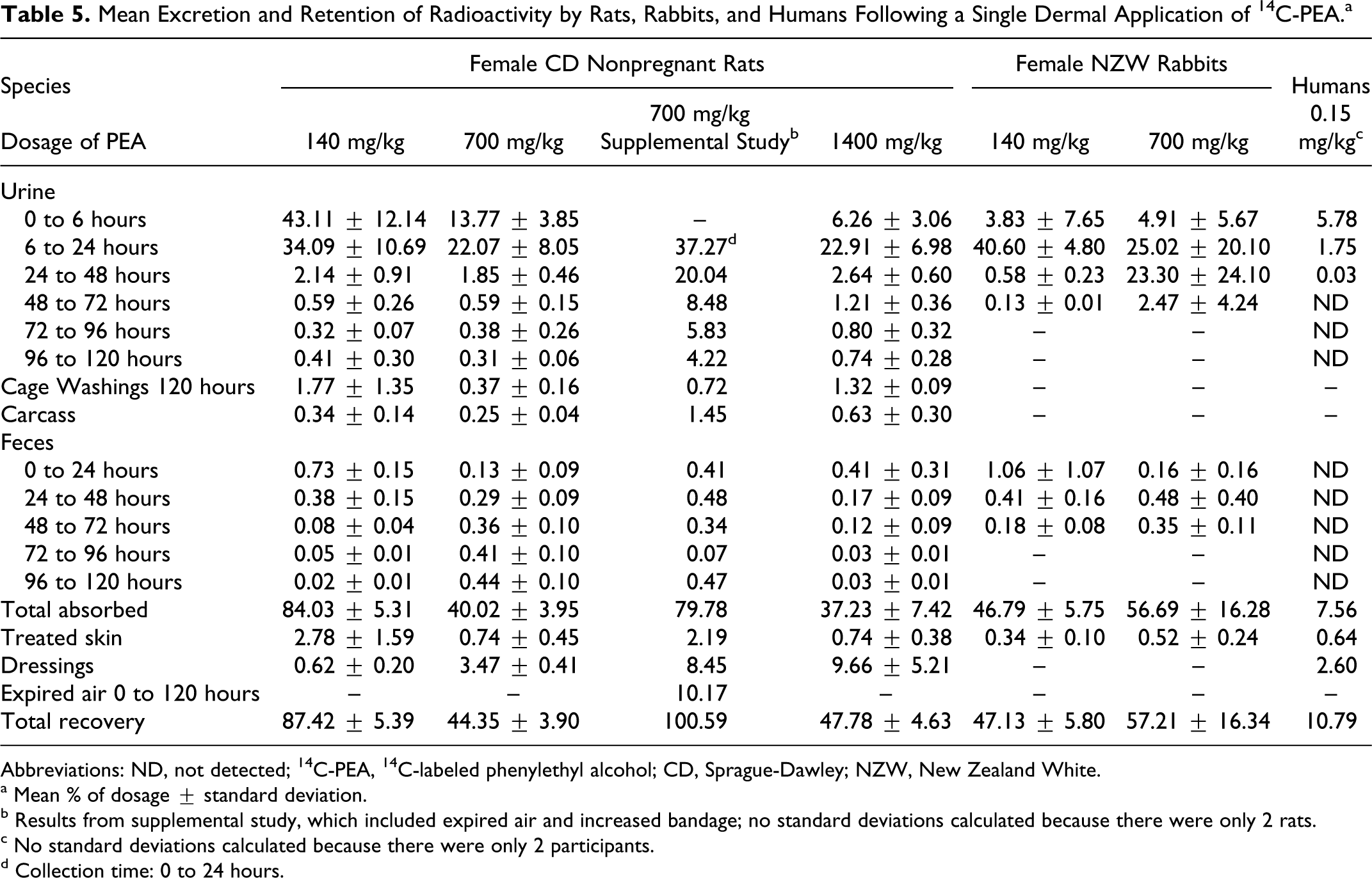
Abbreviations: ND, not detected; 14 C-PEA, 14 C-labeled phenylethyl alcohol; CD, Sprague-Dawley; NZW, New Zealand White.
a Mean % of dosage ± standard deviation.
b Results from supplemental study, which included expired air and increased bandage; no standard deviations calculated because there were only 2 rats.
c No standard deviations calculated because there were only 2 participants.
d Collection time: 0 to 24 hours.
Dermal applications of 140 or 700 mg/kg 14 C-PEA for 5 consecutive days did not appreciably change the excretion pattern observed after a single dosage (data not shown). Most of the 14 C label was still excreted in the urine during the 24 hours after each treatment (63%-68% in the 140 mg/kg group and 32%-34% in the 700 mg/kg group). Mass balance studies revealed 18.7% of the low-dosage and 31.6% of the high-dosage 14 C label were not recovered, presumably due to evaporation from the application site.
