
Abstract
Developmental toxicity studies for pharmaceutical safety testing are designed to evaluate potential adverse effects of drug treatment on pregnancy and on the developing embryo/fetus. Biopharmaceuticals present specific challenges for developmental toxicity testing because the pharmacology of these molecules, which are frequently human-specific proteins, is often restricted to humans and nonhuman primates (NHPs). For those species-restricted molecules, the only option for the evaluation of potential effects on development of the human biopharmaceutical is to use NHPs. This article reviews each of the stages of development in cynomolgus macaques (the most frequently used NHP) and the potential exposure of the embryo, fetus, and infant following administration of a biopharmaceutical during pregnancy and lactation. Because the purpose of the NHP developmental studies is to identify potential human risks, a comparison between macaque and human development and potential exposure has been made when possible. Understanding the potential exposure of the conceptus relative to critical periods in development is essential to designing a scientifically based study that adequately addresses human risks. Some options for NHP study designs, including the option of combining end points into a single study, and the pros and cons of each of the study options have been reviewed. Developmental studies for biopharmaceuticals in NHPs need to be optimally designed on a case-by-case basis taking into consideration the pharmacology of the molecule, the type of molecule (antibody or non-antibody), the potential exposure relative to the development of potential target organs, the clinical use, and the ethical considerations associated with the use of NHPs.
Introduction and Background
Purpose of Developmental Toxicity Testing
Developmental toxicity studies for pharmaceutical safety testing are designed to evaluate potential adverse effects of drug treatment on pregnancy and on the developing embryo/fetus. Adverse effects on development can occur due to direct exposure of the developing embryo/fetus to the drug or can occur indirectly due to toxicity to the mother or to the placenta. The ability of a drug to produce direct toxic effects on the conceptus will depend on the degree of embryo/fetal exposure, potential on-target and off-target toxicities, and the timing of exposure relative to susceptible periods in development.
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S5 (R2) regulatory guidance document 1 describes 3 types of studies that can be conducted to evaluate potential effects of medicinal products on reproduction and development. These studies include a fertility study, an embryo/fetal development (EFD) study, and a pre- and postnatal development (PPND) study. For the purposes of this review, the focus will be on the evaluation of embryo/fetal and pre- and postnatal development.
According to the ICH S5 (R2) guidance document, EFD toxicity studies are generally conducted in 2 mammalian species, with the preferred species being rats and rabbits. 1,2 The EFD study is designed to detect any structural abnormalities arising from drug exposure during the embryonic period. These studies are frequently completed prior to the inclusion of women of childbearing potential in clinical trials. Information generated from either preliminary or definitive EFD studies as well as any other pertinent information are included in the patient informed consent documents for clinical trials. In certain instances, women of childbearing potential can be included in clinical trials without the completion of EFD studies so long as extensive precautions are taken to prevent pregnancy (refer to ICH M3 (R2)). 3
Pre- and postnatal development studies are generally conducted in only a single mammalian species, with the preferred species being the rat. 1,4 The PPND study is designed to detect any structural and/or functional abnormalities arising from drug exposure during the embryonic, fetal, and postnatal periods These studies should be completed by the time of product registration. Information from the PPND study as well as the EFD study is included in the product label which provides information for the prescribing physicians and the patients on the potential use of the drug during pregnancy and lactation.
Considerations for Biopharmaceuticals
Biopharmaceuticals represent a diverse group of molecules that are derived from cell cultures or produced using recombinant DNA technology. Biopharmaceuticals include proteins, peptides, and oligonucleotides. Because of the diversity of biopharmaceuticals, each molecule needs to be considered individually based on its structure, physicochemical properties, and its pharmacology. One feature in common with all of the biopharmaceuticals is their large molecular size (~1000-150 000 Da) relative to that of traditional pharmaceutical drugs (~200-400 Da). The cytokines have molecular weights in the 20 000 to 40 000 Da range, enzymes 60 000 to 100 000 Da, and monoclonal antibodies (mAbs) about 150 000 Da. Some peptides and oligonuleotides may have lower molecular weights but are still larger than most drugs and chemicals. The large molecular size of biopharmaceuticals limits their distribution within the body. Large molecules do not readily diffuse across membranes and so their distribution is largely confined to the blood volume or to the blood and the extracellular fluid volume. The consequence of this limited distribution is that biopharmaceuticals will not enter cells and therefore do not disrupt intracellular mechanisms and do not interact with DNA or other chromosomal material. Large molecules also will not freely diffuse across physiological barriers such as the blood−brain barrier, the blood−retinal barrier, the blood−testis barrier, or the placenta. Significant transport across these barriers will occur only if there are specific mechanisms to transport large molecules across the membranes. One such mechanism is the neonatal Fc receptor (FcRn)-mediated transcytosis of antibodies across the placenta in primates, and across the yolk sac and neonatal gut in rodents. 5 –7
Although large molecules (antibodies and non-antibodies) do not freely penetrate the placenta by simple diffusion, it is possible for small amounts of large molecules to cross the placenta (or other physiological barriers) by paracellular pathways. 8,9 The placenta becomes thinner and more permeable as pregnancy progresses. 10 Some large molecules, which are restricted from crossing the placenta early in pregnancy, may be able to slowly cross the placenta in limited amounts late in pregnancy. Studies evaluating the transfer of proteins in in vitro human placental systems may, therefore, represent a “worse-case” scenario because the placental tissues obtained from full-term pregnancies are at their most permeable.
Because most biopharmaceuticals are proteins or peptides, they are susceptible to proteolytic degradation. Proteolytic degradation in the gastrointestinal tract along with poor absorption across biological membranes makes biopharmaceuticals poor candidates for oral administration. Consequently biopharmaceuticals need to be administered systemically and for some of the smaller molecules, such as insulin, by inhalation.
Many of the recombinant human proteins such as insulin, insulin-like growth factor, growth hormone, and erythropoietin are pharmacologically active in rats and rabbits as well as in humans. Therefore, because the pharmacology is not species restricted, these biopharmaceuticals can be tested in rats and rabbits in accordance with ICH S5 (R2). 1 Molecules that have agonistic activity and are intended to be administered to patients at low levels to restore normal function can show effects on fetal growth when administered to pregnant animals at suprapharmacological doses. This is an expected outcome that is most likely a secondary consequence of the exaggerated pharmacology in the dams. Where studies have been done to measure placental transfer, very little fetal exposure has been demonstrated with these types of agents, 11 thus supporting the hypothesis that the effects are secondary to exaggerated pharmacology in the mothers rather than to direct effects on the conceptus.
The main concern for development toxicity testing for biopharmaceuticals is for those products in which rodents and rabbits are not pharmacologically relevant species. Pharmacological relevance can be determined by a number of methods including target sequence homology between species and cell-based or in vivo functional assays. 12 For some molecules the only pharmacologically relevant species is the nonhuman primate (NHP).
Considerations for the Use of NHPs in Developmental Toxicity Testing
Neither the original ICH S6 guidance document for biotechnology-derived pharmaceuticals 13 nor the ICH S5 (R2) 1 guidance document for the reproductive and developmental toxicity testing of medicinal products specifically addressed how developmental studies should be conducted when NHPs are the only relevant species. Both guidance documents emphasized a case-by-case scientific approach and stated that the actual testing strategy should be determined by the anticipated clinical use, the form of the substance and routes of administration intended for humans, and making use of any existing data on toxicity, pharmacodynamics, kinetics, and similarity to other compounds in structure/activity. The ICH S5 (R2) 1 guidance document provides examples of rodent study designs, but with regard to NHPs it states that they are best used when the objective of the study is to characterize a relatively certain reproductive toxicant, rather than detect a hazard. As discussed in Stewart, 14 >300 fetuses would be required to detect a 10-fold increase in a birth defect that occurred with a background incidence of 3%. Because NHPs have a low background incidence of birth defects (<1%) 15 and produce only a single fetus, it is not possible to power a study to detect rare events. Only those events that occur at an almost certain incidence and that are generally directly related to the pharmacology can be detected. Therefore, NHP development studies are not risk assessment studies in the traditional sense but are rather pharmacological characterization studies. Some of the issues regarding the conduct of NHP reproductive and development studies have been addressed in the draft ICH S6 (R1) 16 addendum.
When NHPs are the only relevant species for nonclinical safety evaluations, the question to be addressed is “how can an NHP study be designed, which optimizes the use of NHPs while obtaining the most valuable information for patients?” and not “how can rodent study designs be adapted for NHPs?” If the NHP study does not provide valuable information for patients, then there is little justification for conducting the study. As stated in ICH S5 (R2), 1 “All persons involved should be willing to discuss and consider variations in test strategy according to the state-of-the-art and ethical standards in human and animal experimentation.” Because the experimental use of NHPs is associated with special ethical concerns, potential strategies for reducing the use of NHPs have recently been considered and proposed. 17,18 These strategies include reducing the number of treatment groups and reducing the number of studies. Because the NHP development studies are not risk assessment studies, it is not necessary to obtain a dose−response relationship. A single high-dose level and a control group may be sufficient to identify any potential effects. 14 Combining developmental study designs into a single study is completely in line with the ICHS5 (R2) 1 regulatory guidance document and is reinforced in the proposed draft addendum to ICHS6 (R1). 16
Although NHPs are most frequently used when they are the only pharmacologically relevant species, it may also be necessary to use NHPs when rodents or rabbits are relevant species but the immunogenicity in those species precludes their use. Nonhuman primates may also be used if there is a specific cause for concern that can only be addressed in NHPs or when the biology in the rodents is different from that in the primates. The predominant NHP species currently used for biopharmaceutical development is the cynomolgus macaque (Macaca fascicularis), constituting the relevant animal model in about 80% of the studies that have been conducted. 18,19
Considerations for Immunogenicity
Because most biopharmaceuticals are human-specific proteins, it is expected that they will be immunogenic in animals but not necessarily in humans. Immunogenicity in animals is, therefore, not expected to be predictive of human immunogenicity, but the development of an anti-drug antibody response in animals can have implications on the study design and on the interpretation of the study results. 20 Anti-drug antibodies can develop in any species but, in general, immune responses toward a human protein are less frequently observed in NHPs than in rodents because of the genetic similarities between humans and NHPs versus humans and rodents.
When an anti-drug antibody response does occur, it can reduce exposure of animals to the test biopharmaceutical and thereby overestimate safety. One strategy for dealing with this is to increase the dosing frequency and/or the dose level to “overcome” the immune response. Using this “dosing through” strategy can result in sustained high exposure of the animals to the biopharmaceuticals during the critical periods of development. Immune responses against the human biopharmaceutical can, however, have adverse consequences in the animals, such as immune complex deposition in tissues, autoimmunity, or hypersensitivity. 21 Because these animal-specific effects may not be relevant to humans, this may overestimate the toxicity of the product.