HPLC analysis of plasma obtained at Cmax for total 14 C label from female CD nonpregnant rats revealed that PAA was the primary entity associated with radioactivity, regardless of the applied dermal dosage. The major metabolite in urine excreted during 24 hours after a single dosage, or 24 hours after the last of 5 consecutive dosages of 14 C-PEA, corresponded chromatographically to phenaceturic acid and it accounted for 73% to 87% of the urinary radioactivity. Hippuric acid accounted for 4% to 6%. In addition to phenaceturic acid, PAA and PEA were also identified in urine after a single dosage of 140 mg/kg 14 C-PEA (at 4% and 2% of the dose, respectively), but after single dosages of 700 mg/kg 14 C-PEA or multiple dosages of 140 or 700 mg/kg 14 C-PEA, the amounts were generally ≤1%.
Quantitative tissue distribution studies were conducted in male Long-Evans rats treated with single 140 mg/kg topical applications of 14 C-PEA for a 6-hour exposure period. The greatest concentrations of radioactivity were detected in the tissues at 3 hours. By 24, 48, and 72 hours post treatment, all tissues, except treated skin and fat, had concentrations less than 1.0 μg/g, or radioactivity was below the limit of accurate detection. The results indicate that 14 C-PEA was rapidly absorbed and eliminated from most tissues (Table 6).
Concentrations of Radioactivity in Selected Tissues of Male Long-Evans Rats Following a Single Dermal Application of 140 mg/kg 14 C-PEA.a
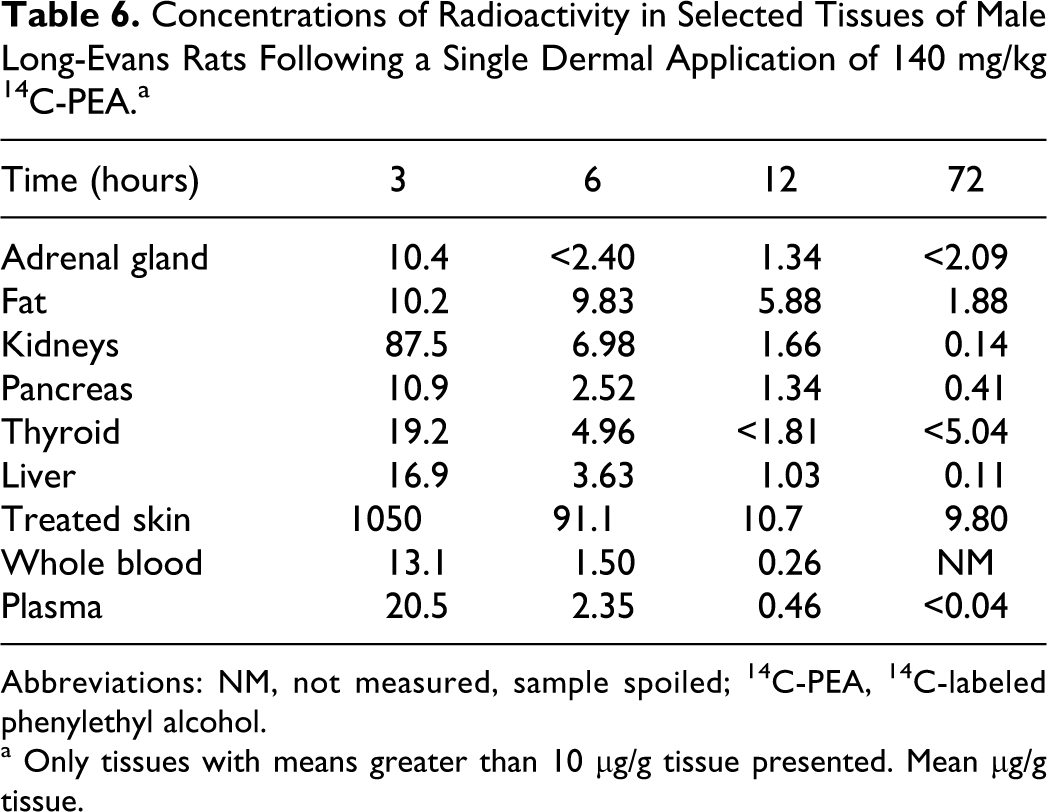
a Only tissues with means greater than 10 μg/g tissue presented. Mean μg/g tissue.
Whole-body autoradiography performed in female CD pregnant rats administered 14 C-PEA daily on GDs 6 through 17 confirmed the results of the quantitative tissue distribution studies in male Long-Evans rats. With 140 mg/kg 14 C-PEA, maximum tissue radioactivity was observed at plasma Cmax (2 hours) after the last of the 10 daily dermal applications. With 700 mg/kg 14 C-PEA, maximal distribution was noted at plasma Cmax (4 hours) after the first dosage. Radioactivity at both dosages was associated with the dermal application site, the gastrointestinal tract, kidneys and urinary tract, liver, blood, and a number of endocrine and secretory organs. Embryos were not clearly evident in rats examined at GDs 5 and 10; however, radioactivity was observed in fetuses of rats from each dosage level at the plasma Cmax after 10 treatments (ie, on GD 15).
Studies in rabbits
When single dosages of 140 or 700 mg/kg 14 C-PEA were applied to the skin of female NZW rabbits, a mean plasma Cmax of 91.2 μg/mL (as 14 C-equivalents) was reached 4 hours after a dosage of 140 mg/kg. Concentrations declined to 40.4 and 1.8 μg/mL at 6 and 12 hours, respectively, and were below the level of detection at 24 hours. The dosage of 700 mg/kg 14 C-PEA produced a mean Cmax of 623 μg/mL at 6 hours and a mean concentration of 275 μg/mL at 24 hours, the final sampling time. The 24-hour plasma concentration for PAA nearly superimposed that of the total 14 C eqivalents at both doses, which indicates that the minimal levels of PEA or additional metabolites are present. The high plasma PAA concentration–time profile that was maintained for 24 hours after dermal application of 700 mg/kg differed markedly from the profile of the 140 mg/kg dosage level. This suggests that this high dose saturates the glycine conjugation pathway by which PAA is excreted.
Means of 45% and 1.7% of the 140 mg/kg 14 C-PEA dosage were excreted in the urine and feces, respectively, during the 72 hours after application. For the 700 mg/kg dosage, the excretion rates were 56% and 0.99% in the urine and feces, respectively, during the same time period. Most of the radioactivity at both dosage levels was excreted from 0 to 24 hours post treatment. Mean radioactivity totals of 47.1% and 57.2% of the 140 and 700 mg/kg dosages, respectively, were recovered (Table 5). The remainder was considered to have been lost via evaporation, as occurred in the rat studies.
Phenaceturic acid was the main component in the urine and accounted for 34% to 36% of the radioactivity at both dosage levels; hippuric acid and a conjugate of PEA each accounted for 2% to 3%. The PAA was not detected at any significant level (<0.3%) in urine from the 140 mg/kg dosage, but it was associated with approximately 6% of the 700 mg/kg dosage excreted in the urine. These results suggest that the pathway for the formation of phenaceturic acid from PAA was becoming saturated at the 700 mg/kg level.
Studies in humans
Examination of the absorption and elimination of 14 C-PEA in 2 human male volunteers after single dermal applications of 10 mg (approximately 0.15 mg/kg) revealed that mean radioactivity concentrations of 3.5 ng/mL were detected in the plasma 15 minutes after application of the radiolabeled PEA. These levels rose to a Cmax of 13.8 ng/mL at 1.5 hours and then declined to 11.7 ng/mL and 3.1 ng/mL at 2 and 4 hours post treatment, respectively. All later samplings were below the limit of accurate detection (3.0 ng/mL). Because of the low level of radioactivity, plasma samples were not subjected to HPLC analyses.
During the 5 days after the single dermal application to the chest, a mean of 7.56% of the dosage was recovered in the urine. It was recovered only during the first 48 hours after application. Radioactivity in the feces was below the limit of detection. Most of the radioactivity was lost from the skin during the 6-hour exposure period, presumably due to evaporation; 2.60% of the dosage was recovered from the dressings and 0.64% from the skin washes at 6 hours (Table 5).
Two metabolites of PEA were detected in the urine. The major metabolite (4.1% of the dosage) was identified as phenylacetylglutamine, the glutamine conjugate of PAA. The second metabolite (2.7% of the dosage) was a glucuronide conjugate of PAA.
Discussion and Conclusions
Comparative pharmacokinetics, absorption, and disposition studies were conducted in pregnant and nonpregnant rats, rabbits, and humans to attempt to determine an appropriate margin of safety for PEA in pregnant women. No observable effect level or no observed adverse effect level (NOAEL) values, as well as calculated margins of safety based on a maximum human exposure of 0.3 mg/kg per day for a 60-kg woman, 11 varied greatly in the 4 rat developmental toxicity studies, although most of the variation was assumed to be due to differences in the routes of administration (gavage vs diet vs dermal application). 4 It was not known whether species differences in absorption, distribution, metabolism, and excretion also played a prominent role in these variations.