Another consequence of anti-drug antibodies against human biopharmaceuticals in animals is that the antibodies that are generated are likely to cross the placenta if they are of immunoglobulin G (IgG) subtype. If these antibodies bind to an endogenous human protein, they could result in adverse effects in the fetuses that may, or may not, be relevant to humans.
Although monomeric antibodies are transported across the placenta, immune complexes do not appear to be transported but instead are trapped in the placenta. 22 This may have implications on the estimation of maternal versus fetal exposure ratios if the assay for detection of the biopharmaceutical cannot distinguish between free and antibody bound mAb.
Developmental Stages and Potential Exposure to Biopharmaceuticals
To design a scientifically based, clinically relevant, nonclinical assessment of potential developmental effects of biopharmaceutical treatment, it is important to understand the timing and extent of exposure relative to susceptible periods of development. The following sections provide a “snapshot” of some of these events. Where possible a comparison has been made between macaques (the most frequently used NHPs) and humans. The discussions on developmental stages and potential exposure to biopharmaceuticals are mostly focused on antibody therapeutics because this is the only class of biopharmaceuticals that are known to produce significant prenatal exposure to the developing fetus. For non-antibody biopharmaceuticals, it can be assumed that potential effects are mostly limited to maternal or placental effects because they are not transported across the placenta. When endogenous IgGs are used as examples, a number of caveats need to be taken into consideration: Not all subclasses of IgGs are transported across the placenta equally, and different pathogens produce different repertoire of IgGs. It is assumed that IgG antibodies detected in fetal blood are of maternal origin because fetuses have a limited ability to generate IgG until after birth. It is also assumed that therapeutic antibodies that have an unmodified Fc fragment will be transported across the placenta in a manner similar to that of naturally acquired antibodies. For the discussions on stages of development, the day of presumed fertilization is considered day 0.
The Embryonic Period
The embryonic period is the period of major organ formation. This period spans from gestation day (GD) 20 to 50 in macaques 23,24 and from days 20 to 56 in humans. 25 The first trimester in macaques 23 is estimated to be from GD 0 to 55 and in humans 25 from GD 0 to 90. Therefore, the embryonic period in macaques continues through to near the end of the first trimester, whereas in humans the embryonic period ends about 4 weeks before the end of the first trimester. Consequently, in humans the first trimester includes not only the embryonic period but also the early fetal period. This is important to consider when the term trimester is used to compare developmental stages across species.
The embryonic period consists of growth that involves rapid cell division, morphogenesis that involves movement and elaboration of cells to form organs, and differentiation that involves the maturation of physiological processes. The sequence and timing of organogenesis is very similar in both humans and in macaques. 23,24 Because rapid development occurs during organogenesis, this period is highly susceptible to teratogenic agents that may cause major congenital anomalies.
The cardiovascular system is the first major system to develop and function in the embryo. The embryonic heart beat can be detected by ultrasound examination as early as GD 22 in both humans and macaques. 26 By the end of the period of major organogenesis, the human and macaque embryo is approximately 30 mm in length from crown to rump, weighs about 8 g, and has distinct human (or NHP) characteristics. At this time, all major organs are in place.
Small molecular weight drugs and chemicals, which may be toxic to the developing embryo and which can diffuse across the placenta during the embryonic period, have the potential to induce teratogenic effects. For this reason a teratogenicity study, which involves dosing pregnant animals during the period of major organogenesis, has become standard evaluation for pharmaceuticals and chemicals.
Very few studies have attempted to collect embryonic blood samples from humans or NHPs for the measurement of drug or antibody concentrations because the blood volume of the embryo is very small and it is technically very difficult to obtain uncontaminated blood samples in quantities sufficient for analysis. As a substitute for blood samples, Jauniaux et al 27 collected coelomic samples by coelocentesis from 6 to 12 weeks (late embryonic period and early fetal period) human pregnancies. The coelomic cavity, which contains the secondary yolk sac, surrounds the amniotic cavity in the first 12 weeks of human pregnancy. This cavity is directly connected to both the fetal gut and the dorsal aorta by the vitelline duct. 28 Substances that are able to cross the trophoblastic barrier may directly enter the fetal circulation at the level of the villous trees or the chorionic plate. Therefore, concentrations of substances in the coelomic cavity may approximate concentrations within the fetus. In this study, the coelomic concentrations of total IgG and IgA increased linearly between 7 and 12 weeks of gestation, with the median coelomic versus maternal concentrations over this period being 3.5% for IgG and less than 1% for IgA. Antigen-specific antibodies to Toxoplasmosis gondii, Cytomegalovirus, and rubella were not detectable in fetal serum before 9 weeks of gestation. If it is assumed that all of the IgG antibodies in the fetal cavities were of maternal origin, then these studies would suggest that low levels of Igs can be transferred to the fetal compartments during the early fetal period. The earliest samples collected in this study were during the final stages of the embryonic period, weeks 7 to 8 at which time Igs were detectable in the coelomic fluid but were at very low levels. In macaques, the earliest time point reported is GD 84 (34 after the end of major organogenesis) at which time the fetal concentrations were about 7% of the maternal concentrations. 29 From the observations of maternal versus fetal concentrations of endogenous antibodies in humans and macaques, a complete absence of embryonic exposure during the period of major organogenesis cannot be assumed. However, because the fetal exposures at the earliest time points measured were low, it can be assumed that the embryonic exposures at earlier time points would also be low. What is important to understand for the evaluation of patient risks are the pharmacology of the molecule and whether a small fraction of a clinically relevant dose could be sufficient to evoke a pharmacological or toxicological effect in the developing embryo. For example, if the antibody inhibits a critical embryonic growth factor and is administered to patients at high doses, as is often the case for the treatment of oncology indications, a potential effect on the developing embryo cannot be entirely excluded. However, if the antibody targets a soluble protein that is not essential for normal development and is administered to patients at a low dose, it is unlikely that a teratogenic effect will occur even if some embryonic exposure did occur.
Although the embryonic period is the most susceptible period for teratogenicity induced by small molecule drugs or chemicals that can readily diffuse across the placenta during the period of major organogenesis, the primate embryo is somewhat protected from direct adverse effects of both antibody and non-antibody biopharmaceuticals because these large molecules do not cross the placenta to any great extent during the embryonic period.
The Fetal Period
The fetal period begins at the end of organogenesis (week 8 in humans and week 7 in macaques) and is marked by closure of the hard palate. During the early fetal period primary ossification centers appear in the skeleton, the early genitalia appear, erythropoiesis shifts from the liver to the spleen, and urine formation begins. This initial period is followed by a period of rapid ossification of the skeleton and sexual differentiation. In macaques, the development for some organ systems during the fetal period is accelerated relative to that of humans 30 (Table 1 ).
Timing of Some Major Postnatal Developmental Events in Macaques and Humans a
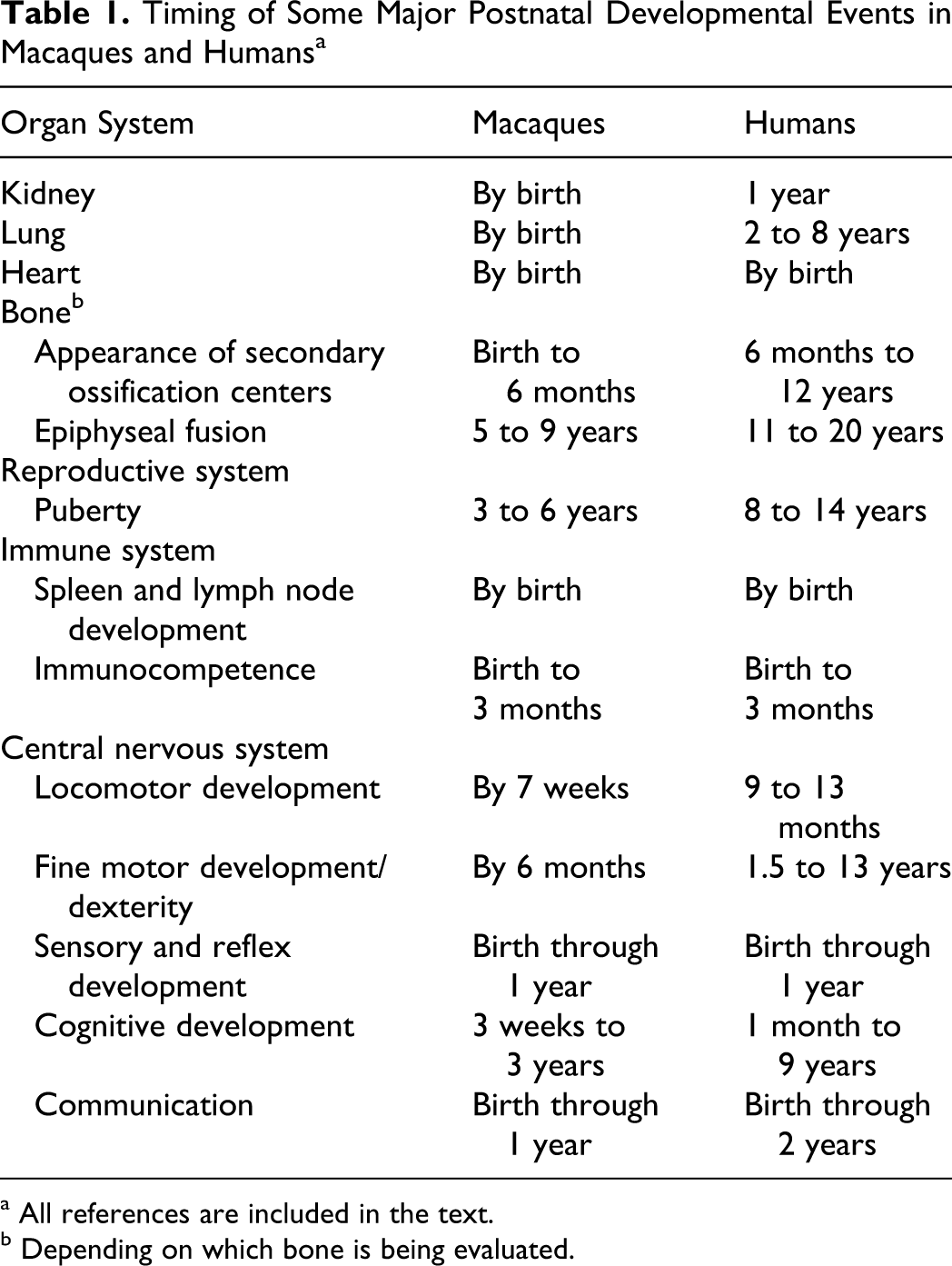
a All references are included in the text.
b Depending on which bone is being evaluated.
The lungs mature during the fetal period and become adapted for gaseous exchange. The bronchioles and the alveoli divide and become more vascularized. The kidneys become fully formed during the fetal period. 31 The development of the human and macaque immune system appears to be similar. 23,24,32 This is especially true when the development of the immune system in humans and macaques is compared as a percentage of the total gestation period. 24 During the fetal period, there is an increase in the number of the various immune cells within the immune organs and gradual aggregations of the cells to acquire their final anatomical locations. Macaque fetuses have been shown to have IgG-, IgM-, and IgA-producing cells as early as GD 70. 23 The IgG- and IgA-secreting cells are predominantly in the gastrointestinal tract, whereas the IgM-secreting cells are in the thymus and spleen.
Numerous studies have evaluated the concentrations of IgGs in human and macaque fetal versus maternal blood samples. In one study, radiolabeled IgG was injected into pregnant women who were at about 12 weeks of gestation and were undergoing elective abortions. 33 Cord blood collected 24 hours after 125I-IgG injection demonstrated concentrations in fetuses that were 2.8% of maternal concentrations. This observation is consistent with the Jauniaux et al 27 study described above, which demostrated a low level of IgG transfer during the early fetal period. A review of the literature of clinical studies in which maternal and fetal serum concentations of IgG were measured showed that fetal IgG concentrations were extremely low at the beginning of pregnancy, increased with gestational age, and that fetal concentrations after the 35th week of pregnancy exceeded maternal concentrations. 34 A similar pattern of IgG distribution has been reported for the macaque. 24,29,35 A number of studies included in the clinical review by Palfi and Selbing 34 further evaluated the concentrations of each of the 4 human IgG subtypes in maternal verus fetal serum. 36 –39 These studies showed that the fetal−maternal ratio of IgG1 antibodies was greater than that for the other isotypes with a general order of IgG1 > IgG4 > IgG3 > IgG2. The fetal versus maternal distribution of the macaque IgG subclasses has not been reported and to date there is only limited information on the distribution of human IgG subtypes in macaques.
Exposure of the conceptus to pharmaceutical agents during the fetal period is less likely to produce a teratogenic effect than exposure during the embryonic period. However, for mAbs, exposure to the conceptus during the fetal period may be high and potential disturbances in growth or normal functional development need to be considered. Therefore, for mAbs, it is important that dosing in an NHP developmental study be extended into the fetal period if there is any possibility that treatment may be administered to patients during pregnancy. This is less of a concern for non-antibody biopharmaceuticals that will likely have only limited access to the conceptus throughout the entire pregnancy.
The Postnatal Period
The transition from the fetal period to the postnatal period involves the transfer of gaseous exchange function from the placenta to the lung. In the cardiovascular system, there is a cessation of umbilical circulation and a closure of the prenatal shunts. The immune system changes from intrauterine maternal influences to infant control. The postnatal period is a period of growth and functional maturation of the various organs. At birth, macaques are as developed, or more developed, than humans depending on the organ system (Table 1). Postnatally macaques grow at a faster rate than humans with 1 macaque year being approximately equivalent to 4 human years based on skeletal growth and sexual maturation. 15
At birth, the lung is more mature in macaques than in humans. 40,41 In humans, over 85% of the alveoli develop after birth and full lung maturity is not reached until about 2 to 8 years of age. 42 Macaques are also more advanced at birth for several of the most commonly measured end points of central nervous system (CNS) functional development. 43
Postnatal development of kidney and heart appears to be very similar in humans and macaques. 31,44 Nephrogenesis is complete by birth and kidney function matures postnatally with increases in glomerular filtration rate and concentrating ability. In a study of cynomolgus macaques from postnatal weeks 4 through 39, the clinical pathology parameters remained almost unchanged during this time period except for a change in γ-glutamyl transferase and an increase in blood urea nitrogen. 45 These findings indicated the early well-developed function of the neonatal macaque kidney. In humans, the kidney is considered to undergo functional development up to 1 year of age. 31
In humans, the stomach of the new born has low levels of acid and pepsin and the levels increase with age as the stomach becomes more glandular. The intestines grow in length and diameter during the postnatal period and mature functionally. By late infancy, most gastrointestinal functions are comparable to that of adults. 46
The main organ systems that continue to undergo growth and maturation during the postnatal period in both macaques and humans are growth of the skeleton, development of central nervous system function, acquisition of immune competence, and sexual maturation.
Skeletal development begins prenatally with the appearance of the primary ossification centers and continues postnatally with the appearance of the secondary ossification centers, longitudinal bone growth at the epiphyseal growth plate, and eventual fusion of the growth plates when the skeleton has reached its adult size. At birth, monkeys have more ossification centers than humans. 47 It has been estimated that bone age in macaques at birth resembles that of a 5- to 6-year-old human and within the first 6 months of age corresponds to that of a 7-year-old human. 48 –51 The fusion of the growth plates occurs at different ages for different bones and between genders but ranges from about 5 to 9 years of age in macaques. 52 –54
Postnatal development of the reproductive system is similar in humans and in NHPs and is characterized by a perinatal episode of reproductive hormone secretion followed by a hiatus in the reproductive endocrine system until the onset of puberty. 55 –57 Sexual maturity occurs at about 3 to 4 years in female cynomolgus macaques and about 4 to 6 years in male cynomolgus macaques. 15 Both in humans and NHPs, kisspeptin is a pivotal regulator of the endocrine onset of puberty. 58 Kisspeptin provides the drive to gonadotropin-releasing hormone system which in turn activates gonadotropin secretion followed by gonadal steroid secretion. The blood−testis barrier becomes established at puberty. Spermatogenesis occurs early in puberty in response to elevated levels of follicle-stimulating hormone and testosterone. Following ovarian steroid hormone secretion, gonadotropin-producing cells of the anterior pituitary acquire the capacity to respond to the stimulatory actions of estradiol after menarche. 59 Ovulation occurs after menarche and secondary sexual characteristics occur at puberty.
The postnatal development of immune systems in both macaques and humans is similar. 23 At birth, the primate immune system is more mature than that of rodents. 32 During the postnatal period immune competence is acquired. At birth, both human and macaque neonates have high serum concentration of IgG and low serum concentrations of IgM and IgA. The IgG in neonatal serum is believed to be mostly of maternal origin. The levels of IgM and IgA gradually increase postnatally. The IgG concentrations decrease over the first 6 months of postnatal life as a result of clearance of maternally derived antibodies, and thereafter increase as the infant begins generating its own IgG. 23
At birth, serum IgG concentrations in the neonates are similar to or exceed maternal concentrations. 29,34,60 In nonclinical toxicology studies, it is not unusual for pharmacologically relevant levels of therapeutic antibodies to be detected in infant serum for 6 months or more after birth, as a result of the high-dose levels that are administered to the dams. Exposure of macaques to antibodies during the fetal period and during the first 3 to 6 months of postnatal life covers many critical periods relevant to human development (Table 1). Because monkeys are born at a more developmentally advanced stage than humans, in some respects, and develop postnatally at a more rapid rate than humans, the combination of exposure during the fetal period and postnatally through 6 months of age is equivalent to exposing human infants through at least 2 years of age that is, through to the end of the infant stage. For mAbs with a long serum half-life, dosing of the macaque infants postnatally may not be essential to mimic human exposure during the early postnatal years.
Considerations for Modified Antibodies
All of the information discussed above regarding placental transfer of antibodies refers to naturally acquired antibodies or to therapeutic mAbs that have sequences similar to that of the natural antibodies. However, genetic engineering enables the development of antibodies that have desirable properties such as enhanced serum half-life or enhanced biodistribution. 61,62 These antibodies may behave very differently from natural antibodies. Because the FcRn receptor is involved in both serum persistence and transport of antibodies across the placenta, modification of the IgG molecule to increase the affinity for FcRn may increase the distribution of the antibody to the fetus. For example, mutations in the CH3 domain of human IgG1 (His-433 to Lys and Asn-434 to Phe) were shown to result in an increase in the affinity of binding of the antibody to human FcRn and to an increase in the transfer rate of the antibody across the human placenta. 63
Embryo/Fetal Development Study Designs in Macaques
Basic Study Design
The basic EFD study design in macaques is based on the ICHS5 (R2) rodent (Figure 1 ). The purpose of the EFD study (previously referred to as a segment II study or a teratology study) is to detect adverse effects on the pregnant female and development of the embryo and fetus consequent to exposure of the female from implantation to closure of the hard palate. This dosing period includes the period of major organogenesis (the embryonic period), the period of high susceptibility to toxic chemicals. Toxicity parameters evaluated in these studies include enhanced toxicity relative to that in nonpregnant females, embryo/fetal death, altered growth, and structural changes.
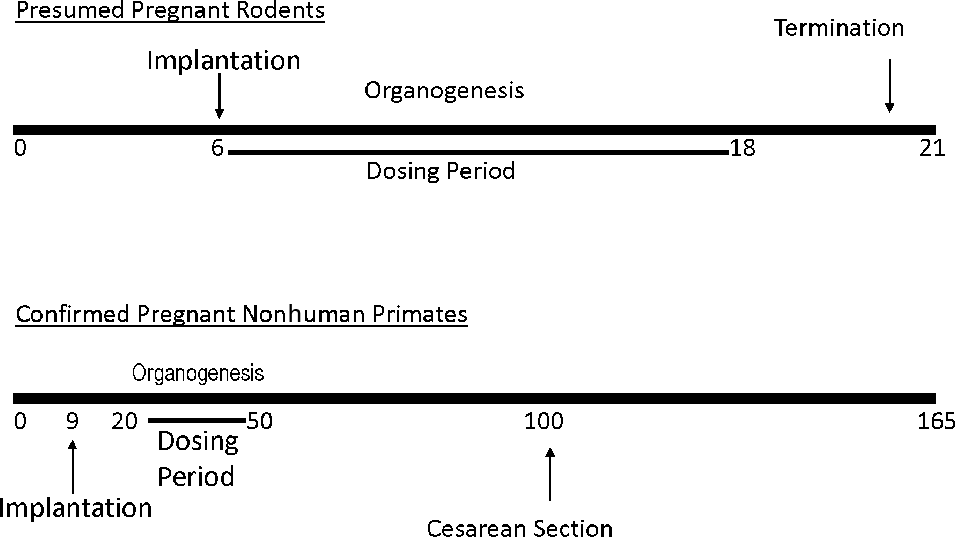
Comparison of basic rodent versus macaque embryo/fetal developmental study designs.