Results from the present studies suggest that species differences in dermal absorption were major factors in calculating a realistic margin of safety for PEA. Absorption through rodent skin is, in most cases, higher than through human skin, although there is no consistent pattern between the compounds.12–14 This higher absorption in rodent skin may be due to differences in morphology of the individual skin layers, or immunological or metabolic factors.14–16 In addition, the percentage of absorption of a limited source of test article depends on the concentration and length of exposure and necessitates that (1) the exposure remains constant during the exposure period and (2) reflects actual use conditions. 17
To meet these criteria, single-dosage and repeat-dosage dermal absorption studies were used to explore exposure in rats. The same procedures were followed as in the developmental toxicity studies, except that the applied material was 14 C-PEA, applied in dosages of 140, 700 or 1400 mg/kg per day. The PEA was readily absorbed from the skin and excreted primarily in the urine. There was no clear effect of dosage on the percentage of material absorbed, as measured by excretion of radioactivity (84% of the 140 mg/kg dosage was recovered; 79% in the 700 mL/kg dose from the supplemental study) with significant amounts of PEA lost due to evaporation, despite occlusion of application sites with foil and a porous bandage.
The rapid excretion of PEA was confirmed in a quantitative tissue distribution study in male rats, in which essentially all absorbed PEA from dosages of 140 mg/kg were eliminated from tissues by 12 hours. Some radioactivity remained in (decreasing order) treated skin, fat, kidneys adrenal glands, pancreas, and liver.
Autoradiography of pregnant rats reflected the distribution observed in quantitative studies in male rats. However, radioactivity was only observed in fetuses during peak blood levels in dams administered repeated dosages of 140 or 700 mg/kg PEA for 10 consecutive days, and then only in low or moderate concentrations.
The pharmacokinetics of PEA in rabbits after single topical applications of 140 or 700 mg/kg was similar to that of rats (about 50% of the applied PEA was absorbed and subsequently metabolized, mostly to PAA, as in rats). However, in rabbits at a dosage of 700 mg/kg, the concentration–time profile of PAA was markedly different (prolonged, high levels for 24 hours), which was unlike the profile in rats. These results suggested a dose-dependent saturation of the glycine conjugation pathway in rabbits, which was confirmed by the presence of PAA in the urine.
A dermal absorption study in humans under simulated exposure conditions, in which 10 mg of radiolabeled PEA was applied to the chest, revealed that essentially all absorbed PEA was excreted in the urine within 24 hours. An average of only 7.6% of the total dosage was absorbed, compared to 77% in the rat (after 24 hours). Based on the results presented here, the rat and human dermal absorption data were used to revise the development toxicity margin of safety. This revision takes into account the estimated daily human exposure 11 and the percentage of applied dermal dose that is absorbed by humans. These are then compared to the rat maternal and developmental NOAEL 4 for dermal application and the percentage of applied dermal dose that is absorbed by rats. The margin of safety is calculated to be greater than 2600 based on the following:
human exposure: 0.3 mg/kg per day × 7.6% absorbed by humans = 0.02 mg/kg per day;
NOAEL rat: 70 mg/kg per day × 77% absorbed by rats = 53.9;
53.9/0.02 = a margin of safety of 2695.
However, adverse developmental effects of the fetus are often considered to be due to excessively high peak exposures to the noxious agent. 18 Therefore, calculation of peak plasma concentrations is also of interest in the estimation of a developmental toxicity margin of safety. As such, the margin of safety based on peak plasma concentration yielded a margin of safety greater than 6000 as described below.
Cmax human plasma level from 0.15 mg/kg dermal simulated dose: 0.014 μg/mL;
Interpolated rat peak plasma level at 70 mg/kg (NOAEL dosage): approximately 90 μg/mL*
90/0.014 = a margin of safety of 6428
(*This figure was derived from the rat peak plasma level of 180 ug/mL at a dose of 140 mg/kg/d, 2 times greater than the NOAEL, which is interpolated to be 90 ug/mL at the NOAEL.)
Based on the accumulated conservative data and the calculations presented above, it is concluded that PEA, under the declared levels of use as a fragrance ingredient, would not produce developmental or reproductive effects in humans.
Footnotes
Authors’ Note
Author David R. Hawkins has retired and can be reached at the following address: Suffield, Norwich NR11 7EP, UK.
Declaration of Conflicting Interests
Valerie T. Politano, Gretchen Ritacco and Anne Marie Api are employees of the Research Institute for Fragrance Materials, an independent research institute supported by the manufacturers of fragrances and consumer products containing fragrances.
Funding
This research was supported by the Research Institute for Fragrance Materials, an independent research institute that is funded by the manufacturers of fragrances and consumer products containing fragrances.