The basic macaque study design involves dosing from GD 20 to GD 50 (period of major organogenesis/embryonic period), with fetuses being harvested for gross examination on GD 100 (65 days before parturition). The macaque study design is, therefore, similar to that of rodents; but for practical purposes, there are a few differences in the study designs between rodent and macaque. One difference in the study design between the rodent and the macaque is that in macaques dosing is not initiated at implantation, which occurs at about GD 9 64 but is delayed until after implantation has occurred and the pregnancy can be confirmed by ultrasound examination. This is necessary because macaques have a naturally low fertility rate and high spontaneous abortion rate such that only a small fraction of mating will result in a successful pregnancy. Therefore, to avoid dosing many more animals than are needed for the study, it is preferable to wait until pregnancy is confirmed by ultrasound examination, before dosing is initiated. Pregnancy can be detected by ultrasound examination or by measurement of serum monkey chorionic gonadotropin ([mCG]; ≥1 ng/mL) by 3 days after implantation. 65 The advantage of using ultrasound for the determination of pregnancy is that this provides a direct real-time readout. Measurement of serum concentrations of mCG can be evaluated retrospectively to confirm that monkeys that were diagnosed as pregnant by ultrasound prior to treatment initiation remained pregnant until the time of dosing initiation. 66 This is important because implantation-related uterine changes can be observed sonographically in animals that do not progress to viable pregnancy. 65
For the basic macaque EFD study, GD 100 has been established as a suitable time for the fetal examinations. Gestation day 100 was selected based on the fact that by this time the fetuses are sufficiently developed for adequate anatomical and skeletal evaluations. 23 Given that the purpose of an embryo/fetal study is to evaluate maternal effects and any gross morphological defects in the fetuses following maternal dosing during the embryonic period, this basic study design meets these objectives. For small molecule drugs that can diffuse across the placenta, developmental toxicity can occur due to direct embryonic exposure to the drug during the period of major organogenesis or secondary to maternal toxicity. For large, non-antibody biopharmaceuticals, any potential effects would most likely be related to maternal toxicity because as described previously large molecules do not freely diffuse across the placenta and are therefore not expected to produce significant direct embryonic or fetal exposure.
One example of a non-antibody biopharmaceutical class that produces maternal effects that adversely affect pregnancy is the type I interferons (IFNs). These molecules can only be evaluated in NHPs due to the species-restricted pharmacology. In vitro studies using human full-term placenta demonstrated that IFN-α does not cross in vitro the full-term human placenta. 67 Administration of IFN-α and other types of IFNs resulted, in most studies, in a dose-dependent increase in abortions but with no signs of teratogenicity 68 (Table 2 ). This effect was subsequently shown to be associated with a decrease in progesterone levels in the mothers. 30 Although a treatment-related increase in abortion rates was demonstrated in these studies, the use of NHPs to evaluate aborifacient potential is not generally recommended. Abortion rates of up to 20% or greater can occur in control groups of macaques, making it difficult to evaluate whether abortions in treated animals are treatment related. 15
Embryo−Fetal Losses in Macaques Dosed With Human Type I Interferons During Pregnancy a
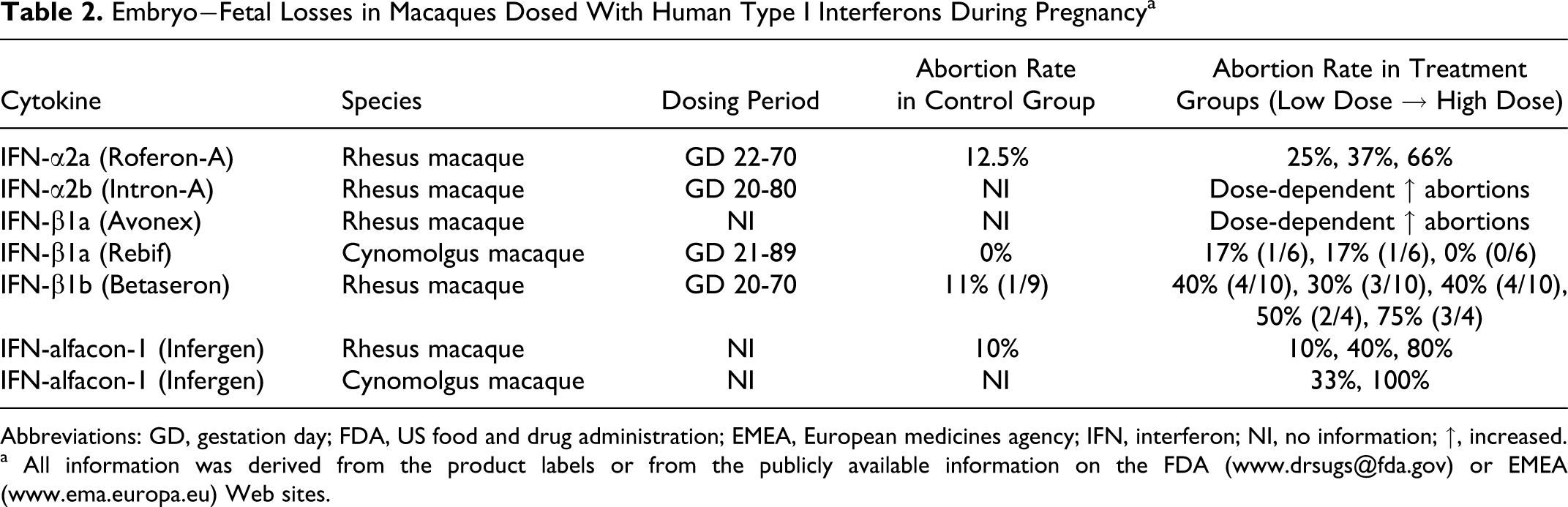
Abbreviations: GD, gestation day; FDA, US food and drug administration; EMEA, European medicines agency; IFN, interferon; NI, no information; ↑, increased.
a All information was derived from the product labels or from the publicly available information on the FDA (www.drsugs@fda.gov) or EMEA (www.ema.europa.eu) Web sites.
Antibody biopharmaceuticals are expected to be transferred from the mother to the fetus during pregnancy. However, as described previously, this transfer does not appear to occur consistently throughout pregnancy but occurs predominantly during the later part of pregnancy in humans and NHPs (Figure 2). Because of this differential transfer throughout pregnancy, the likelihood of significant exposure occurring during major organogenesis is low and the likelihood of inducing dysmorphogenic effects from direct embryonic exposure is low.
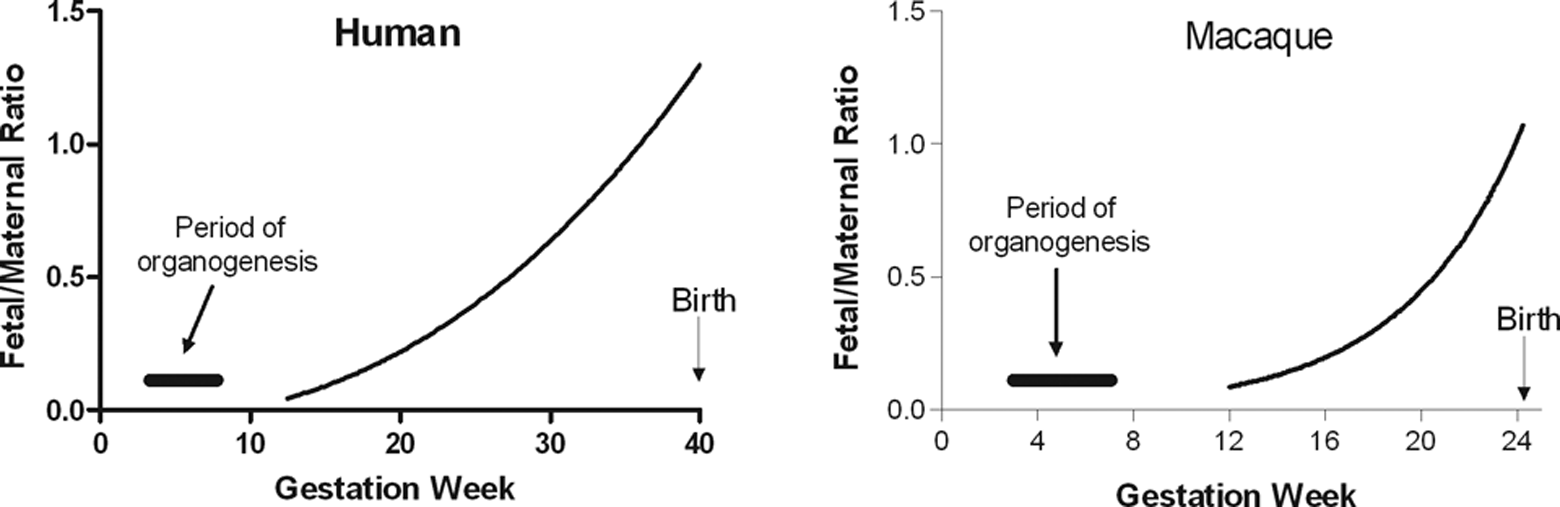
Simulation of fetal−maternal concentration ratios with gestational age in humans and macaques. The human simulation was based on data from Palfi and Selbing, 34 where a regression of f/m IgG = 0.399 − 0.059 × GA + 0.003 × GA2 − 2.065-5 × GA3 was proposed. The macaque simulation is based on data from Fujimoto et al 29 and Coe et al. 35
In the basic macaque EFD study design, there is a 50-day treatment-free period before the harvesting of the fetuses by cesarean section (GD 100). However, the mothers may still have significant levels of antibody in their serum at the time of the cesarean section because antibodies have long serum half-lives. Although significant embryonic exposure is not expected during the period of major organogenesis, fetal exposure is expected to begin shortly after organogenesis and to increase as pregnancy progresses. Therefore, because the mothers remain exposed to the antibody during the fetal period, it is likely that the fetuses will also become exposed to the antibody. However, as illustrated in Figure 3, the fetal exposure is expected to be only a fraction of the maternal exposure between GD 50 and GD 100. Although the exposure in the fetuses will be low relative to the maternal exposure, the fetuses may still be exposed to pharmacological levels of the antibody if mothers were dosed with high multiples of the pharmacological dose.
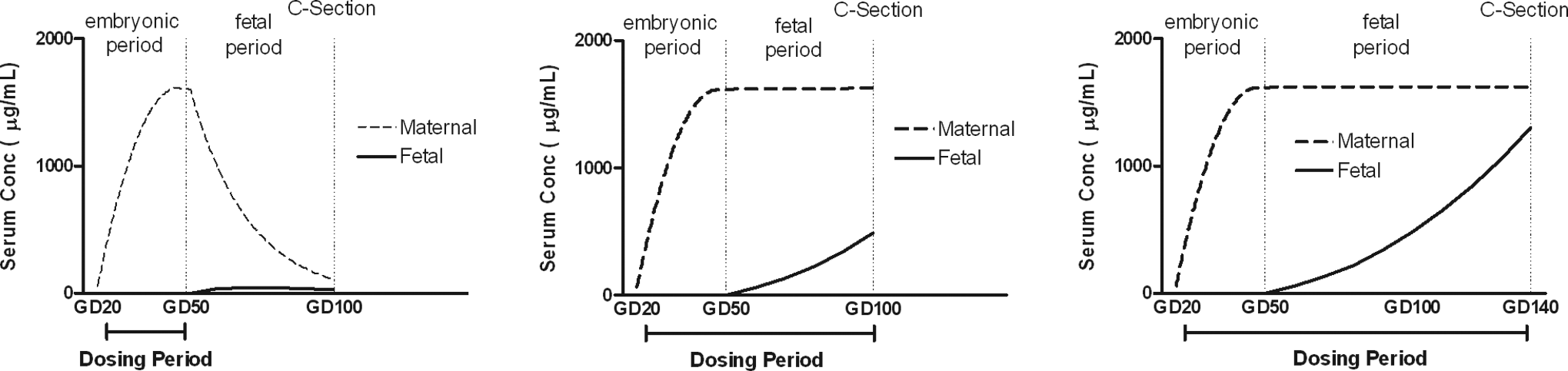
Hypothetical representations of maternal versus fetal serum concentrations of mAb. Assumptions are that the mAb in monkeys has a serum half-life of 14-days and that fetal−maternal concentration ratios are 1% on GD 50 (end of organogenesis), 30% on GD 100, and 80% on GD 140. Assumptions for maternal versus serum ratios are based on data from Fujimota et al 29 ; Coe et al 35 and from the marketed mAbs. mAbs indicate monoclonal antibodies; GD, gestational day; C-Section; Cesarean section.
Expanded EFD Studies
Although the fetal period is a period of growth rather than major organ development, the organ systems continue to grow and mature during the fetal period. For immune-modulating biopharmaceuticals, it has been recommended that the dosing period be extended into the fetal period. 19,30 This extended dosing into the fetal period is particularly important for antibody or antibody-like immune-modulating biopharmaceuticals, because the FcRn-mediated placental transfer of molecules that contain the Fc domain of IgG increase during the fetal period. Therefore, to optimize fetal exposure but without extending the duration of the study, a GD 20 to GD 100 dosing period with cesarean sections on GD 100 could be used (Figure 4). A comparison of hypothetical curves that show potential maternal versus fetal exposure in a study with dosing from GD20 to GD50 versus a study with dosing from GD20 to GD100 shows that fetal exposure is enhanced with the extended maternal dosing regimen (Figure 3). Although a standard EFD study only includes gross morphological examination of organs, histopathological examination of potential target tissues could also be included in an EFD study to enhance the overall evaluation.
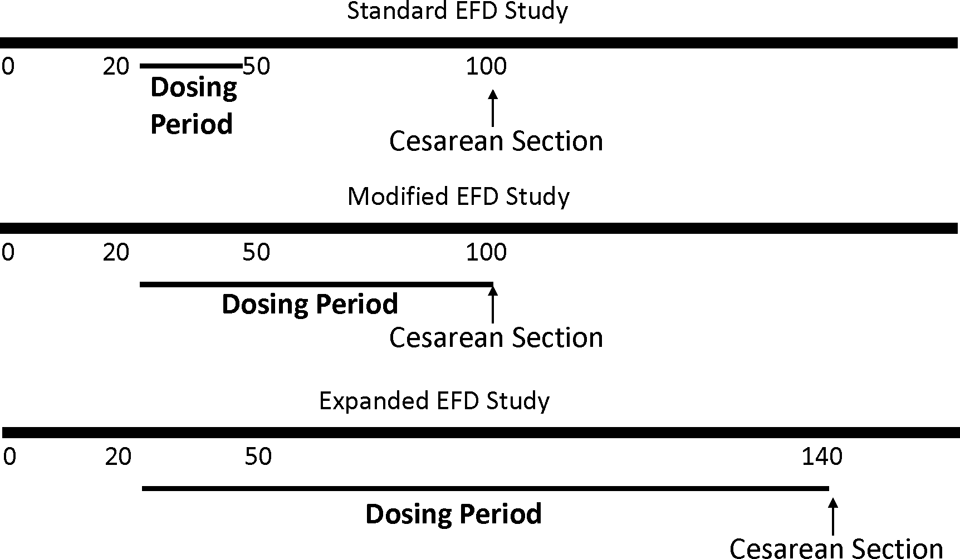
Examples of options for embryo/fetal development studies in macaques. EFD indicates embryo/fetal development.
An additional option for expanding the EFD study is to delay the cesarean sections until GD 140 or GD 150, with dosing up to the time of the cesarean sections (Figure 4). During the additional 40 or 50 days of maternal exposure, the fetal exposure will also increase such that by GD 140 or GD 150 the fetal exposure will approach that of the mothers (Figure 3). The advantage of this study design is that a greater period of fetal development will be evaluated and the fetuses will be exposed to high concentrations of the biopharmaceutical antibody. Terminating the pregnancies on GD 140 or 150, rather than allowing them to go through to full term, has the advantage that the wave of late pregnancy losses that occur post GD 150 is avoided (Figure 5). With cesarean sections at GD 140 or GD 150, 10% to 15% fewer animals are required at the start of the study than when pregnancies are taken to full term. Very few pregnancy losses occur between GD 100 and GD 150, so delaying the cesarean sections to GD 140 or GD 150 does not increase the number of animals but it does increase the duration of the study by 40 or 50 days.
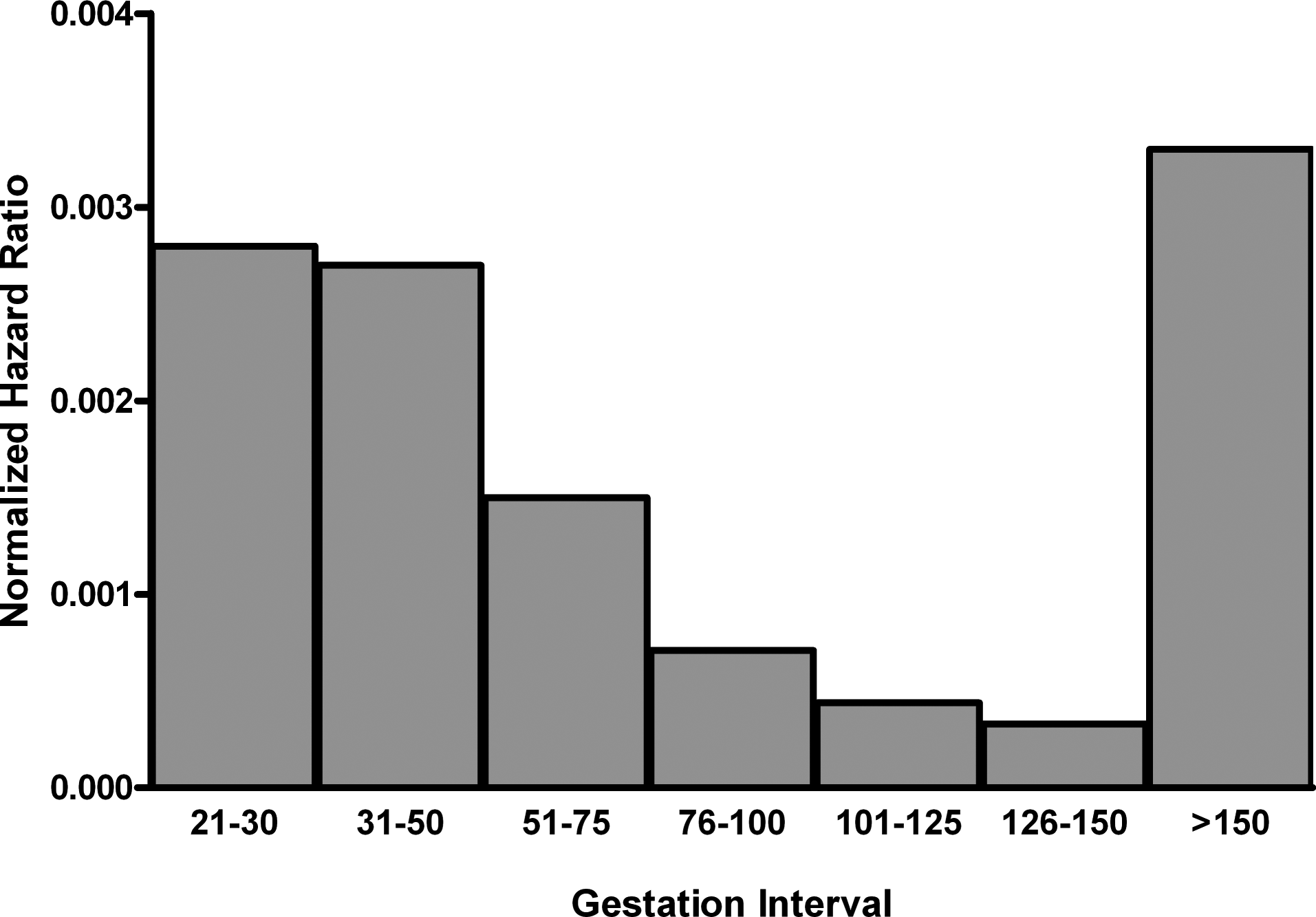
Hazard ratio during the prenatal and postnatal period until day 7 postpartum, normalized by interval duration (gestation days). The hazard ratio is the ratio of the number of losses during an interval to the number of fetuses/live infants at the start of each interval, that is, the conditional failure rate. The length of each interval varies between 10 and 25 days. To account for these differences, the hazard ratios shown have been divided by interval length (days; modified from Jarvis et al). 69 The duration of the >150 category was taken to be 10 days and corresponds to an average gestation period of 160 days. 57
The disadvantage of all the EFD studies is that functional consequences are not being measured. The EFD studies can only measure structural changes. A few examples of some effects that may be detectable in EFD studies following exposure during the fetal period are altered architecture of the lymphoid compartments, altered skeletal growth, altered hematopoiesis, altered sexual differentiation, and altered overall growth.
The expanded EFD study with histopathological examination of tissues and other potential end points such as clinical pathology or biomarkers clearly goes beyond the scope of the basic teratology study which is designed only to detect gross morphological changes. This, however, is appropriate when NHPs are the only possible species. For NHP studies, it is important that the maximum amount of information be obtained from each animal and that the number of animals used is the optimal required to obtain a meaningful assessment of potential hazards. It is also important that each study is tailored for the molecule that is being evaluated and for the clinical indication. To date, basic or expanded EFD studies in NHPs have been conducted for most of the approved species-restricted biopharmaceuticals (Table 2). For late stage oncology indications, an EFD study may be the only study that is required for registration of the product. 70
Pre- and Postnatal Development Study Designs in Macaques
The ICH S5 (R2) guidance document describes a prenatal and postnatal study design in which pregnant rodents are dosed from implantation through weaning and the pups are observed through sexual maturity. Rodent PPND studies are generally conducted after an EFD study has been completed. Information from the EFD study can be used to aid in the design of the PPND study. For NHP studies, this sequential segmented approach may not be practical from a timing perspective and from an animal usage perspective. Historically for some of the approved biopharmaceuticals, a single study has been initiated with separate cohorts being assigned for fetal examinations and postnatal examinations (Table 3 ).
Species-Restricted Antibody and “Antibody-Like” Biopharmaceuticals Tested in NHPs a
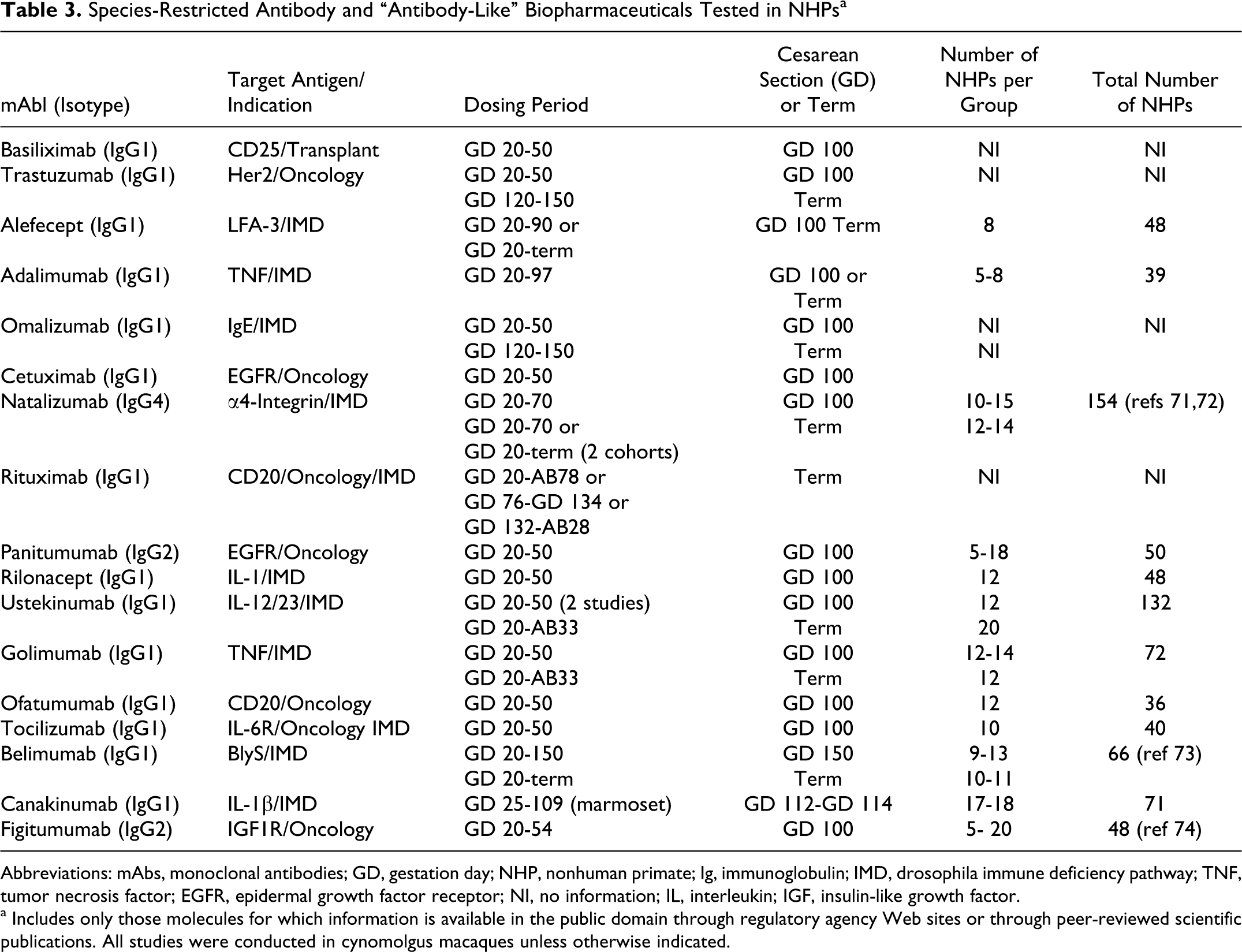
Abbreviations: mAbs, monoclonal antibodies; GD, gestation day; NHP, nonhuman primate; Ig, immunoglobulin; IMD, drosophila immune deficiency pathway; TNF, tumor necrosis factor; EGFR, epidermal growth factor receptor; NI, no information; IL, interleukin; IGF, insulin-like growth factor.
a Includes only those molecules for which information is available in the public domain through regulatory agency Web sites or through peer-reviewed scientific publications. All studies were conducted in cynomolgus macaques unless otherwise indicated.
Duration of Postnatal Dosing
When to start dosing in a macaque PPND study
Although dosing is initiated at the beginning of organogenesis in rodent PPND studies, it may not be necessary to initiate dosing at the beginning of organogenesis in NHPs. The NHP studies that have been conducted to support the approved biopharmaceuticals have initiated dosing anywhere from GD 20 to GD 120 (Table 3). The rationale for each of these study designs is not available in the publicly available information but is most likely based on the intended clinical use, the pharmacology of the biopharmaceutical, and the overall objective of the study.
If separate EFD and PPND studies are conducted in NHPs and the dosing in the EFD study was from GD 20 to GD 50, then dosing in a follow-on PPND study could potentially begin after GD 50 if there were no treatment-related effects in the EFD study. 66 The advantage of delaying dosing in a PPND study until after GD 50 is that the wave of early abortions will be avoided and fewer pregnant animals will be needed to obtain the desired number of infants versus initiating dosing on GD 20. In 138 female cynomolgus macaques with dosing initiated at GD 70, prenatal loss rate was below 10%. 19 This compared with 20% or greater when dosing is initiated on GD 20. One disadvantage of initiating dosing in the PPND study on the day that dosing is discontinued in the EFD study is that it may be possible that events that are initiated in the EFD study may not be realized until later in development and therefore there is a small risk that some information could be lost with this 2 study approach. However, to minimize this risk the EFD study could include more extensive evaluations than the standard gross examination and the dosing period in the EFD and PPND studies could overlap to minimize the possibility of missing critical information. For example, an expanded embryo/fetal development (eEFD) study could be conducted with dosing starting at GD 20 and ending somewhere between GD 100 and GD 150, and the PPND study could have dosing from GD 50 to birth (Figure 6). Having an adequate overlap in the dosing periods is also important to ensure that the fetus is exposed to high concentrations of the antibody during gestation. Many biopharmaceuticals have a long serum half-life and therefore require a number of doses before they attain steady state. This is particularly true when an extravascular route of administration is being used.
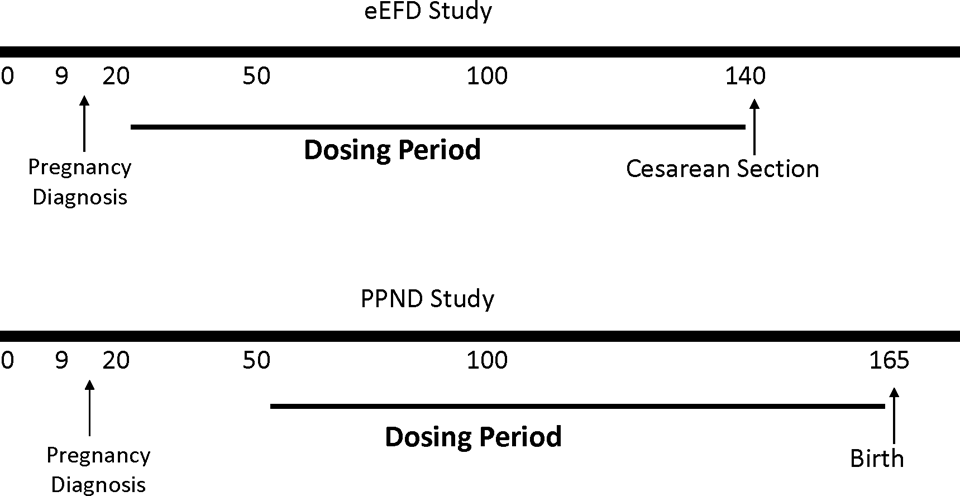
Option for evaluation effects on embryo/fetal development and pre- and postnatal development in separate studies eEFD, expanded embryo/fetal development; PPND, pre and postnatal development..
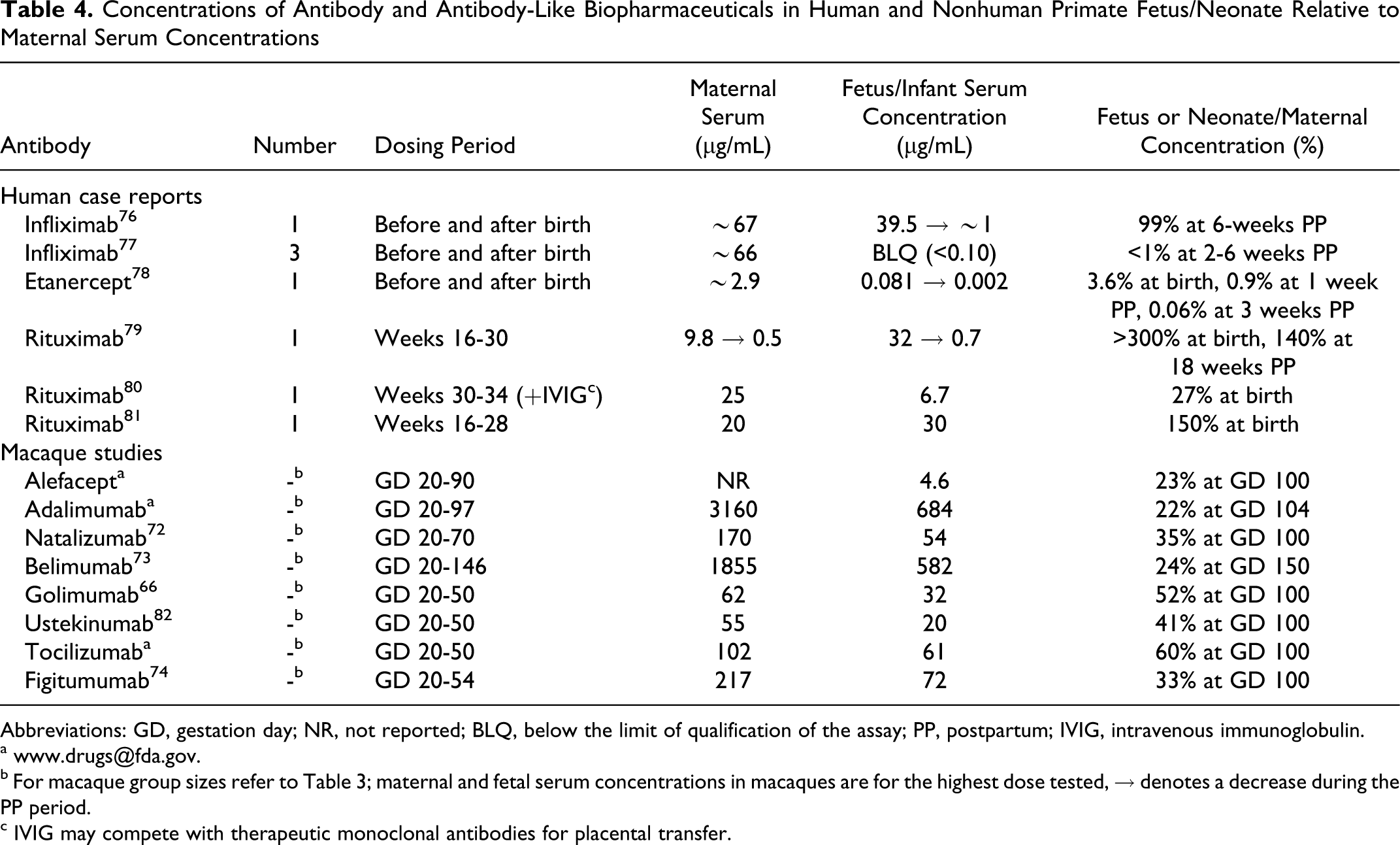
One major disadvantage of initiating dosing in the PPND study late in gestation is that placental transfer/equilibrium may not be complete within the time frame of the study. For example, when CD4-IgG1 was administered to pregnant rhesus macaques at GD 150 and GD 160, cord blood collected 24 hours after dose initiation showed fetal concentrations of CD4-IgG that were only 3% of the maternal concentrations. 75 This contrasts with the similar neonatal and maternal exposures obtained for IgG1 mAbs, when dosing is initiated early in pregnancy (Table 4 ). 66,71,73,82
Concentrations of Antibody and Antibody-Like Biopharmaceuticals in Human and Nonhuman Primate Fetus/Neonate Relative to Maternal Serum Concentrations
Abbreviations: GD, gestation day; NR, not reported; BLQ, below the limit of qualification of the assay; PP, postpartum; IVIG, intravenous immunoglobulin.
b For macaque group sizes refer to Table 3; maternal and fetal serum concentrations in macaques are for the highest dose tested, → denotes a decrease during the PP period.
c IVIG may compete with therapeutic monoclonal antibodies for placental transfer.
When to stop dosing in a PPND study
In the rodent ICH S5 (R2) PPND study design, the dams are dosed until weaning. The reason for this is that small molecule pharmaceuticals can be secreted in breast milk and once ingested can be absorbed across the gastrointestinal tract.
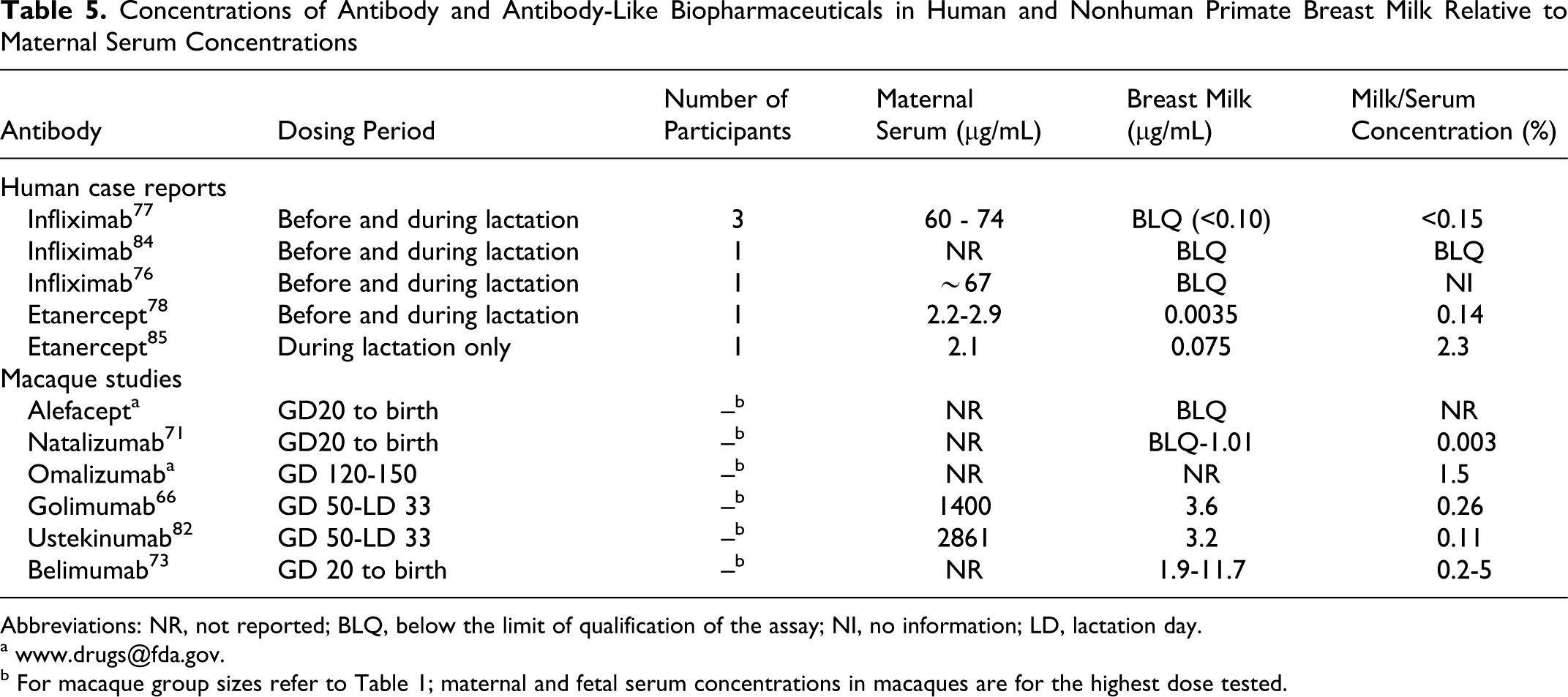
In humans, the predominant antibodies in breast milk are the secretory IgA antibodies that are generated locally within the mammary gland. 83 IgA in mammary secretions is not absorbed into the infant’s blood. It is resistant to proteolytic degradation and plays a local protective role in the lumen of the intestine. Immunoglobulin G antibodies are not resistant to proteolytic degradation. A few studies conducted in humans and macaques have shown low-level transfer of therapeutic IgG antibodies into breast milk (Table 5 ). However, the infants from mothers treated with therapeutic antibodies during pregnancy, and in some cases also during lactation, show a decrease in serum concentrations during the postpartum lactation period, 66,71,73,76,78,82 suggesting that the therapeutic antibodies present in the milk have little or no contribution to systemic exposure in the infants. This contrasts with rodents in which FcRn expressed in the neonatal gut transfers IgG molecules secreted in the rodent mammary secretions across the rodent gut. 86 This is the primary route for IgG acquisition in rodent neonates. 87,88 Although FcRn has been shown to be present in the gastrointestinal tract of human fetuses, 89 it appears that FcRn has little or no function on IgG transport across the gut of the infant primate.
Concentrations of Antibody and Antibody-Like Biopharmaceuticals in Human and Nonhuman Primate Breast Milk Relative to Maternal Serum Concentrations
Abbreviations: NR, not reported; BLQ, below the limit of qualification of the assay; NI, no information; LD, lactation day.
b For macaque group sizes refer to Table 1; maternal and fetal serum concentrations in macaques are for the highest dose tested.
Most biopharmaceutical mAbs have long serum persistence such that mAb remains detectable in the maternal serum for days to weeks after discontinuation of treatment. This may also result in the secretion of the mAb in the breast milk even when dosing is discontinued at parturition. The immediate postnatal period is a critical period in the bonding between mother and neonate in NHPs. Frequent disruption of this bonding process can potentially lead to rejection of the infant by the mother. With only a single infant per dam and a successful pregnancy rate of about 65%, the loss of a single infant could jeopardize the outcome of the study. For NHP studies, it is preferable to limit the amount of handling that occurs and the amount of maternal−infant separation to ensure a successful study outcome. Therefore, it is recommended that for NHP studies dosing of the dams be discontinued at term unless there is a specific reason to believe that the biopharmaceutical may have maternal effects that might alter breast milk production.
The Enhanced PPND Study Design
An enhanced PPND (ePPND) study design has recently been described 14 and is suggested as an acceptable option in the ICH S6 (R1) addendum. The ePPND study involves dosing of pregnant macaques from GD 20 to birth and evaluation of the infants postnatally for morphological and functional development. This study essentially combines all the components of the EFD and the PPND studies into a single study with the exception that fetal examinations are not included. Fetal examinations are an important component of rodent EFD studies because rodents tend to cannibalize malformed or nonviable fetuses, resulting in a loss of critical information. 90 Nonhuman primates do not cannibalize malformed or nonviable fetuses, and, therefore, there is no loss of information on potential teratogenicity if fetal examinations are excluded from NHP studies. The only potential information that could be lost is if there is a developmental delay during pregnancy that is not evident at birth. Slight developmental delays that occur in utero but that do not translate to abnormalities in the infants are unlikely to be of clinical concern. Therefore, there is not a strong rationale that fetal examinations are important for NHP studies. Fetal viability can be monitored throughout gestation by ultrasound examination. The ultrasound examination can also measure fetal growth and heart beat. However, measurement of in utero growth rate is not important if the infants are normal at birth.
Recommended end points for an ePPND study are offspring viability and survival, external malformations, skeletal effects (eg, by X-ray) and, ultimately, visceral morphology at necropsy. Histopathology, although not generally included in rodent developmental studies, can be included in NHP developmental studies to enhance the value of the study. Because there are likely to be more infants in an ePPND study than in a general toxicity study, it may not be practical or necessary to conduct histopathology on all tissues from all animals. Other end points in the offspring can also be evaluated if relevant for the pharmacological activity (eg, immune function or neurobehavioral assessment). The duration of the postnatal phase will be dependent on the additional end points considered relevant for the pharmacological activity and the clinical indication. Generally, a large array of parameters are available for postnatal infant evaluation comprising both general and specific parameters (Table 6 ). The duration of the postnatal observation phase to some extent is also dictated by the choice of parameters, for example minimal infant age is 6 to 9 months if a learning/memory test is required.
Parameters for Evaluation of Postnatal Development in the Cynomolgus Macaques

Abbreviations: TDAR, T cell-dependent antibody response; NK, natural killer cell; DTH, delayed-type hypersensitivity test; DEXA, dual-energy X-ray absorptiometry; CT, computed tomography; NHP, nonhuman primate; ECG, electrocardiography; TK, toxicokinetics.
a This test is problematic in NHP. In case of continued maternal dosing, milk can be collected for further analysis.
In the ePPND study design, it is essential that a sufficient number of infants are available for postnatal examination. Because of the number of early and late pregnancy loses that occur in an ePPND study, a larger number of pregnant females need to be initiated on study versus a PPND study in which dosing is initiated after the period of major organogenesis. However, because the EFD study is eliminated, the overall number of animals used for the ePPND assessment is fewer than when separate EFD and PPND studies are conducted.
Group Size Considerations for NHP Developmental Toxicity Studies
An analysis performed to explore the influence of initial group size on the potential for data interpretation in macaque studies using the end points of embryo/fetal survival and infant survival (postpartum day 7) indicated that average survival rate until postnatal day 7 was 65%. 69
It was observed that EFD studies with initial control group sizes of 16 and 20 have an 80% probability of having 13 and 16 ongoing pregnancies at GD 100, respectively. For ePPND studies with initial control group sizes of 16, 20, or 28, there is an 80% likelihood of having 9, 11, or 16 infants at day 7 postpartum, respectively. An ePPND study initiated with a group size of 20 could detect a 3-fold increase of test item-related pregnancy or infant loss at 81% statistical probability. For designing and managing primate developmental toxicity studies, this type of analysis provides an objective tool to facilitate decisions by either supplementing groups with additional pregnant animals or stopping a group because an adverse effect on offspring survival has already been adequately revealed.
Importantly, the analysis is confined to embryo/fetal survival and infant survival until day 7 of age and does not address other pivotal developmental toxicity parameters for example morphogenesis, growth, fetal, and infant function. These data provide an objective approach that facilitates the analysis and interpretation of pre- and postnatal loss of data in macaque developmental toxicity studies and will aid in rationalizing adequate group sizes and study designs. Such options are also in the interest of animal welfare. An important question that remains to be considered is an agreement on the statistical power demanded for an NHP developmental toxicity study because this will determine the required group size.
Advantages and Disadvantages of the ePPND Study Versus Separate EFD and PPND Studies
The current version of ICH M3 guidance document states that “monoclonal antibodies for which embryo/fetal exposure during organogenesis is understood to be low in humans based on current scientific knowledge, the developmental toxicity studies can be conducted during Phase III. The completed reports should be submitted with the marketing application.” The reason for this exception for mAbs is that most mAbs can only be tested in NHPs and the risk of inducing a teratogenic effect due to embryonic exposure that could not be predicted from the known pharmacology of the molecule is low. Because only a small number of women of childbearing potential are included in phases I and II clinical trials, extensive precautions are taken to avoid pregnancy, and because an NHP EFD study provides only very limited information on the effects on pregnancy and morphological development it is appropriate that this study be eliminated in favor of a more extensive ePPND evaluation, once proof of concept has been established clinically. The main issue is for exposure during the fetal period, that is, following the period of major organogenesis. Exposure during the fetal period has traditionally only been evaluated as part of a PPND study which is completed prior to product registration. Because of the ethical consideration for the use of NHPs in research, the cost of the studies, and the duration of a PPND or ePPND study (1-3 years), it is desirable that clinical proof of concept is obtained before the NHP study is initiated. This recommendation is no different from other types of therapeutics in which the PPND evaluation is conducted during phase III.
During the course of clinical development, patients can be informed of potential risks based on information in the scientific literature or information on products with a similar pharmacological profile. Information generated from genetically modified rodents may be very helpful in understanding potential hazards that could occur during development. The disadvantage of the ePPND study approach is that no product-specific information is available during the course of development and a large study needs to be committed to with no prior experience with the molecule in pregnant animals.
According to the current ICHS6 (R1) addendum, the ePPND study design can also be considered for non-antibody biopharmaceuticals. However, if the biopharmaceutical is expected to cross the placenta, the study should be initiated prior to phase III with an interim report being available prior to phase III clinical studies.
The main advantage of the ePPND study is that all stages of development can be evaluated in a single study versus 2 separate studies. This may reduce the total number of NHPs needed for the evaluation of development. However, because there will be early and late pregnancy losses, this needs to be taken into consideration when determining the number of animals that will be needed. Based on Monte Carlo simulations of pre- and postnatal loss of data until postpartum day 7, for an initial vehicle group size of 20 the most likely outcomes were 11 to 15 live infants at day 7, with 90% of the simulations yielding 10 to 16 live infants at day 7. 69 It is, therefore, recommended that group sizes of at least 20 pregnant animals per group be used for an ePPND study.
Even though the ePPND study has many advantages, there may be instances in which it is desirable to have some information on product-specific potential developmental effects prior to phase III. One situation may be when information in genetically deficient animals suggests that absence of the target pathway results in embryo/fetal toxicity or embyo/fetal lethality. Although the informed consent document for such an agent will provide strong warnings against becoming pregnant while on treatment and women would be required to take precautions against becoming pregnant, excessive precautionary statements in the absence of product-specific information may deter women of childbearing potential from participating in clinical trials and may discourage investigators from recruiting women of childbearing potential into clinical trials. Although a developmental study in NHPs with the product showing no abnormalities will not eliminate the risk, it will provide information that is useful to both patients and clinicians, should an inadvertent pregnancy occur during a clinical trial.
Because of the large commitment in terms of number of animals and costs of conducting an ePPND study to support clinical trials, it may be desirable to obtain some preliminary information on NHPs prior to making such a large commitment. A small-scale expanded EFD study could be considered to identify any obvious treatment-related adverse effects. Because this study is not intended to detect subtle changes and will be followed by a more extensive PPND study later in development, the study could be conducted with a small number of animals per group and as few as 2 groups. The advantage of conducting such a small-scale study is not only that it may identify any major hazards but also that any information obtained from this study can be used to help design the follow-on PPND study. If no obvious treatment effects are observed in an eEFD study with dosing from GD 20 to GD 140 (avoids late pregnancy losses), then a PPND study could be conducted with dosing from GD 50 to term. Because this PPND study design avoids the early pregnancy losses, it could be conducted with fewer animals per group, when compared with an ePPND study. If a potential adverse effect is observed in the eEFD study, then the more extensive ePPND study may be warranted. The decision on whether to conduct a single study versus a split study design, therefore, should be based on the significance of the potential theoretical risks.
Future Directions
The developmental studies conducted to date in NHPs for biopharmaceuticals have been modeled mostly on the study designs outlined in the ICHS5 (R2) guidance document. The study designs outlined in the guidance document were proposed with a primary focus on rodent and rabbit studies for small molecule drugs. What is clear from the experience that has been gained with biopharmaceuticals tested in NHP development studies is that a standard approach cannot be applied to all biopharmaceuticals and that the testing strategy needs to be adapted for the type of molecule, the pharmacology of the molecule, and the timing of potential fetal exposure relative to organ development.
Historically mAb therapies were limited to the treatment of serious life-threatening diseases such as oncology or organ transplant. For those indications, nonclinical evaluations of potential effects on development were not conducted or an EFD study was conducted in which dosing was limited to the embryonic period (per ICH S5). More recently indications have expanded into less severe patient populations, some of which require chronic therapy and include women of childbearing potential. To support clinical trials and registration of these agents, an increasing number of NHP studies have been required and the scope of these studies has increased. What is clear from NHP studies is that if a greater amount of information is obtained from each animal, then fewer animals will be needed overall to obtain the necessary information to inform patients of potential adverse effects on pregnancy and on development. This objective can be met by combining studies into a single study and increasing the number of in-life and postnatal evaluations. Histopathology may be included in NHP developmental studies to enhance the value of the study. The recently proposed ePPND study provides an option for combining studies into a single study and reducing the overall NHP numbers while still obtaining relevant information to inform patient of potential risks. The optimization of NHP studies is an evolving area that is likely to change as more biopharmaceutical are tested in NHPs and more information is gathered that will aid in the understanding of potential risks of biopharmaceutical exposure during pregnancy and during postnatal development.
At the time of this writing, the ICH S6 regulatory guidance document was undergoing an addendum and was at step 3 in the addendum process. It is anticipated that a number of changes will be made to this addendum prior to finalization, which may affect the future directions of developmental toxicity testing for biopharmaceuticals in NHPs.
Conclusion
Developmental toxicity studies in NHPs need to be designed on a case-by-case basis based on the intended clinical use, the type of molecule, the pharmacology of the molecule, and the potential exposure of the fetus relative to the development of potential target organs of toxicity. To support chronic clinical indications, a single well-designed study that evaluates all stages of development, and focuses on potential target organs, may be the most appropriate design in terms of providing clinically relevant information as well as from an animal usage perspective.
Footnotes
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) received no financial support for the research and/or authorship of this article.